
Abstract
Glucocorticoids such as dexamethasone (DEX) are commonly used clinically but can induce skeletal muscle atrophy with prolonged or high-dose exposure. Trichosanthes kirilowii Maximowicz (TK), a traditional medicinal herb, has known pharmacological effects, but its impact on muscle atrophy remains unclear. This study investigated the anti-atrophic potential of TK in DEX-induced muscle atrophy models. In C2C12 cells, TK improved cell viability and restored myotube diameter and number, downregulated atrophy markers MuRF-1 and Atrogin-1, and upregulated myogenic markers MyoD, MyoG, and MHC. TK also restored Sirtuin 1 (SIRT1) expression suppressed by DEX, and inhibition of SIRT1 with EX527 reduced TK’s beneficial effects, indicating a SIRT1–dependent mechanism. TK rescued DEX-induced reductions in phosphorylated AKT and mTOR, supporting its role in muscle anabolism. In vivo, oral TK administration attenuated DEX-induced weight loss, preserved gastrocnemius muscle mass, and improved motor performance. Molecular docking analysis revealed that key TK-derived compounds, including 5-dehydrokarounidiol, steryl glucoside, and isomultiflorenol, exhibited moderate binding affinity (− 6.3 to − 6.6 kcal/mol) toward the allosteric site of SIRT1. These findings suggest that TK protects against muscle atrophy by modulating SIRT1–related anabolic and catabolic pathways. TK may offer therapeutic potential for sarcopenia and glucocorticoid-induced muscle wasting.
Similar content being viewed by others


Introduction
Skeletal muscle plays a vital role in locomotion, metabolism, and overall physical function1. Loss of muscle mass and strength, regardless of its cause, significantly impairs quality of life and increases the risk of disability, metabolic disorders, and mortality2,3. Among the various causes of muscle atrophy, prolonged exposure to glucocorticoids, whether exogenously administered as pharmacological agents or endogenously elevated due to chronic stress or disease, has been identified as a major contributor to muscle wasting. Glucocorticoid-induced muscle atrophy is characterized by increased protein degradation and decreased protein synthesis, resulting in a negative protein balance4,5. Clinical studies have shown that patients undergoing long-term glucocorticoid therapy experience significant declines in mobility and muscle function, along with increased frailty and metabolic complications6,7. In addition, conditions associated with endogenous hypercortisolemia, such as Cushing’s syndrome, are linked to severe muscle loss and reduced physical performance8. These findings highlight the need for effective therapeutic strategies to prevent or reverse glucocorticoid-induced muscle wasting.
At the molecular level, dexamethasone (DEX), a potent synthetic glucocorticoid, promotes muscle catabolism by activating the ubiquitin–proteasome pathway. Specifically, DEX upregulates muscle-specific E3 ubiquitin ligases such as muscle RING finger-1 (MuRF-1) and atrogin-1, which are central regulators of proteolysis. Simultaneously, DEX suppresses anabolic and myogenic signals by downregulating myosin heavy chain (MHC) and myogenic differentiation factor (MyoD), further impairing muscle integrity and regeneration.
Recent studies have drawn attention to the role of Sirtuin 1 (SIRT1), a NAD⁺-dependent deacetylase, in the regulation of muscle homeostasis9,10. SIRT1 has been shown to exert protective effects against muscle atrophy by modulating oxidative stress responses, mitochondrial biogenesis, and inflammation. In C2C12 myoblasts, SIRT1 enhances proliferation and inhibits apoptosis, in part through the AKT-Nrf2 signaling and Wnt pathways, underscoring its therapeutic potential in muscle-wasting conditions11,12.
Trichosanthes kirilowii Maximowicz (TK) is a traditional herbal medicine used in East Asia, primarily for treating respiratory disorders13. Its seeds are rich in pharmacologically active constituents, including flavonoids, triterpenoids, and ribosome-inactivating proteins, which have demonstrated anti-inflammatory, anticancer, immunomodulatory, and antioxidant effects14,15,16,17. However, the potential of TK in the context of muscle atrophy has not yet been investigated.
In this study, we investigated the protective effects of TK against DEX-induced muscle atrophy using both in vitro and in vivo mouse models. Furthermore, we explored whether the muscle-preserving effects of TK are mediated through a SIRT1–dependent mechanism. By combining cellular and animal-level approaches, this study aimed to elucidate the potential of TK as a therapeutic candidate for glucocorticoid-induced muscle wasting and to clarify its underlying molecular mechanisms.
Materials and methods
Mouse C2C12 cells and differentiation
We seeded mouse C2C12 muscle myoblasts (ATCC, Gaithersburg, USA) on various cell culture plates (i.e., 6-, 24-, and 96-wells) and 100 mm2 dishes according to the experimental purpose. We cultured the cells in proliferation media (DMEM + 10% fetal bovine serum + 1% penicillin/streptomycin) using 2% horse serum prior to differentiation. We performed media change or subculture once every two days and differentiation for six days. Figure 1 A presents the experimental timeline.
TK extraction and DEX treatments
Dried Trichosanthes kirilowii (TK) seeds were procured from Green M.P. Pharmaceutical Co., Ltd. (Sungnam, Republic of Korea). The product is officially registered with the Korean Ministry of Food and Drug Safety (MFDS) under item standard code 200,906,696, ensuring product traceability and batch consistency. The same batch was used for all experiments in this study.
For extraction, 30 g of dried TK seeds were boiled in 300 mL of distilled water for 3 h at 105°C using a heating mantle (Misung Scientific Co. Ltd., Gyeonggido, Republic of Korea). After boiling, the mixture was filtered through filter paper (Hyundai Micro, Seoul, Republic of Korea) to remove particulate matter. The filtrate was then lyophilized and stored at − 80°C until use. Next, we froze the resulting filtrate at −70°C and subsequently lyophilized it using a freeze dryer (Ilshin BioBase Co., Ltd., Gyeonggido, Republic of Korea) to obtain the dry extract in a powdered form. We prepared 10- and 20-mg/mL TK stocks in phosphate buffered saline (PBS). After stabilization and differentiation, we treated the C2C12 cells with various TK concentrations, 100 µM DEX (Sigma-Aldrich, St. Louis, USA) alone, or a mixed of TK and DEX for 24 h.
Cell viability assay
Cell viability was evaluated using a CCK-8 assay (Dojindo, Kumamoto, Japan) in both undifferentiated and differentiated C2C12 cells. We initially seeded the C2C12 cells onto 96-well plates containing culture medium and subsequently treated them with varying TK concentrations ranging from 1 to 400 µg/mL. After a 24 h incubation period, we added the CCK-8 solution, prepared by mixing with a 10% culture medium, to each well. Next, we incubated the plates for an additional 4 h, and measured the optical density (OD) at 450 nm using an Epoch microplate reader (BioTek, Winooski, VT, USA). We calculated the cell viability by comparing the OD of each sample with that of the control using the following formula: Cell viability (%) = (OD of each sample ÷ Mean OD of blank group) × 100.
Live–dead and TUNEL assays
To visualize apoptosis, we imaged the cells using a confocal microscope following fluorescent staining. Live–dead assay was performed on undifferentiated C2C12 myoblasts using a Live & Dead Cell Imaging Kit (Thermo Fisher Scientific, Massachusetts, USA) according to the manufacturer’s protocol. In contrast, TUNEL assays were conducted on differentiated C2C12 myotubes using a TUNEL Assay Kit (Abcam, Cambridge, UK) in accordance with the manufacturer’s instructions. After staining, C2C12 cells were observed under a confocal microscope (Nikon, Tokyo, Japan) at 100× magnification. Live and dead cells were quantified by counting the number of green- and red-stained cells, respectively, per microscopic field using ImageJ software (version 1.37, National Institutes of Health, Bethesda, MD, USA). The results were expressed as the number of live cells/field and dead cells/field at 100× magnification.
Flow cytometry
To analyze the percentage of apoptotic cells, we performed flow cytometry on undifferentiated C2C12 myoblasts using the APC Annexin V Apoptosis Detection Kit with propidium iodide (PI; BioLegend, San Diego, CA, USA), according to the manufacturer’s instructions. After two washes, we separated the cells from the plate using 0.025% trypsin. Next, we stained the cells with Annexin V and PI in a binding buffer at 25°±1℃ in the dark. After 15 min, we analyzed the stained cells using an Accuri™ C6 Plus Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and carried out data processing using the FlowJo software (BD Biosciences).
Real-time PCR
Total RNA was extracted from differentiated C2C12 myotubes in each culture well using TRIzol reagent (Thermo Fisher Scientific, MA, USA), following the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized from the extracted RNA using AccuPower RT PreMix and oligo(dT) primers (Bioneer, Daejeon, Republic of Korea). Quantitative real-time PCR was then performed using iQ™ SYBR® Green Supermix (Bio-Rad) on a CFX Connect Real-Time PCR Detection System (Bio-Rad), with gene-specific primers and synthesized cDNA. Relative gene expression levels were calculated using the ΔΔCt method, with GAPDH as the internal control. The primer sequences used in this study are listed in Table 1.
Western blotting
Western blot analysis was performed using differentiated C2C12 myotubes. Following treatment, the cells were homogenized in RIPA buffer (Gen-DEPOT, Barker, TX, USA) supplemented with protease and phosphatase inhibitors (Millipore, Burlington, MA, USA). Protein concentrations in the lysates were determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific), following the manufacturer’s instructions. Equal amounts of protein were separated by electrophoresis on 10–12% SDS–polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked with 5% Difco™ Skim Milk (BD Biosciences) in Tris-buffered saline (Bio-Rad, San Diego, CA, USA) containing 0.1% Tween 20 (Sigma-Aldrich) and incubated overnight at 4 °C with the appropriate primary antibodies. After three washes, the membranes were incubated with HRP-conjugated secondary antibodies for 2 h at 4 °C. Protein bands were detected using an enhanced chemiluminescence (ECL) system (Bio-Rad) and visualized using a charge-coupled device (CCD) imager (Amersham Imager 600; GE Healthcare Life Sciences, Uppsala, Sweden). Detailed information on the primary and secondary antibodies is provided in Table 2.
Immunocytochemistry
Immunocytochemistry was performed using differentiated C2C12 myotubes. Cells were fixed with 4% paraformaldehyde at 25 °C and stored in PBS at 4 °C. For fluorescent staining, the cells were permeabilized with 0.2% Triton X-100. After two washes, the cells were incubated with 2% (v/v) normal goat serum for 1 h at room temperature to block nonspecific binding. Subsequently, the cells were incubated overnight at 4 °C with primary antibodies. After primary antibody incubation, the cells were treated with fluorescently labeled secondary antibodies (Jackson ImmunoResearch, Pennsylvania, USA) for 2 h at 25 °C. The antibodies employed, including both primary and secondary, are detailed in Table 3. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and the samples were imaged using a confocal microscope (Nikon, Tokyo, Japan). Fluorescence intensity was quantified using ImageJ software (NIH, Frederick, MD, USA).
Dexamethasone-induced muscle atrophy mice model
Eight-week-old male C57BL/6 mice maintained in a specific pathogen-free conditions (Samtako Bio, Osan, Republic of Korea) were used for the experiment. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Jaseng Animal Care and Use Committee (Approval No. JSR-2024-12−001-A). The experiments were conducted in accordance with the ARRIVE guidelines and with applicable institutional and national regulations. Mice were randomly divided into five groups (n = 6/group). (1) Sham group, which received an intraperitoneal (i.p.) injection of physiological saline containing 20% sulfobutyl ether-β-cyclodextrin (SBE-β-CD) and 5% dimethyl sulfoxide (DMSO, Sigma-Aldrich); (2) Control group, which received DEX (Sigma-Aldrich, 25 mg/kg, i.p.) once daily; (3–5) TK groups, which received DEX (25 mg/kg, i.p.) along with oral administration of TK at doses of 50, 100, or 200 mg/kg, respectively. Both TK and DEX were administered once daily for 14 consecutive days. Body weight was monitored every other day throughout the experimental period. At Day 14, mice were anesthetized with 2–3% isoflurane (JW Pharmaceutical, Gwacheon-si, Republic of Korea) gas and perfused through the cardiovascular system with 0.9% saline followed by 4% paraformaldehyde (Biosesang, Yongin, Republic of Korea). Mice that remained alive after perfusion were euthanized by carbon dioxide asphyxiation, and the right gastrocnemius muscle (GCM) was immediately harvested and weighed using an electronic balance.
Hematoxylin & eosin (H&E) staining
After weighing, the right GCM was immediately immersed in 4% paraformaldehyde (Biosesang, Korea) at room temperature for 48 h for fixation. Fixed tissues were then processed through a graded ethanol series, cleared in xylene, and embedded in paraffin. Paraffin blocks were sectioned using a HistoCore MULTICUT microtome (LEICA, Wetzlar, Germany) at a thickness of 5 μm. Sections were floated on a 40 °C water bath for 2–3 min, mounted onto coated slides, and dried on a hotplate at 37°C. For histological assessment of muscle morphology and quantification of fiber cross-sectional area (CSA), sections were stained with hematoxylin and eosin (H&E). Briefly, slides were immersed in hematoxylin for 2 min 30 s, rinsed under running tap water for 2 min, and counterstained with eosin for 50 s. Subsequently, slides were dehydrated through graded ethanol (70–100%), cleared in xylene, and coverslipped. Stained sections were examined using a Nikon bright-field microscope at ×200 magnification for general morphology and at ×400 magnification for quantitative analysis. Three non-overlapping fields were randomly captured from the central region of the transverse section of each muscle at ×400 magnification. This region, corresponding to the mid-belly of the GCM, was selected to minimize variability across samples. The CSA of individual muscle fibers was measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA) after scale calibration. Only intact fibers with clearly defined borders were included in the analysis. A total of approximately 200 fibers per group (n = 6) were evaluated. The mean fiber CSA (µm²) was calculated for each mouse and used as the unit of statistical analysis.
Treadmill exhaustion test
A treadmill exhaustion test was conducted as previously described with slight modifications18. Prior to testing, mice underwent a 3-day acclimation period during which they were individually placed on a motorized treadmill (Rodent Treadmill NG, Ugo Basile, Varese, Italy) and allowed to run at a speed of 10 m/min on a 0° incline for 5 min per day. During acclimation, the shock grid was activated to encourage consistent running behavior. For the exhaustion test, the treadmill incline was set at 10°, and the initial running speed was 10 m/min. The speed was increased by 2 m/min every 2 min until reaching a maximum of 46 m/min. Mice were placed individually on the treadmill lanes, and the test was initiated with the activation of both the running belt and the electric stimulus grid.
Molecular docking analysis
Molecular docking was performed to evaluate the potential binding affinity between SIRT1 and TK-derived compounds; a total of 104 compounds were collected from the literature19,20,21,22,23,24, and 96 ligands with available 3D conformer structures in PubChem validated by CAS numbers where necessary were selected for docking analysis. The crystal structure of human SIRT1 was obtained from the RCSB Protein Data Bank (PDB ID: 4ZZJ), which contains the SIRT1/activator/substrate complex. To isolate the target protein for docking, the activator molecule ((3 S)−1,3-dimethyl-N-[3-(1,3-oxazol-5-yl)phenyl]−6-[3-(trifluoromethyl)phenyl]−2,3-dihydropyrido[2,3-b]pyrazine-4(1 H)-carboxamide, abbreviated as 4TQ) and the substrate p53 were removed using PyMOL (https://pymol.org/2/), resulting in a clean SIRT1 protein structure used as the receptor.
Candidate compounds were selected based on previously published references, and only those with available 3D conformer files from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) were included in the analysis. In cases where compound names differed across sources, CAS numbers were cross-referenced to ensure accurate compound identification. Ligand and receptor structures were prepared for docking by adding polar hydrogen atoms, assigning Kollman charges to the receptor, and converting the files to PDBQT format using AutoDockTools. Water molecules were removed prior to docking. Targeted docking was performed by focusing on the known allosteric activator site of SIRT1. The grid box was defined based on the 4TQ binding site observed in the 4ZZJ crystal structure, with the center coordinates set at X: −0.558, Y: 44.823, Z: −0.327, and a box size of 30 × 30 × 30 Å. This region corresponds to the known allosteric activator site of SIRT1 and was confirmed to be spatially distant from the p53 substrate-binding site, minimizing the likelihood of inhibitory interactions. Docking simulations were performed using AutoDock Vina (version 1.1.2), and binding affinities (kcal/mol) were recorded for each compound. Docking poses were visualized and analyzed using PyMOL, and a schematic diagram of ligand–receptor interactions was constructed to illustrate key binding residues.
Statistical analyses
We statistically analyzed the data and expressed them as the mean ± SD using the GraphPad Prism program (Graphpad Software Inc, San Diego, CA, USA). We performed group comparisons using one-way ANOVA, followed by Tukey’s post hoc test to identify significant differences. In the analysis of body weight changes over time, a two-way repeated-measures ANOVA (factors: time × treatment) was performed to account for within-subject variability, followed by Tukey’s multiple-comparison test. A p-value < 0.05 was considered statistically significant.
Results
TK enhances cell viability while alleviating DEX-induced cytotoxicity and apoptosis in C2C12 cells
To evaluate the protective effect of TK in differentiated C2C12, we first established an in vitro model of DEX-induced muscle atrophy using differentiated C2C12 myotubes (Fig. 1A). We first evaluated its effects on C2C12 cell proliferation and survival under both normal and DEX-induced muscle atrophy conditions (Fig. 1B, C). As a prerequisite to modeling glucocorticoid-induced muscle atrophy, we treated differentiated C2C12 myotubes with increasing concentrations of DEX (10–200 µM). As shown in Supplementary Fig. S1, DEX treatment resulted in a dose-dependent decrease in cell viability, with statistically significant cytotoxicity observed from 10 µM. Among the tested concentrations, 100 µM DEX consistently induced moderate but reproducible cytotoxic stress without causing excessive cell death, making it an appropriate concentration for subsequent experiments. This choice is also supported by previous studies that used similar DEX concentrations to induce muscle atrophy in vitro25.
Following this, we conducted a CCK-8 assay to assess the protective effects of TK in C2C12 myoblasts, both in the presence and absence of DEX-induced stress (Fig. 1B). Starting at a concentration of 50 µg/mL, TK treatment alone significantly enhanced C2C12 myoblast viability with maintained protective effect even in the presence of DEX. Under differentiated conditions, 200- and 400-µg/mL TK treatments alone significantly increased cell viability. When co-treatment with DEX, TK exhibited a protective effect against DEX-induced cytotoxicity, with a significant increase in cell viability observed from 50 µg/mL (Fig. 1C). Based on the cell viability assay results, TK concentrations of 100, 200, and 400 µg/mL were selected as optimal for subsequent experiments due to their consistent cytoprotective effects. To further validate the protective efficacy and assess the potential cytotoxicity of these selected doses, we additionally performed Live/Dead staining assay (Fig. 1D). DEX-treated cells exhibited significantly increased cell death compared to the BLK group, indicated by a reduced number of live cells and an increased number of dead cells per field (Fig. 1E, F). Co-treatment with TK (100, 200, and 400 µg/mL) demonstrated a dose-dependent protective effect: the number of live cells increased and dead cells decreased accordingly. Notably, in the TK200 and TK400 groups, we observed a significant reduction in DEX-induced cell death, highlighting the protective role of TK in preserving cell viability under stress conditions.
We next evaluated whether TK could prevent apoptosis in DEX-treated myotubes using Annexin V/PI and TUNEL staining. To determine whether the cell death was due to apoptosis or necrosis, Annexin V/PI staining followed by flow cytometry was performed. DEX treatment significantly increased the proportion of Annexin V-positive cells, indicating an increase in early apoptotic populations. This increase was significantly reduced by TK treatment in a concentration-dependent manner (Fig. 1G, H). Next, TUNEL staining was performed to visualize apoptosis of C2C12 myotubes. DEX treatment significantly increased TUNEL-positive staining, indicating elevated apoptosis in C2C12 myotubes (Fig. 1I, J). However, TK treatment effectively reduced TUNEL-positive myotubes in dose-dependent manners, suggesting that TK mitigates DEX-induced DNA fragmentation and protects against apoptotic cell death.
TK regulates DEX-induced muscle atrophy by regulating atrophy- and differentiation-related markers in C2C12 myotubes
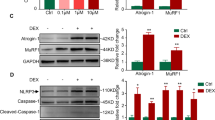
To investigate how TK affects muscle atrophy markers, we analyzed MuRF-1 and Atrogin-1 expression both at the mRNA and protein levels in DEX-treated atrophic C2C12 myotubes using real-time PCR and Western blotting. Real-time PCR results revealed that DEX treatment significantly increased MuRF-1 and Atrogin-1 mRNA expressions, confirming muscle atrophy induction (Fig. 2A, B). However, TK co-treatment led to statistically significant and dose-dependent MuRF-1 and Atrogin-1 mRNA level reductions at all tested concentrations. At the protein level, our western blot analysis revealed that MuRF-1 and Atrogin-1 expression was significantly upregulated upon the DEX treatment (Fig. 2C-E). While TK 100 µg/mL treatment exhibited a trend toward reduced expression, variability among samples resulted in no statistically significant difference at this concentration. However, TK treatment at 200 and 400 µg/mL significantly suppressed MuRF-1 and Atrogin-1 protein expressions, indicating dose-dependent atrophy-related protein accumulation inhibition. Next, we analyzed MHC, MyoD, and MyoG expression levels, key muscle differentiation markers, using western blot analysis (Fig. 2F–I). Western blot results showed that DEX treatment significantly reduced the expression of MHC, MyoD, and MyoG, confirming its inhibitory effect on muscle differentiation. MHC and MyoD expression were not significantly restored by TK at 100 µg/mL, but were significantly increased at 200 and 400 µg/mL. In contrast, MyoG expression showed a significant, dose-dependent increase starting from the lowest TK concentration (100 µg/mL). To further elucidate the anabolic mechanisms underlying the protective effects of TK, we analyzed the phosphorylation levels of key signaling molecules involved in muscle cell survival and growth, namely AKT and mTOR. Western blot analysis showed that DEX treatment significantly reduced the phosphorylation of both AKT and mTOR, indicating suppression of anabolic signaling pathways (Fig. 2J-L). Notably, p-AKT levels were not significantly altered by TK at 100 µg/mL, but showed a significant increase at 200 and 400 µg/mL compared to the DEX group. In contrast, p-mTOR levels were significantly increased in a dose-dependent manner starting from 100 µg/mL, indicating that p-mTOR elevation by TK occurs at lower doses than p-AKT under these conditions. These findings suggest that TK co-treatment restores p-AKT and p-mTOR phosphorylation, implicating the AKT/mTOR pathway in its anti-atrophic effects.
TK restores SIRT1 expression and prevents DEX-induced myotube atrophy in a SIRT1-dependent manner
To determine whether the anti-atrophic effects of TK are mediated through the SIRT1 modulation, we referred to previous studies that utilized EX527 to evaluate SIRT1–dependent mechanisms26. In parallel, we also sought to verify whether EX527 selectively inhibits SIRT1 under our experimental conditions. C2C12 myotubes were treated with EX527 at concentrations of 0, 10, 25, 50, and 100 µM for 24 h, followed by western blot analysis to assess the protein expression levels of SIRT1, SIRT2, and SIRT3 (Supplementary Fig. S2). The results showed a clear dose-dependent reduction in SIRT1 protein levels, whereas SIRT2 and SIRT3 did not exhibit consistent or dose-dependent changes across the same concentration range. These findings indicate that, under our experimental conditions, EX527 exerts a more selective inhibitory effect on SIRT1.
We first assessed C2C12 cell viability in the presence and absence of EX527. TK treatment significantly enhanced the viability of DEX-treated undifferentiated C2C12 cells (Fig. 3A). However, when SIRT1 was inhibited by EX527, TK failed to restore cell viability, indicating that its cytoprotective effect relies on SIRT1 signaling. We observed a similar trend in differentiated C2C12 myotubes, where TK significantly improved cell survival in DEX-treated myotubes (Fig. 3B). However, upon EX527 treatment, we could not observe the dose-dependent protective effect of TK.
We examined SIRT1 expression levels in DEX-treated C2C12 myotubes using ICC assays. Our confocal images revealed that SIRT1 predominantly localized in the nuclei of C2C12 myotubes under healthy conditions. However, its expression was markedly downregulated upon DEX treatment. Notably, co-treatment with TK dose-dependently restored SIRT1 expression (Fig. 3C, D). Our quantitative fluorescence analysis confirmed a significantly reduced SIRT1 expression in the DEX-treated group compared to the BLK group, while TK treatment led to a dose-dependent and statistically significant increase in SIRT1 levels. To further confirm that the protective effects of TK are more closely associated with SIRT1 involvement, we performed additional immunocytochemical staining for SIRT2 and SIRT3 (Supplementary Fig. S3). The results showed that SIRT2 expression was not significantly altered by DEX or TK treatment, and although SIRT3 expression was reduced by DEX, TK administration did not significantly restore it. These findings suggest that the protective effects of TK are more strongly linked to SIRT1 regulation than to SIRT2 or SIRT3.
However, when treated with EX527, overall SIRT1 expression levels were markedly reduced, and the concentration-dependent increase observed with TK treatment under normal conditions was attenuated. While the difference between TK100 and TK200 was no longer significant, TK400 still showed a significant increase in SIRT1 expression compared to the DEX-only group.
In addition, immunofluorescence imaging was used to quantitatively analyze myotube morphology by measuring the average diameter and total number of myotubes under both EX527–treated and untreated conditions (Fig. 3E, F). DEX treatment significantly reduced both myotube diameter and number, indicating the induction of muscle atrophy. Co-treatment with TK dose-dependently reversed these changes, restoring myotube thickness and number. However, the protective effects of TK were significantly attenuated in the presence of EX527. Under SIRT1 inhibition, TK treatment did not produce statistically significant improvements in either myotube diameter or myotube number compared to the DEX-only group. Specifically, the increase in myotube diameter observed with TK100 was no longer significant under EX527 treatment, and significant improvements were observed only at TK200 and TK400. In the case of myotube number, none of the TK-treated groups showed statistically significant differences from the DEX group.
To further support the ICC findings, we analyzed SIRT1 protein expression using western blotting (Fig. 3G, H). SIRT1 levels were markedly decreased following DEX treatment, whereas TK administration restored SIRT1 protein expression in a dose-dependent manner. However, under EX527 co-treatment, the TK-induced upregulation of SIRT1 was significantly suppressed. Notably, a statistically significant increase in SIRT1 expression was observed only in the TK400 group, suggesting that EX527 markedly interfered with SIRT1 restoration at lower TK doses.
TK exhibits weakened anti-atrophic and myogenic effects under EX527 treatment
To elucidate the role of SIRT1 in mediating the anti-atrophic and myogenic effects of TK, we examined the expression of muscle atrophy-related markers (MuRF-1 and Atrogin-1), myogenic differentiation markers (MyoD and MyoG), and anabolic signaling proteins (p-AKT and p-mTOR) under EX527 treatment. Real-time PCR analysis revealed that the TK-induced suppression of MuRF-1 and Atrogin-1 mRNA expression was significantly attenuated when co-treated with EX527 (Fig. 4A, B). While TK treatment alone led to a dose-dependent downregulation of these genes, the co-treatment with EX527 diminished this effect. Specifically, MuRF-1 mRNA expression was significantly increased only at the 400 µg/mL dose of TK compared to the DEX group, while Atrogin-1 expression showed no significant difference across all TK groups relative to DEX.
At the protein level, the inhibitory effects of TK on MuRF-1 and Atrogin-1 expression were noticeably reduced in the presence of EX527 (Fig. 4C–E). DEX treatment significantly increased MuRF-1 expression, and TK at 100 and 200 µg/mL failed to produce a meaningful suppressive effect. Only TK at 400 µg/mL exhibited a statistically significant reduction in MuRF-1 levels (p < 0.01), indicating a partial attenuation of TK’s protective effect against DEX-induced muscle atrophy under SIRT1 inhibition. Similarly, TK-induced suppression of Atrogin-1 expression was also diminished under EX527 treatment. Although a decreasing trend was observed at the highest dose (TK400), the inter-sample variability was relatively high, and no statistically significant difference was found compared to the DEX group. These results suggest that the presence of EX527 compromises the anti-atrophic actions of TK at the protein level, and that the effect on Atrogin-1 appears less consistent than that observed for MuRF-1 under these conditions.
Additionally, MyoD and MyoG protein levels were significantly decreased by DEX treatment and were not restored by TK under EX527 co-treatment (Fig. 4F-H). We further assessed the p-AKT and p-mTOR, which were both significantly downregulated by DEX treatment (Fig. 4I-K). TK treatment failed to significantly reverse this suppression at all tested concentrations when co-treated with EX527, indicating a disruption of TK-mediated anabolic signaling. These results demonstrate that inhibition of SIRT1 with EX527 compromises the beneficial actions of TK on muscle atrophy and regeneration, suggesting that TK’s therapeutic effects are at least partly mediated via the SIRT1 signaling pathway. To validate whether the effects of TK are associated with SIRT1 deacetylase activity, we further examined the acetylation status of SIRT1 downstream targets, FOXO1 and FOXO3, under conditions with or without EX527 co-treatment.
Under EX527-untreated conditions, DEX treatment significantly increased the acetyl-FOXO1/FOXO1 protein ratio compared with the blank. TK co-treatment reduced this elevation in a dose-dependent manner, with statistically significant decreases observed in the TK200 and TK400 groups (Supplementary Fig. S4A-C). In contrast, when EX527 was co-administered, overall acetyl-FOXO1/FOXO1 levels were elevated across all groups. Notably, the combination of DEX and TK under EX527 treatment led to a further increase in the acetyl-FOXO1/FOXO1 ratio compared with the corresponding groups without EX527, indicating that the inhibitory effect of TK on FOXO1 acetylation was abolished by SIRT1 inhibition.
The acetyl-FOXO3/FOXO3 ratio exhibited a pattern similar to that of acetyl-FOXO1/FOXO1. In the absence of EX527, DEX treatment increased the acetylated FOXO3 fraction, whereas TK co-treatment tended to reduce this level, although the differences were not statistically significant (Supplementary Fig. S4D-F). Under EX527 co-treatment, acetyl-FOXO3/FOXO3 expression was elevated in all experimental groups. Specifically, a significant increase was observed in the EX527-treated blank group compared with its non-treated counterpart, and the EX527 + DEX group showed a significant elevation relative to the DEX-only condition. These results suggest that EX527 counteracted the deacetylating influence of TK, maintaining high acetylation levels of FOXO3 regardless of TK treatment.
TK administration restores muscle mass and improves motor endurance in DEX-induced muscle atrophy mice model
The in vivo efficacy of TK was evaluated using a DEX-induced muscle atrophy mouse model. Mice received intraperitoneal injections of DEX (25 mg/kg) once daily for 14 consecutive days, along with oral administration of TK extract at doses of 50, 100, or 200 mg/kg/day (Fig. 5A). Efficacy was assessed by measuring GCM muscle mass and performing a treadmill exhaustion test to evaluate muscle strength and endurance on the day of sacrifice.
To evaluate systemic effects and overall condition, body weight was monitored every two days during the 14-day treatment period (Fig. 5B). The DEX control group showed a significant reduction in body weight beginning on day 4 compared to the sham group, with this difference persisting throughout the study period. In contrast, the TK groups demonstrated a trend toward increased body weight, although the changes were not statistically significant. However, a significant increase was observed in the TK200 group on day 6 compared to the DEX group.
At the time of sacrifice, GCM were excised and weighed to assess muscle mass preservation. As expected, DEX administration induced a significant reduction in GCM mass (Fig. 5C, D), consistent with muscle atrophy. However, TK treatment significantly increased GCM mass compared to the DEX group at all tested doses. To assess functional recovery, treadmill exhaustion tests were conducted on day 14 (Fig. 5E, F). The DEX control group exhibited a marked decrease in both running time and distance compared to the sham group, indicating significant impairment in muscle strength and endurance. TK administration led to a significant improvement in these parameters, with both the TK100 and TK200 groups showing enhanced running performance relative to the DEX group. Consistent with these functional improvements, histological analysis revealed that the mean cross-sectional area of GCM fibers, which had been reduced by DEX treatment, was significantly increased by all three concentrations of TK, as demonstrated by H&E staining (Fig. 5G, H). These findings collectively indicate that TK exerts a protective effect against DEX-induced muscle atrophy, improving both muscle function and morphology.
Molecular docking predicts moderate binding of TK-derived compounds to the allosteric site of SIRT1
To evaluate the potential of TK-derived compounds as SIRT1-binding molecules, molecular docking was performed using the SIRT1 crystal structure (PDB ID: 4ZZJ), focusing on the known allosteric activator binding site. Among the screened compounds, three—5-dehydrokarounidiol, steryl glucoside, and isomultiflorenol—were selected based on their relatively favorable binding energies. 5-dehydrokarounidiol (− 6.61 ± 0.31 kcal/mol), steryl glucoside (− 6.59 ± 0.28 kcal/mol), and isomultiflorenol (− 6.34 ± 0.30 kcal/mol) all fell within the range typically considered moderate binders (− 6 to − 7 kcal/mol), indicating their potential as SIRT1 modulators (Fig. 6A–C).
The RMSD values, reflecting the conformational stability of docking poses, were lowest for steryl glucoside (1.57 ± 0.80 Å), suggesting a relatively consistent binding mode. In contrast, 5-dehydrokarounidiol and isomultiflorenol showed higher RMSD values (3.89 ± 3.68 Å and 3.63 ± 2.23 Å, respectively), indicating greater variation in predicted binding conformations (Table 4).
Hydrogen bond analysis, performed using PLIP, confirmed that all detected hydrogen bonds had donor–acceptor distances within the standard threshold of 3.5 Å. Steryl glucoside exhibited the highest average number of hydrogen bonds (1.89 ± 1.96; range: 1.82–3.39 Å), followed by 5-dehydrokarounidiol (1.56 ± 0.53; 1.90–3.49 Å), whereas isomultiflorenol formed the fewest hydrogen bonds (0.78 ± 0.97; 1.92–2.50 Å). Hydrophobic interactions were observed for all compounds, with steryl glucoside displaying the greatest average number of hydrophobic contacts (6.89), followed by isomultiflorenol (5.50) and 5-dehydrokarounidiol (5.44). Additionally, salt bridges were identified in several steryl glucoside binding poses, further contributing to its broader interaction profile. These findings suggest that hydrophobic interactions, rather than extensive hydrogen bonding, may play a predominant role in stabilizing ligand–receptor binding within the SIRT1 allosteric site.
Analysis of interacting amino acid residues revealed that all three ligands commonly engaged with residues such as THR219, ILE223, ASN226, ILE227, and GLU230, which are located near the known allosteric binding region. Steryl glucoside uniquely formed contacts with LYS235 and LTS233, while isomultiflorenol interacted with HIS473, suggesting slight variations in binding orientation and pocket occupancy.
Discussion
This study demonstrates the protective effects of TK against DEX-induced muscle atrophy in both undifferentiated and differentiated C2C12 myotubes. The rationale for conducting experiments under both cellular states stems from the biological characteristics of C2C12 cells: in the undifferentiated state, they function as muscle progenitor cells (myoblasts), while upon differentiation, they form myotubes that more closely resemble mature muscle fibers in both structure and function. Evaluating TK’s effects across both stages allowed us to assess whether TK not only enhances stress resistance and viability in proliferating myoblasts but also confers protection against DEX-induced atrophic changes in differentiated myotubes.
By comparing TK’s effects on cell viability under both conditions, we identified 100, 200, and 400 µg/mL as the optimal concentration range. These doses were selected based on their reproducible ability to alleviate DEX-induced cytotoxicity and to exert consistent cytoprotective effects in both undifferentiated and differentiated C2C12 cells. While CCK-8 assay results indicate an improvement in cell viability, it is important to note that this assay reflects overall mitochondrial activity and cellular metabolic status, and cannot distinguish between enhanced proliferation and metabolic activation.
To assess TK-related inhibitory mechanisms in DEX-induced muscle atrophy, we first investigated apoptosis, a key muscle degeneration pathway27. Apoptosis is among the primary mechanisms underlying DEX-induced muscle atrophy, leading to myofiber loss and the functional decline of the skeletal muscles28. Numerous studies described that muscle atrophy is closely associated with significant apoptotic myonucleus loss. Excessive apoptosis in muscle cells accelerates myofiber degradation, a critical factor in muscle atrophy28,29. Glucocorticoids, such as DEX, activate the mitochondrial apoptotic pathway by increasing pro-apoptotic protein (e.g., Bax) and decreasing anti-apoptotic protein (e.g., Bcl-2) levels, thereby promoting programmed cell death30. Apoptosis-executing protein activation (e.g., that of Caspase-3) leads to structural damage and functional impairment in the muscle cells31,32. To assess apoptosis levels, we performed a TUNEL assay to detect DNA fragmentation, a hallmark of apoptosis, using the TdT enzyme to incorporate labeled deoxynucleotides at the 3′-hydroxyl ends of DNA strands. Furthermore, we conducted Annexin V/PI staining, a widely used method for identifying apoptotic cell populations. Our results demonstrated that TK treatment significantly and dose-dependently reduced TUNEL- and Annexin V-positive cell numbers, indicating that TK effectively inhibited DEX-induced apoptosis. This result is particularly significant as it suggests that TK not only enhances cell viability but it is also pivotal for preventing muscle atrophy progression and muscle cell function maintenance.
Muscle atrophy is primarily driven by increased protein degradation, often regulated by the ubiquitin-proteasome system (UPS)33. MuRF-1 and Atrogin-1 are key E3 ubiquitin ligases that mediate muscle protein degradation and are highly upregulated under catabolic conditions, such as glucocorticoid-induced muscle atrophy34. Increased expression of these markers indicates muscle wasting, their suppression is thus a critical indicator of muscle-preserving effects. In this study, TK treatment significantly downregulated MuRF-1 and Atrogin-1 expression, suggesting its potential to inhibit proteolytic pathways and prevent excessive muscle degradation. Beyond protein degradation, muscle differentiation and myotube formation are crucial in maintaining muscle integrity. MHC is a structural protein essential for myofiber function, while MyoD and MyoG are key transcription factors involved in the regulation of myogenic differentiation and muscle regeneration35. DEX-induced atrophy disrupts the expression of these markers, leading to impaired muscle formation and regeneration. Our results demonstrated that TK treatment restored MHC, MyoD, and MyoG expression, thereby promoting myotube formation and muscle differentiation and further reinforcing its protective role against muscle atrophy. In addition, TK treatment reversed the DEX-induced suppression of anabolic signaling pathways, as indicated by restored phosphorylation of AKT and mTOR. Oral administration of TK at 50, 100, and 200 mg/kg significantly attenuated DEX-induced muscle atrophy in vivo, preserving gastrocnemius muscle mass and enhancing physical performance. These findings provide important evidence that TK mitigates glucocorticoid-induced muscle wasting by suppressing catabolic processes and promoting muscle differentiation, supporting its potential as a therapeutic agent for muscle preservation.
Finally, we focused on SIRT1, a central regulator involved in muscle protection and the inhibition of atrophy-related mechanisms36. Based on our initial findings demonstrating the cytoprotective and anti-apoptotic effects of TK (Fig. 1), we selected SIRT1 as a key molecular target for further mechanistic investigation. This decision was also supported by previous studies highlighting the pivotal role of SIRT1 in muscle remodeling and in the regulation of apoptosis and catabolic signaling pathways. For instance, the natural antioxidant resveratrol has been reported to activate SIRT1, thereby suppressing apoptosis and catabolic processes in skeletal muscle under stress conditions37. Similarly, myricanol has been identified as a SIRT1 activator, preventing DEX-induced muscle wasting26. Moreover, fucoxanthin reportedly enhances mitochondrial function, regulates autophagy and apoptosis, and restores protein degradation balance through SIRT1 activation38. Based on these results, we hypothesized that SIRT1 modulation might mediate the TK-related muscle-protective effects. To validate this, we assessed whether TK would influence SIRT1 expression in DEX-induced muscle atrophy using ICC and further evaluated cell viability under conditions with or without EX527, a selective SIRT1 inhibitor.
In our study, we also analyzed the protein expression levels of SIRT1, SIRT2, and SIRT3 following EX-527 treatment to explore isoform-specific effects under our experimental conditions (Supplementary Fig. S2). The results demonstrated a clear dose-dependent decrease in SIRT1 protein abundance, whereas SIRT2 and SIRT3 did not exhibit consistent or dose-dependent changes. Although we did not directly measure SIRT enzyme activities, prior biochemical evidence indicates that EX-527 inhibits SIRT1 approximately 100-fold more potently than SIRT2 and SIRT3 by exploiting SIRT1’s unique NAD⁺-binding channel39. Consistent with this, other studies have reported that EX-527 shows minimal inhibitory effects on SIRT2/3 at concentrations typically used for SIRT1 inhibition40,41. Furthermore, to assess whether TK-mediated effects are functionally linked to SIRT1 deacetylase activity, we examined the acetylation status of FOXO1 and FOXO3, two downstream targets of SIRT1. TK treatment attenuated DEX-induced FOXO1/3 acetylation in the absence of EX-527, whereas this deacetylation effect was abolished under EX-527 co-treatment, supporting that TK’s regulatory influence on FOXO acetylation is dependent on SIRT1 activity (Supplementary Fig. S4). In line with these findings, our data showing a dose-dependent decrease in SIRT1 abundance—with minimal changes in SIRT2 and SIRT3—are consistent with the literature-supported SIRT1-selective inhibition profile of EX-527. Nevertheless, because our analysis was limited to protein expression and acetylation status, further investigations using direct enzymatic activity assays will be necessary to confirm SIRT isoform selectivity and to delineate the precise role of SIRT1 in mediating TK-induced muscle protection under glucocorticoid exposure.
Our results supported the protective role of TK against muscle degeneration, positioning SIRT1 upregulation as a potential mechanism underlying TK-mediated muscle protection.
However, our study has several limitations. Although previous studies have suggested that pathways such as AMPK and SIRT1 are primarily involved in regulating mitochondrial biogenesis and energy homeostasis under stress rather than directly promoting proliferation42,43, our study did not include direct experimental validation of these processes. Markers such as Ki67 for proliferation or phospho-AMPK for metabolic activation were not assessed. Therefore, future studies incorporating such specific markers will be essential to elucidate the precise mechanisms underlying TK’s observed cytoprotective effects. In our experiments, we used C2C12 myoblasts and myotubes, which are widely applied in vitro models for muscle research. Nonetheless, these models do not fully replicate the complex physiological environment of skeletal muscles in living organisms. In addition, while we identified 100, 200, and 400 µg/mL as optimal TK concentrations, we did not extensively evaluate long-term cytotoxicity and dose-response effects. Further investigations assessing chronic TK exposure and its effects on cell metabolism, mitochondrial function, and oxidative stress markers would be necessary to gain deeper insights into TK-related safety and long-term efficacy. Specifically, while we identified SIRT1 involvement as a potential mechanism, this conclusion is primarily based on results under pharmacological inhibition using EX527. While EX527 has been widely used to probe SIRT1–dependent pathways, pharmacological inhibition alone may have limitations in fully delineating direct mechanistic involvement. Therefore, genetic approaches such as Sirt-1 knockdown via siRNA or the use of Sirt-1 knockout models would provide more definitive evidence of the causal role of Sirt-1 in mediating TK’s effects and should be considered in future studies. Moreover, identifying the active constituents responsible for the observed protective effects of TK is essential for understanding its mechanism of action. While we did not conduct additional phytochemical profiling in the present study, previous analyses have reported that TK, particularly its root extract, contains high levels of antioxidant polyphenols including gallic acid, caffeic acid, 3,5-dihydroxybenzoic acid, and flavanones44. Notably, the polyphenol content was significantly higher in the root extract (133.85 mg/g) compared to the fruit extract (53.5 mg/g). The same study also demonstrated superior free radical scavenging and lipid oxidation inhibitory activities of the root extract compared to the synthetic antioxidant BHT, along with excellent biocompatibility.
Several of these polyphenols, such as gallic acid and caffeic acid, have independently been shown to upregulate SIRT1 and exert anti-apoptotic or anti-atrophic effects in various biological systems. In addition, other well-known dietary polyphenols, including resveratrol, quercetin, and catechins, have been reported to activate SIRT1 and contribute to cellular protection against oxidative stress, inflammation, and muscle degeneration45,46. These findings provide a plausible biochemical rationale to hypothesize that the SIRT1–associated effects observed in our study may, at least in part, be attributed to polyphenolic constituents naturally present in TK.
To further investigate the potential link between TK and SIRT1 modulation, we conducted an in silico molecular docking analysis to identify TK-derived compounds capable of interacting with the SIRT1 allosteric site. Among the screened candidates, 5-dehydrokarounidiol, steryl glucoside, and isomultiflorenol exhibited moderate binding affinities (− 6.3 to − 6.6 kcal/mol), suggesting their potential as SIRT1 modulators. While these results provide preliminary support for a SIRT1–mediated mechanism, it is important to note that molecular docking is based on static structural models and does not reflect the dynamic nature of protein–ligand interactions in a physiological environment. Factors such as protein flexibility, solvent effects, and conformational changes are not adequately captured in docking simulations.
To better assess the stability and functional relevance of these interactions, future studies should incorporate molecular dynamics simulations and biophysical assays. Furthermore, experimental validation through techniques such as binding affinity assays, SIRT1 activity measurements, or reporter-based functional analyses will be essential to confirm these computational predictions. Such efforts will be critical for elucidating the mechanistic role of individual TK compounds and for advancing TK as a promising candidate for the treatment of glucocorticoid-induced muscle atrophy.
Conclusion
TK protects against DEX-induced muscle atrophy by inhibiting apoptosis, reducing atrophy-related factors, and promoting differentiation, with SIRT1 involvement likely playing a key role. Further studies, including genetic validation and long-term safety assessment, are needed to confirm its therapeutic potential.

Cytoprotective effect of TK against DEX-induced apoptosis of C2C12 cell. (A) Schematic representation of the experimental timeline for TK treatment and analysis in both C2C12 myoblasts and myotubes. (B) CCK assay results highlighting myoblast cell viability in undifferentiated C2C12 cells treated with TK alone or in combination with DEX. (C) CCK assay results presenting viability in differentiated C2C12 myotubes treated with TK alone or in combination with DEX. (D) Live/Dead staining images of C2C12 myoblasts treated with the following: Blank, DEX, DEX + TK 100, 200, and 400 µg/mL. White and yellow scale bars: 200 and 100 μm, respectively. (E and F) The number of green+ and red+ cells result quantifications. The data are expressed as the mean ± standard deviation (SD). (G) Representative flow cytometry dot plots displaying Annexin V and PI (X- and Y-axes, respectively) staining for Blank, DEX, and TK (100, 200, and 400 µg/mL) + DEX groups, indicating apoptotic and necrotic myoblast populations. (H) Quantification of Annexin V-positive apoptotic cell populations in each treatment group. (I) Representative images of TUNEL assay in C2C12 myotubes submitted to Blank, DEX, and TK (100, 200, and 400 µg/mL) + DEX treatments. (J) Quantification of TUNEL-positive intensity in C2C12 myotubes across each treatment group. The data are expressed as the mean ± SD. Statistical significance was assessed using one-way ANOVA with Tukey’s post hoc analysis as follows: #### p < 0.0001 vs. the BLK group; * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001 vs. the DEX group.

Effects of TK on muscle atrophy markers in DEX-treated atrophic C2C12 myotubes. (A, B) MuRF-1 and Atrogin-1 mRNA expression quantifications in DEX-treated atrophic C2C12 myotubes with TK co-treatment, determined by real-time PCR analysis. (C–E) Representative western blot images and quantification of the muscle atrophy-related proteins MuRF-1 and Atrogin-1, with expression levels normalized to β-actin across five treatment groups. (F–I) Representative western blot images and quantification of the muscle differentiation related proteins MHC, MyoD, and MyoG with expression levels normalized to β-actin across five treatment groups. (J–L) Representative western blot images and quantification of the anabolic signaling related proteins p-AKT and p-mTOR with expression levels normalized to β-actin across five treatment groups. The data are expressed as the mean ± SD. Statistical significance was assessed using one-way ANOVA followed by Tukey’s post hoc test as follows: # p < 0.05, ## p < 0.01, ### p < 0.001, and #### p < 0.0001 vs. the BLK group; * p < 0.05, *** p < 0.001, and **** p < 0.0001 vs. the DEX group.

SIRT1–dependent protective effects of TK in DEX-induced C2C12 myotubes. (A) C2C12 myoblast viability assay in the presence and absence of EX527. (B) C2C12 myotube viability assay in the presence and absence of EX527. (C) Representative confocal images showing co-expression of SIRT1 (red) and MHC (green) in C2C12 myotubes across five treatment groups, comparing conditions with and without EX527. (D) Quantitative analysis of SIRT1 fluorescence intensity in C2C12 myotubes across five treatment groups, under conditions with and without EX527. (E) Quantification of average myotube diameter (µm) across the same five groups, comparing the effects of TK with or without EX527 co-treatment. (F) Quantification of average myotube number per field across treatment groups under both EX527–treated and untreated conditions. (G, H) Representative western blot images (G) and quantification (H) of SIRT1 protein levels in each treatment group, with or without EX527 co-treatment. The data are expressed as the mean ± SD. White scale bar: 100 μm. Statistical significance was assessed using one-way ANOVA with Tukey’s post hoc analysis as follows: ### p < 0.001 and #### p < 0.0001 vs. the BLK group; * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001 vs. the DEX group.

EX527 treatment attenuates the anti-atrophic and myogenic effects of TK. (A, B) Relative mRNA expression levels of MuRF-1 (A) and Atrogin-1 (B) in C2C12 myotubes treated with DEX (100 µM), TK (100, 200, or 400 µg/mL) with EX527 (100 µM) for 24 h. (C–E) Representative western blot images and quantification of muscle atrophy-related markers MuRF-1 and Atrogin-1 protein levels in the same treatment groups. (F–H) Representative western blot images and quantification of myogenic differentiation markers MyoD and MyoG in C2C12 myotubes under the same treatment conditions. (I–K) Representative western blot images and quantification of anabolic signaling markers phospho-AKT and phospho-mTOR. The data are expressed as the mean ± SD. Statistical significance was assessed using one-way ANOVA with Tukey’s post hoc analysis as follows: # p < 0.05, ### p < 0.001, and #### p < 0.0001 vs. the BLK group; * p < 0.05 and ** p < 0.01 vs. the DEX group.

TK attenuates DEX-induced muscle atrophy and improves functional performance in vivo. (A) Schematic overview of the DEX-induced muscle atrophy model and TK treatment. (B) Body weight (g) changes monitored every two days during the 14-day period across groups. (C) Representative images of dissected GCM in each group. (D) Quantification of GCM mass in each group, calculated as GCM weight (mg) normalized to body weight (g) on day 14. (E, F) Treadmill exhaustion test results on day 14: running time (min) (E) and running distance (m) (F). Quantification of muscle fiber cross-sectional area (CSA) from GCM sections (G). Representative H&E-stained cross section of gastrocnemius muscle (H). The data are expressed as the mean ± SD. Statistical significance was assessed using one-way ANOVA with Tukey’s post hoc analysis and two-way repeated-measures ANOVA with Tukey’s post hoc analysis as follows: ### p < 0.001 and #### p < 0.0001 vs. the sham group; * p < 0.05 and ** p < 0.01 vs. the control group.

Molecular docking predicts moderate binding of TK-derived compounds to the allosteric site of SIRT1. (A–C) Predicted binding poses of three selected TK-derived compounds—5-dehydrokarounidiol, steryl glucoside, and isomultiflorenol—within the allosteric binding pocket of SIRT1 (PDB ID: 4ZZJ).
Data availability
Data is provided within the manuscript or supplementary information files.
References
Frontera, W. R. & Ochala, J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int. 96, 183–195. https://doi.org/10.1007/s00223-014-9915-y (2015).
Choi, J. W. et al. Effect of low muscle mass on total mortality related to metabolic disease in chronic kidney disease patients. Sci. Rep. 14, 22837. https://doi.org/10.1038/s41598-024-73903-w (2024).
Jun, L., Robinson, M., Geetha, T., Broderick, T. L. & Babu, J. R. Prevalence and Mechanisms of Skeletal Muscle Atrophy in Metabolic Conditions. Int. J. Mol. Sci. https://doi.org/10.3390/ijms24032973 (2023).
Sato, A. Y. et al. Glucocorticoids induce bone and muscle atrophy by Tissue-Specific mechanisms upstream of E3 ubiquitin ligases. Endocrinology 158, 664–677. https://doi.org/10.1210/en.2016-1779 (2017).
Schakman, O., Kalista, S., Barbe, C., Loumaye, A. & Thissen, J. P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell. Biol. 45, 2163–2172. https://doi.org/10.1016/j.biocel.2013.05.036 (2013).
Crawford, B. A., Liu, P. Y., Kean, M. T., Bleasel, J. F. & Handelsman, D. J. Randomized placebo-controlled trial of androgen effects on muscle and bone in men requiring long-term systemic glucocorticoid treatment. J. Clin. Endocrinol. Metab. 88, 3167–3176. https://doi.org/10.1210/jc.2002-021827 (2003).
Pofi, R., Caratti, G., Ray, D. W. & Tomlinson, J. W. Treating the side effects of exogenous Glucocorticoids; can we separate the good from the bad? Endocr. Rev. 44, 975–1011. https://doi.org/10.1210/endrev/bnad016 (2023).
Corrigendum to. Thigh muscle fat infiltration is associated with impaired physical performance despite remission in cushing’s syndrome. J. Clin. Endocrinol. Metab. 107, e447. https://doi.org/10.1210/clinem/dgab590 (2022).
Juan, C. G., Matchett, K. B. & Davison, G. W. A systematic review and meta-analysis of the SIRT1 response to exercise. Sci. Rep. 13, 14752. https://doi.org/10.1038/s41598-023-38843-x (2023).
Schenk, S. et al. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J. Clin. Invest. 121, 4281–4288. https://doi.org/10.1172/JCI58554 (2011).
Kim, S. M. et al. Mealworm hydrolysate ameliorates dexamethasone-induced muscle atrophy via sirtuin 1-mediated signaling and Akt pathway. NPJ Sci. Food https://doi.org/10.1038/s41538-025-00432-9 (2025).
Zhao, Y. et al. l-arginine alleviates LPS-induced oxidative stress and apoptosis via activating SIRT1-AKT-Nrf2 and SIRT1-FOXO3a signaling pathways in C2C12 myotube cells. Antioxid. (Basel) https://doi.org/10.3390/antiox10121957 (2021).
Park, H. J. & Park, S. H. Root extract of trichosanthes Kirilowii suppresses metastatic activity of EGFR TKI-Resistant human lung cancer cells by inhibiting Src-Mediated EMT. Nutr. Cancer. 75, 1945–1957. https://doi.org/10.1080/01635581.2023.2272345 (2023).
Xu, L. et al. Influence of different extraction methods on the chemical composition, antioxidant activity, and overall quality attributes of oils from trichosanthes Kirilowii Maxim seed. Food Control. 142, 109201. https://doi.org/10.1016/j.foodcont.2022.109201 (2022).
Hu, Z. et al. Microwave-assisted extraction, characterization and Immunomodulatory activity on RAW264.7 cells of polysaccharides from trichosanthes Kirilowii Maxim seeds. Int. J. Biol. Macromol. 164, 2861–2872. https://doi.org/10.1016/j.ijbiomac.2020.08.072 (2020).
Lee, J. H., Kim, B., Ko, S. G. & Kim, W. Analgesic effect of SH003 and trichosanthes Kirilowii Maximowicz in Paclitaxel-Induced neuropathic pain in mice. Curr. Issues Mol. Biol. 44, 718–730. https://doi.org/10.3390/cimb44020050 (2022).
Park, S. M. et al. Anti-tumor effects of the ethanolic extract of trichosanthes Kirilowii seeds in colorectal cancer. Chin. Med. 14, 43. https://doi.org/10.1186/s13020-019-0263-8 (2019).
Castro, B. & Kuang, S. Evaluation of muscle performance in mice by treadmill exhaustion test and whole-limb grip strength assay. Bio Protoc. https://doi.org/10.21769/BioProtoc.2237 (2017).
Akihisa, T. et al. Five D:C-friedo-oleanane triterpenes from the seeds of trichosanthes Kirilowii Maxim. And their anti-inflammatory effects. Chem. Pharm. Bull. (Tokyo). 42, 1101–1105. https://doi.org/10.1248/cpb.42.1101 (1994).
Dat, N. T., Jin, X., Hong, Y. S. & Lee, J. J. An Isoaurone and other constituents from trichosanthes Kirilowii seeds inhibit hypoxia-inducible factor-1 and nuclear factor-kappaB. J. Nat. Prod. 73, 1167–1169. https://doi.org/10.1021/np900820p (2010).
Huang, Y., He, P., Bader, K. P., Radunz, A. & Schmid, G. H. Seeds of trichosanthes kirilowii, an energy-rich diet. Z. Naturforsch C J. Biosci. 55, 189–194. https://doi.org/10.1515/znc-2000-3-409 (2000).
Moon, S. S., Rahman, A. A., Kim, J. Y. & Kee, S. H. Hanultarin, a cytotoxic Lignan as an inhibitor of actin cytoskeleton polymerization from the seeds of trichosanthes Kirilowii. Bioorg. Med. Chem. 16, 7264–7269. https://doi.org/10.1016/j.bmc.2008.06.032 (2008).
Wu, S., Xu, T. & Akoh, C. C. Effect of roasting on the volatile constituents of trichosanthes Kirilowii seeds. J. Food Drug Anal. 22, 310–317. https://doi.org/10.1016/j.jfda.2013.12.005 (2014).
Yu, X. et al. Trichosanthis fructus: botany, traditional uses, phytochemistry and Pharmacology. J. Ethnopharmacol. 224, 177–194. https://doi.org/10.1016/j.jep.2018.05.034 (2018).
Han, D. S., Yang, W. S. & Kao, T. W. Dexamethasone treatment at the myoblast stage enhanced C2C12 myocyte differentiation. Int. J. Med. Sci. 14, 434–443. https://doi.org/10.7150/ijms.18427 (2017).
Shen, S. et al. Myricanol rescues dexamethasone-induced muscle dysfunction via a Sirtuin 1-dependent mechanism. J. Cachexia Sarcopenia Muscle. 10, 429–444. https://doi.org/10.1002/jcsm.12393 (2019).
Marzetti, E. & Leeuwenburgh, C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp. Gerontol. 41, 1234–1238. https://doi.org/10.1016/j.exger.2006.08.011 (2006).
Dupont-Versteegden, E. E. Apoptosis in skeletal muscle and its relevance to atrophy. World J. Gastroenterol. 12, 7463–7466. https://doi.org/10.3748/wjg.v12.i46.7463 (2006).
Schwartz, L. M. Skeletal muscles do not undergo apoptosis during either atrophy or programmed cell Death-Revisiting the myonuclear domain hypothesis. Front. Physiol. 9, 1887. https://doi.org/10.3389/fphys.2018.01887 (2018).
Clarisse, D., Offner, F. & De Bosscher, K. Latest perspectives on glucocorticoid-induced apoptosis and resistance in lymphoid malignancies. Biochim. Biophys. Acta Rev. Cancer. 1874, 188430. https://doi.org/10.1016/j.bbcan.2020.188430 (2020).
Laane, E. et al. Dexamethasone-induced apoptosis in acute lymphoblastic leukemia involves differential regulation of Bcl-2 family members. Haematologica 92, 1460–1469. https://doi.org/10.3324/haematol.10543 (2007).
Wang, D., Muller, N., McPherson, K. G. & Reichardt, H. M. Glucocorticoids engage different signal transduction pathways to induce apoptosis in thymocytes and mature T cells. J. Immunol. 176, 1695–1702. https://doi.org/10.4049/jimmunol.176.3.1695 (2006).
Kitajima, Y., Yoshioka, K. & Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: from basic science to disorders. J. Physiol. Sci. 70, 40. https://doi.org/10.1186/s12576-020-00768-9 (2020).
Gumucio, J. P. & Mendias, C. L. Atrogin-1, MuRF-1, and sarcopenia. Endocrine 43, 12–21. https://doi.org/10.1007/s12020-012-9751-7 (2013).
Zanou, N. & Gailly, P. Skeletal muscle hypertrophy and regeneration: interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell. Mol. Life Sci. 70, 4117–4130. https://doi.org/10.1007/s00018-013-1330-4 (2013).
Yang, L. et al. SIRT1 signaling pathways in sarcopenia: novel mechanisms and potential therapeutic targets. Biomed. Pharmacother. 177, 116917. https://doi.org/10.1016/j.biopha.2024.116917 (2024).
Sin, T. K. et al. SIRT1-dependent myoprotective effects of Resveratrol on muscle injury induced by compression. Front. Physiol. 6, 293. https://doi.org/10.3389/fphys.2015.00293 (2015).
Zhiyin, L., Jinliang, C., Qiunan, C., Yunfei, Y. & Qian, X. Fucoxanthin rescues dexamethasone induced C2C12 myotubes atrophy. Biomed. Pharmacother. 139, 111590. https://doi.org/10.1016/j.biopha.2021.111590 (2021).
Gertz, M. et al. Ex-527 inhibits sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. U S A. 110, E2772–2781. https://doi.org/10.1073/pnas.1303628110 (2013).
Hong, J. Y. & Lin, H. Sirtuin modulators in cellular and animal models of human diseases. Front. Pharmacol. 12, 735044. https://doi.org/10.3389/fphar.2021.735044 (2021).
Solomon, J. M. et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 26, 28–38. https://doi.org/10.1128/MCB.26.1.28-38.2006 (2006).
Deshpande, V. V., Bockrath, M., Glazman, L. I. & Yacoby, A. Electron liquids and solids in one dimension. Nature 464, 209–216. https://doi.org/10.1038/nature08918 (2010).
Maurange, C., Cheng, L. & Gould, A. P. Temporal transcription factors and their targets schedule the end of neural proliferation in drosophila. Cell 133, 891–902. https://doi.org/10.1016/j.cell.2008.03.034 (2008).
Zhoh, C. K., Uhm, T. Y. & Kim, J. C. Antioxidants effectiveness of trichosanthes Kirilowii Maximowicz extracts. J. Korean Industrial Eng. Chem. 18, 625–629 (2007).
Chung, S. et al. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch. Biochem. Biophys. 501, 79–90. https://doi.org/10.1016/j.abb.2010.05.003 (2010).
Sarubbo, F., Esteban, S., Miralles, A. & Moranta, D. Effects of Resveratrol and other polyphenols on Sirt1: relevance to brain function during aging. Curr. Neuropharmacol. 16, 126–136. https://doi.org/10.2174/1570159X15666170703113212 (2018).
Acknowledgements
The authors have no acknowledgments to declare.
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number RS-2023-KH139336).
Author information
Authors and Affiliations
Contributions
I-HH: funding acquisition, project administration, resources, supervision, writing–review and editing. Hyunseong Kim: conceptualization, data curation, formal analysis, investigation, methodology, software, visualization, writing–original draft. JYH: formal analysis, validation, visualization, writing–review and editing. Hyun Kim: formal analysis, methodology, visualization. JL: formal analysis, visualization. CY: formal analysis, methodology, validation. W-JJ: formal analysis, methodology, validation. YJL: validation, writing–review and editing. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, H., Hong, J.Y., Yeo, C. et al. Trichosanthes kirilowii Maximowicz attenuates dexamethasone induced atrophy in C2C12 myotubes through a Sirtuin 1 associated mechanism. Sci Rep 15, 44601 (2025). https://doi.org/10.1038/s41598-025-28257-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28257-2
