
Abstract
Acute myocardial infarction (AMI) stands as a major contributor to mortality and disability worldwide, with obesity playing a significant role in its development. This study aims to investigate potential biomarkers associated with obesity-related genes (ORGs) in patients with AMI. We downloaded four training gene expression datasets and one validation dataset from the Gene Expression Omnibus (GEO) database, and extracted ORGs from the GeneCards database. Feature genes were identified using machine learning techniques, and their diagnostic potential was evaluated through receiver operating characteristic (ROC) curves. To assess the identified critical pathways, Gene Set Enrichment Analysis (GSEA) was performed. Levels of immune infiltration were analyzed using the CIBERSORT algorithm and single-sample Gene Set Enrichment Analysis (ssGSEA). A signature comprising five genes (IL1RN, TLR2, NFKBIA, MMP9, and ITLN1) was established as a diagnostic biomarker for AMI, achieving an area under the curve (AUC) of 0.924, which was confirmed in the GSE59876 dataset (AUC = 0.825). According to the diagnostic model, comparisons between high- and low-risk groups revealed six distinct immune cell types and thirteen differing immune functions. Validation through reverse transcription quantitative polymerase chain reaction (RT-qPCR) reaffirmed the differential expression of the signature genes in AMI and control samples. Our findings provide crucial insights into the roles of ORGs in AMI and may facilitate the identification of valuable biomarkers for AMI diagnosis.
Similar content being viewed by others


Introduction
The rates of incidence and mortality associated with cardiovascular diseases are increasing, with acute myocardial infarction (AMI) being the most lethal and presenting considerable health risks to patients1,2. AMI signifies a severe progression of ischemia and hypoxia in myocardial tissue resulting from the blockage of coronary arteries; this condition may lead to localized or extensive damage and necrosis of myocardial cells, causing serious complications, including cardiogenic shock, heart failure, and cardiac arrest3,4. Furthermore, some cases of AMI may exhibit no symptoms during their initial stages. Consequently, accurate and timely diagnosis, along with effective reperfusion therapies such as thrombolysis or percutaneous coronary intervention (PCI), is vital in reducing the extent of AMI and enhancing patient outcomes5,6. At present, the identification of AMI typically relies on alterations in cardiac biomarkers. Standard biomarkers used in clinical settings consist of cardiac troponin T, cardiac troponin I, creatine kinase-MB (CK-MB), and myoglobin7. Nevertheless, these biomarkers are primarily released from necrotic cardiomyocytes within 2–4 h after the onset of AMI, and their concentrations can also be elevated in patients suffering from chronic kidney disease, heart failure, sepsis, and thyroid disorders, particularly among the elderly8. Hence, it is essential to discover additional, more specific biomarkers for the diagnosis of AMI.
Invasive coronary angiography is considered the “gold standard” for the detection of acute myocardial infarction (AMI). Nonetheless, this procedure comes with significant costs and may carry risks for patients9. Due to advancements in gene chip technology and transcriptome sequencing techniques, an increasing number of gene chip applications and bioinformatics analyses are being adopted in cardiovascular research and clinical practice, aiding in the discovery of new biomarkers for the early diagnosis and prognosis of various diseases10,11,12. Conventional methods typically concentrate on a limited number of genes or proteins; in contrast, bioinformatics allows for the evaluation of complex biological systems as cohesive entities. Its advantages in fields such as disease forecasting, personalized medicine, and drug development are becoming more widely acknowledged in clinical environments13,14. For example, Chen et al. identified PRF1 and TBX21 as novel biomarkers for diagnosis as well as potential therapeutic targets through the analysis of microarray expression profiles in patients with AMI15. Similarly, Kiliszek et al. employed a microarray technique to showcase that during ST-segment elevation myocardial infarction (STEMI), numerous genes show altered expression patterns, including those associated with various pathways related to platelet functionality, lipid and glucose metabolism, and the stability of atherosclerotic plaques16. Additionally, Liu et al. conducted a weighted gene co-expression network analysis (WGCNA) on GSE4648 and highlighted the diagnostic potential of ten hub genes, including Socs3, Hspa1b, Atf3, Il1b, Cxcl1, Selp, Ptgs2, Cxcl2, S100a8, and Myd88, using both bioinformatics and laboratory methods17.
In recent years, the issues of obesity and being overweight have generated increasing concerns. The prevalence of these conditions is on the rise and has been linked to type II diabetes mellitus, metabolic syndrome, various cancers, hypertension, and cardiovascular diseases within the general population18,19,20. However, the connection between being overweight or obese and acute myocardial infarction (AMI) remains a topic of debate. Mehta et al. indicated that obese individuals experiencing AMI have a lower risk of mortality compared to those with a normal body mass index (BMI)21. Conversely, Yusuf et al. found that abdominal obesity elevates the risk of AMI across different ages and genders in all regions22. Nonetheless, there has been a limited number of studies that have examined the influence of obesity-related genes (ORGs) on the diagnosis, risk assessment, and prognosis of AMI thus far.
In this study, our goal is to carefully organize multiple microarray datasets, including AMI and control samples, using data from the Gene Expression Omnibus (GEO) database. The main objective is to identify potential biomarkers associated with ORG for the diagnosis of AMI by combining bioinformatics analysis and machine learning methods.
Materials and methods
Data acquisition, processing and identification of differentially expressed genes
We conducted a comprehensive screening of the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) using the search terms “AMI” or “acute myocardial infarction,” “array” type, and “Homo sapiens.” The inclusion criteria for datasets required that each group contain at least three patients and three controls, and that gene symbols and Entrez IDs were available in the annotated platforms (GPL). Ultimately, five datasets were selected (Table S1). The datasets GSE48060, GSE60993, GSE66360, and GSE97320, encompassing 90 samples from AMI and 81 control samples, were combined to create a metadata file. This metadata served as the training cohort for the analysis of differentially expressed genes (DEGs) and the development of a diagnostic model. Additionally, GSE59867, which includes 111 AMI samples and 46 control samples, was selected for independent validation. To preprocess the data and eliminate batch effects, the ComBat function from the SVA package was employed23, followed by principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE) to verify the removal of batch effects. The “limma” package in R was applied for background correction, inter-array normalization, and analysis of differential expression between AMI and control samples. A false discovery rate adjusted p-value of < 0.05 and | log2 (fold change) | >0.8 were established as the criteria for identifying DEGs. We identified 1556 obesity-related genes (ORGs) from the GeneCards database (https://www.genecards.org/) using the term “obesity” with a relevance score of ≥ 5 as the screening criteria. Finally, the differentially expressed obesity-related genes (DE-ORGs) were derived by intersecting the DEGs with ORGs.
Screening of candidate diagnostic biomarkers
To discover genes with diagnostic capabilities, three machine learning techniques were employed to forecast disease status. The least absolute shrinkage and selection operator (LASSO) is a regression analysis technique that incorporates regularization to enhance predictive accuracy24,25. The LASSO methodology was implemented with the “glmnet” package in R to detect genes significantly linked to the differentiation between AMI and normal samples. Support vector machine recursive feature elimination (SVM-RFE) was employed to identify the features with the greatest discriminative ability26. In the case of random forest, we assessed the error rates for 1 to 500 trees, with the optimal number of trees identified as the count yielding the lowest error rate while maintaining the best stability27. The selection of candidate genes was based on the combined outcomes of the three machine learning methods.
Function analysis and protein-protein interaction network of DEGs
In order to enhance our understanding of the biological functions associated with the chosen DEGs, we conducted an analysis utilizing Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways through the “clusterProfiler” package. Concurrently, we developed a protein-protein interaction (PPI) network by employing the STRING database (http://string-db.org), applying a minimum interaction score set to a medium confidence level of 0.4 as our selection criterion and excluding isolated nodes from the network.
Construction of obesity-related genes model
The expression levels of the candidate genes were obtained from both the training and validation cohorts, and the feature value for each sample was computed using the following formula: \(Feature{\text{ }}value{\text{ }} = {\text{ }}\beta _{0} + {\text{ }}\beta _{1} ^{*} X_{1} + {\text{ }}\beta _{2} ^{*} X_{2} + {\text{ }}...{\text{ }} + {\text{ }}\beta _{n} ^{*} X_{n}\). In this equation, β0 denotes the Y-intercept, βn signifies the binary logistic regression coefficient for the n-th gene, Xn represents the expression measurement of the n-th gene, and n indicates the total number of genes included in the regression model. To assess the diagnostic efficacy of the feature genes, the receiver operating characteristic (ROC) curve was generated utilizing the pROC package.
Immune cells infiltration, immune function and correlation analysis
The immune gene dataset and its corresponding annotations were obtained from the ImmPort database (https://www.immport.org/). To assess the infiltration proportions of 22 different immune cell types across all samples in the training cohort, the CIBERSORT algorithm was employed. The immune gene-annotated pathways were evaluated in each sample using the ssGSEA algorithm. Subsequently, based on the median feature values, samples within the training cohort were categorized into low-risk and high-risk groups. The Wilcoxon test, utilizing the “ggplot2” package in R, was conducted to analyze the variations in immune cell types and immune functions between these two risk groups28. To determine the relationships between diagnostic genes and the differing immune cells or functions, Spearman correlation analysis was executed. Furthermore, the genes distinguishing the low-risk from the high-risk groups underwent KEGG pathway enrichment analysis via the gene set enrichment analysis (GSEA) method29. Multiple-testing corrections were applied to the comparisons of immune-cell and immune-function, as well as to the GSEA results. An adjusted p-value < 0.05 were considered significant.
Real-time qPCR
Blood samples from five patients diagnosed with AMI and five control subjects were obtained from Huaihe Hospital at Henan University in China. The criteria for excluding participants from this study included the presence of cancer, autoimmune disease, serious infectious disease, advanced liver and kidney failure, hematological disease, and a prior history of cardiovascular disease (Table S 2). Prior to sample collection, informed consent was secured from all participants, both patients and healthy individuals. The study protocols involving human blood were carried out in alignment with the principles outlined in the Declaration of Helsinki and received approval from the Medical School’s Ethics Committee at Henan University, China (HUSOM-2018-282). Peripheral blood mononuclear cells (PBMCs) were isolated using a PBMC separation solution. Total RNA was extracted employing Trizol reagent (Takara, Dalian, China) as per the manufacturer’s guidelines. Following this, the extracted RNA underwent reverse transcription (RR036A, Takara) to create complementary DNA (cDNA). Real-time PCR was performed with the TB GreenTM premix Ex TaqTM (RR420A, Takara) using the ABI Prism 7900 System, adhering to the following protocol: the denaturation step was carried out for 10 s at 95 °C, the annealing phase took 20 s at 60 °C, the extension phase lasted for 30 s at 72 °C, and a total of 40 cycles were executed30. Gene expression levels were analyzed using the 2−ΔΔCt method, with GAPDH being the endogenous control. The primer sequences are detailed in Supplementary Table S3.
Statistical analysis
Statistical analyses for the bioinformatics section of this research were performed using R (version 4.4.2). To assess the diagnostic effectiveness of the biomarkers and the diagnostic model, ROC curve analysis was applied. The correlation between the expression levels of model genes and the presence of infiltrating immune cells was evaluated utilizing Spearman’s correlation coefficient. All statistical tests were two-sided, and a p-value of less than 0.05 was considered statistically significant.
Results
Screening of DEGs in AMI
Following the standardization of the original data from the four data sets, t-SNE and PCA mappings were created, demonstrating that batch differences were effectively eliminated and the data remained stable (Fig. 1A-D). Next, the DEGs within the training cohort were analyzed utilizing the limma package after addressing batch effects. In comparison to the control group, 157 genes showed significant expression changes in the AMI patients group, featuring 142 genes up-regulated and 15 down-regulated. These DEGs were then represented in both a volcano plot and a cluster heatmap (Fig. 1E, F).

Normalization of the dataset and analysis of differential gene expression. (A-D) Normalization and batch effect correction in four microarray datasets GSE48060, GSE60993, GSE66360, and GSE97320. (E, F) Volcano map of differentially expressed genes (DEGs) after screening (adjusted p < 0.05 and |log2FC|>0.8) (E) and cluster heatmap of DEGs (F) between acute myocardial infarction (AMI) and control samples using the merged dataset (AMI = 90, control = 81) derived from GSE48060, GSE60993, GSE66360, and GSE97320.
Identification of differentially expressed obesity-related genes and enrichment analysis
The overlap of 157 genes with differential expression and those associated with human obesity indicated the identification of 18 differentially expressed obesity-related genes (DE-ORGs), which include IL1RN, SERPINA1, TLR2, NFKBIA, PYGL, IL1B, MMP9, DGAT2, TLR4, NLRP3, ITLN1, CEBPB, CD163, ALDH2, STEAP4, IRS2, SLC7A7, and PTGS2 (Fig. 2A). The heatmap indicated that these 18 genes formed a cluster characterized by elevated expression in AMI samples while exhibiting lower expression in control samples, as evaluated using the training database (Fig. 2B). To further explore the pathophysiological roles of these DE-ORGs, enrichment analyses were conducted using clusterProfiler, involving Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Disease Ontology (DO). The GO analysis showed that the DE-ORGs were predominantly implicated in the response to lipopolysaccharide, inflammatory responses, and membrane rafts (Fig. 2C). KEGG analysis revealed that these DE-ORGs participated in various signaling pathways, such as those related to lipids and atherosclerosis, the IL-17 signaling pathway, and the TNF signaling pathway. Moreover, they also played roles in the NF-kappa B signaling pathway and toll-like receptor signaling pathway (Fig. 2D). Additionally, the DO analysis indicated that the 18 DE-ORGs were primarily enriched in conditions such as pancreatitis, fatty liver disease, and lipid storage disease (Fig. 2E). Considering the strong association between pancreatitis, lipid metabolism disorders, and cardiovascular disease31,32,33, these findings underscored a significant link between DE-ORGs and AMI, emphasizing that DE-ORGs primarily regulate inflammatory responses and lipid storage.

Differentially expressed obesity-related genes (DE-ORGs) and functional enrichment. (A) Identification of 18 DE-ORGs by overlapping the 157 differentially expressed genes with 1556 obesity-related genes. (B) Expression heatmap of 18 DE-ORGs in AMI patients and controls. (C) GO enrichment analysis of DE-ORGs. (D) KEGG enrichment analysis of DE-ORGs. (E) DO enrichment analysis of DE-ORGs.
Diagnostic features biomarkers were determined by machine learning
A total of three machine learning algorithms were utilized to identify diagnostic signature biomarkers within the 18 DE-ORGs mentioned above. By applying LASSO analysis, we discovered 9 feature genes: IL1RN, SERPINA1, TLR2, NFKBIA, IL1B, MMP9, ITLN1, ALDH2, and PTGS2 (Fig. 3A, B). Support vector machine (SVM) is supervised machine learning method widely used for classification and regression tasks. To mitigate the risk of overfitting, a recursive feature elimination (RFE) algorithm was employed to extract the most relevant genes from the meta-data cohort. Subsequently, SVM-RFE was utilized to identify the features with the highest discriminative power. The SVM-RFE algorithm indicated that the model’s prediction error was minimized when n = 16, enhancing its predictive capacity (Fig. 3C, D). Consequently, we identified sixteen feature genes including IL1B, ITLN1, NFKBIA, PTGS2, MMP9, TLR2, IL1RN, ALDH2, SLC7A7, CEBPB, TLR4, IRS2, CD163, PYGL, NLRP3, and DGAT2. Random forest analysis revealed that the model reached a stable state at ntree = 500 (Fig. 3E). Following this, we selected genes with importance scores exceeding 5, leading us to eight genes: MMP9, SERPINA1, IL1RN, TLR2, IRS2, NFKBIA, ITLN1, and DGAT2 (Fig. 3F). Finally, we intersected the findings of the three machine learning algorithms (Fig. 3G), resulting in the identification of five key genes: IL1RN, TLR2, NFKBIA, MMP9, and ITLN1. The distribution of these genes among differentially expressed genes is depicted in Fig. 3H. An analysis of protein-protein interactions suggests that these selected candidate genes can create an interaction network that revolves around IL1RN (Fig. 3I).

Identification of diagnostic signature genes. (A, B) The variation curve of regression coefficient (A) and root mean square (RMS) error (B) as a function of Log (λ) in Lasso regression. (C, D) The results of support vector machine-recursive feature elimination (SVM-RFE) algorithm, with the broken line chart showing the number of genes corresponding to the lowest error rate (C) and the highest accuracy (D) for AMI. (E) Identification of the AMI-specific genes using random forest approach, illustrating the impact of the number decision trees on the error rate; the x-axis represents the number of decision trees, while the y-axis denotes the error rate. (F) The most importance genes selected by random forest, with the x-axis indicating the importance index and the y-axis listing the genes. (G) Venn diagram of five candidate genes. (H) Distribution of five candidate genes in a volcano map of differentially expressed genes (DEGs) between AMI and control samples. (I) Protein-protein interaction network based on candidate genes.
Obesity-related genes model for diagnosis of AMI and validation
The five identified genes were utilized to develop a diagnostic model employing a binary logistic regression algorithm within the training cohort. The feature value is calculated using the formula: feature value = -23.3899 + 1.3338 * IL1RN + 0.4214 * TLR2 + 0.6228 * NFKBIA + 0.3462 * MMP9 + 0.6269 * ITLN1. The ROC curve generated from the diagnostic model for acute myocardial infarction (AMI) was employed to assess the model’s diagnostic performance. The results indicated that the area under the curve (AUC) for this model was 0.924 in the training dataset and 0.825 in the validation dataset, reflecting a high level of diagnostic accuracy (Fig. 4A, B). Subsequently, a nomogram was developed utilizing the “rms” package to forecast the occurrence of AMI, incorporating IL1RN, TLR2, NFKBIA, MMP9, and ITLN1. The “Points” were illustrated independently as scores for the five key DE-ORGs, while the “Total Points” represented the cumulative score of these DE-ORGs (Fig. 4C). To evaluate the nomogram’s predictive performance, the AUC was utilized, demonstrating superior predictive accuracy when compared to the five previously mentioned DE-ORGs (Fig. 4D and Supplementary Figure S1A-E). The calibration curve revealed a minimal discrepancy between the actual and predicted incidences of AMI (Fig. 4E). Additionally, the Decision Curve Analysis (DCA) illustrated that the diagnostic model performs effectively, yielding significant net benefits (Fig. 4F). Lastly, we assessed the expression levels of IL1RN, TLR2, NFKBIA, MMP9, and ITLN1 in both AMI and healthy controls using the validation dataset GSE59876. Notable alterations were observed in the expression of IL1RN, TLR2, NFKBIA, and MMP9 between the AMI and control cohorts, whereas ITLN1 did not show significant variation (Fig. 4G). RT-qPCR was conducted to further validate the expression levels of the diagnostic biomarkers, and the findings confirmed that the expression levels of IL1RN, TLR2, NFKBIA, MMP9, and ITLN1 were in agreement with those of the training sets (Fig. 4H and Supplementary Figure S1F-J).

Development and validation of a diagnostic model for predicting AMI. (A, B) ROC curves for the diagnostic model in both the training cohort (A) and the validation cohort (B). (C) Nomogram designed for predicting AMI within the training cohort. (D) The ROC curves of the diagnostic model, revealing AUC of 0.933 for the training set and 0.882 for the testing set. (E, F) Calibration curve (E) and DCA curve (F) of the nomogram model in the training cohort. (G) Validation of the expression of diagnostic biomarkers using the GSE59867 dataset (AMI = 111, control = 49). (H) Diagnostic biomarkers validated in peripheral blood samples from AMI patients (n = 5) and controls (n = 5) through RT-qPCR.
Correlation analysis between immune cells and high-risk and low-risk populations
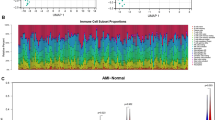
The samples in the training dataset were categorized into low-risk (n = 85) and high-risk (n = 86) groups based on the median feature value obtained from the diagnostic model. A total of six immune cell subsets exhibited distinct infiltration patterns between these two groups, including resting memory CD4 T cells, gamma delta T cells, resting NK cells, monocytes, activated mast cell, and neutrophils (Fig. 5A). The heatmap depicting differential immune cell expression was generated using the “pheatmap” package in R (Supplementary Figure S2A). The relationship between immune cells and genes of the diagnostic model was assessed across different risk groups. The genes IL1RN, TLR2, NFKBIA, and MMP9 demonstrated a significant positive correlation with neutrophils in both risk categories. Additionally, genes IL1RN, TLR2, and NFKBIA exhibited positive correlations with activated mast cells, while IL1RN, TLR2, NFKBIA, and MMP9 revealed a notable negative correlation with memory resting CD4 T cells and gamma delta T cells within the high-risk group (Supplementary Figure S2B, C). Moreover, a significant variance was detected among thirteen immune functions, the most pronounced of which was related to CCR, CD8 + T cells, cytolytic activity, macrophages, MHC class I, neutrophils, NK cells, parainflammation, T cell co-inhibition, T cell co-stimulation, Th1 cells, Th2 cells, and tumor-infiltrating lymphocytes (TIL) (Fig. 5B). The correlation analysis between diagnostic marker genes and immune function categories was performed using the Spearman method. In the high-risk group, IL1RN and TLR2 displayed a notably positive correlation with macrophages and immature dendritic cells (iDCs), while showing a significant negative correlation with T cell co-stimulation, Th2 cells, Th1 cells, helper T cells, checkpoints, and type II interferon (IFN) responses (Fig. 5C, D). Additionally, the findings from the GSEA enrichment analysis revealed that the high-risk group showed significant enrichment in pathways such as chemokine signaling, FC-gamma-R-mediated phagocytosis, leishmania infection, and Toll-like receptor signaling, while ribosome enrichment was most pronounced in the low-risk group (Fig. 5E). This observation implies that these immune functions could play crucial roles during key moments in the pathophysiology of AMI.

Correlation of immune cells and immune functions with high- and low-risk groups. (A, B) Differential analysis of immune cell infiltration (A) and 29 immune functions (B) across these groups. (C, D) Heatmap of the correlation between diagnostic genes and 29 immune functions in the low-risk group (C) and the high-risk group (D). (E) GSEA results for the pathways in the high-risk group. ** p < 0.01, *** p < 0.001; ns, no significant.
Discussion
AMI represents the most severe form of coronary artery disease, resulting in millions of fatalities each year across both developed and developing nations34,35. The evolution of AMI is notably rapid, often leading to delays in treatment. Even with advancements in reperfusion techniques and pharmacological therapies, AMI continues to pose a significant challenge to global health, impacting over 7 million individuals globally on an annual basis36. In industrialized nations, obesity has emerged as a critical health concern, with its prevalence escalating among both adults and children37. This issue goes beyond aesthetics; it considerably heightens the likelihood of serious health complications, such as AMI22,38,39. The link between obesity and coronary artery disease (CAD) largely arises from atherosclerosis, which is intensified by excess fat, especially in the abdominal region40. The American Heart Association recognizes obesity as a significant modifiable risk factor for CAD. In a study based on population data, independent associations were found between being overweight or obese and the early onset of AMI41,42. Moreover, various long-term longitudinal studies have confirmed that obesity serves as an independent risk factor for coronary atherosclerosis43,44. Nevertheless, there has been a scarcity of reports focusing on the impact of obesity-related genes in the diagnosis and risk assessment of AMI. To address this shortcoming, the present study established a new diagnostic signature related to DE-ORGs utilizing datasets obtained from the GEO database. Through comprehensive analysis of transcriptome alongside clinical data, this signature demonstrated promising discriminatory efficacy in both training and validation cohorts. It offers remarkably high accuracy in predicting AMI, with AUC values ranging from 0.747 to 0.962. Furthermore, this signature is correlated with the infiltration of immune cells and varying patterns of immune function. These findings provide new perspectives that enrich the discourse on obesity-related genes and cardiovascular disease (CVD), suggesting valuable clinical applications for the early detection and risk classification of AMI.
In this study, we developed a robust diagnostic scoring system composed of five specific genes: IL1RN, TLR2, NFKBIA, MMP9, and ITLN1. Previous studies have revealed certain associations between these genes and the development and pathogenesis of cardiovascular disease (CVD). For example, IL1RN (interleukin 1 receptor antagonist) is essential in modulating inflammatory responses by suppressing the activities of interleukin 1 (IL-1), a significant pro-inflammatory cytokine45,46. An analysis comparing gene expression levels in 92 patients with AMI and 57 control participants demonstrated that IL1RN, along with IL1B and AQP9, exhibited significant dysregulation in AMI. Functional enrichment analysis associated these genes with immune-related pathways, such as leukocyte migration and cytokine production. The research proposed that the FSTL3-miR-330-3p-IL1B/IL1RN axis could represent novel RNA regulatory pathways involved in the progression of AMI. These results underscored IL1RN’s dual function in inflammation and tissue repair, indicating its potential as a diagnostic or therapeutic target47. TLR2 (toll-like receptor 2), a well-conserved member of the TLR family, has been linked to the regulation of ventricular remodeling following AMI48. Notable cardioprotective benefits, including the preservation of cardiac function and the reduction of ischemia-reperfusion injury, were reported in a mouse model of AMI after the inhibition of TLR248,49. NFKBIA, referred to as NF-kappa B inhibitor alpha, is a protein that plays a crucial role in the regulation of the NF-kappa B signaling pathway, which is essential for controlling the expression of genes involved in inflammation, immune response, and cell survival50. Chronic inflammation is a key characteristic observed in conditions like atherosclerosis, heart failure, and myocardial infarction51,52. By modulating this pathway, NFKBIA can affect the onset and progression of these diseases. MMP9, known as Matrix Metalloproteinase-9, functions as an enzyme that is integral to the degradation of the extracellular matrix. This enzyme plays a significant role in various physiological and pathological processes, encompassing tissue remodeling and inflammation. Its function is closely linked to numerous diseases, particularly those associated with cardiovascular conditions53,54. Increased levels of MMP9 are commonly identified in individuals diagnosed with atherosclerosis55, wherein plaque accumulation occurs in the arteries, resulting in narrowing and diminished blood flow. This plaque accumulation is aggravated by inflammatory processes within the arterial walls, while MMP9 further promotes the degradation of the extracellular matrix in these regions, potentially leading to plaque instability and a heightened risk of rupture53. In the case of myocardial infarction, or a heart attack, serum levels of MMP9 rise markedly within 6 h of onset, peaking between 24 and 48 h thereafter. There is a significant correlation between MMP9 levels and the extent of myocardial infarction as well as the reduction in left ventricular ejection fraction. In clinical settings, the simultaneous assessment of MMP9 and cardiac troponin I (cTnI) enhances the diagnostic sensitivity for early acute coronary syndrome (ACS) (< 4 h) from 72% to 89%56,57. ITLN1 (also referred to as “omentin”) is a highly prevalent mRNA and protein found in visceral adipose tissue and has been linked to the pathophysiology of obesity as well as other metabolically associated diseases, being characterized as a “novel adipokine”58,59. In their research utilizing the ApoE −/− mouse model, Lin et al. demonstrated that administering ITLN1 could elevate collagen levels in the coronary vascular plaques of these mice, diminish the size of necrotic cores, and prevent plaque rupture. This suggests that ITLN1 plays a role in modulating macrophage functionality, impeding the release of inflammatory factors and apoptosis, while also enhancing the stability of atherosclerotic plaques via integrin receptors α v β 3 and α v β 560. In a separate study, Bai et al. reported that plasma concentrations of ITLN1 in individuals with coronary heart disease (CAD) were markedly lower compared to healthy controls (61.21 ± 10.21 ng/dL vs. 95.22 ± 12.21 µg/L, p < 0.0001), and a negative correlation was found between ITLN1 levels and the severity of CAD61. Nevertheless, our findings revealed that the mRNA expression of ITLN1 in the group of patients with acute myocardial infarction (AMI) was significantly elevated compared to the control group. The reasons for this discrepancy with existing literature may relate to differences in sample sizes or individual patient variations, which require further investigation. Thus far, the aforementioned findings partially correspond with the outcomes of the present study, wherein TLR2 and MMP9 were found to be upregulated in AMI. Notably, the abnormal expression of IL1RN, TLR2, NFKBIA, MMP9, and ITLN1 is strongly associated with clinical outcomes in CVD patients. Additionally, this novel diagnostic signature consisting of five DE-ORGs demonstrated excellent predictive capability in both the training cohort (AUC = 0.924) and external validation cohort (AUC = 0.825), offering valuable insights for the prompt diagnosis of AMI. Huang et al. created a logistic regression diagnostic model for AMI, achieving AUC values of 0.794 in the training set and 0.745 in the testing set62. Meanwhile, Chen et al. established a random forest diagnostic model for AMI, with AUC values of 0.855 (training set) and 0.731 (testing set)63. In comparison to these prior studies, our diagnostic model demonstrated commendable accuracy, indicating that it is more robust and universally applicable. This may provide significant insights into new molecular subtypes and improve early diagnostic assessments of AMI, ultimately facilitating tailored treatment and management approaches for future patients.
However, it is important to recognize the constraints of our research. First, the original case numbers in each dataset were somewhat limited, prompting us to utilize multiple datasets. Second, because the study was retrospective and certain critical clinical data were missing in the GEO datasets, comparing the diagnostic model’s predictive value with that of traditional biomarkers in diagnosing AMI is not feasible. Third, verification of the expression levels of IL1RN, TLR2, NFKBIA, MMP9, and ITLN1 should be conducted using RT-qPCR or Western blot in datasets that include larger sample sizes. Lastly, this investigation relied on bioinformatics analysis, highlighting the necessity for larger, prospective clinical validation and comparison with established cardiac biomarkers, such as troponins and CK-MB, to evaluate in vivo outcomes.
Conclusion
Through three machine learning algorithms, LASSO, RF, and SVM-RFE, we identified five genes associated with obesity: IL1RN, TLR2, NFKBIA, MMP9, and ITLN1. These genes may serve as potential therapeutic targets and biomarkers for the development of AMI. More importantly, the present study created an innovative diagnostic model aimed at the early AMI detection, centered on these five genes, thereby providing fresh insights into the underlying mechanisms of AMI and presenting an intriguing avenue for future research initiatives.
Data availability
All data generated or analyzed during this study are included in this published article. The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
References
Zalewski, J. et al. Complicating acute myocardial Infarction. Current status and unresolved targets for subsequent research. J Clin. Med 10. (2021).
Udell, J. A. et al. Prevention of heart failure after acute myocardial infarction. Lancet 406, 1154–1170 (2025).
Liakos, M. & Parikh, P. B. Gender disparities in Presentation, Management, and outcomes of acute myocardial infarction. Curr. Cardiol. Rep. 20, 64 (2018).
Bakaeen, F. G. et al. : The American association for thoracic surgery expert consensus document: coronary artery bypass grafting in patients with ischemic cardiomyopathy and heart failure. J. Thorac. Cardiovasc. Surg. 162 829–850.e821. (2021).
Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury. N Engl. J. Med. 357, 1121–1135 (2007).
Pan, A. et al. MiRNA biomarkers: advancing early detection of acute myocardial infarction. Mikrochim Acta. 192, 500 (2025).
Morrow, D. A. et al. National academy of clinical biochemistry laboratory medicine practice guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Circulation 115, e356–375 (2007).
Wang, C. & Jing, Q. Non-coding RNAs as biomarkers for acute myocardial infarction. Acta Pharmacol. Sin. 39, 1110–1119 (2018).
Anders, K. et al. Coronary artery bypass graft (CABG) patency: assessment with high-resolution submillimeter 16-slice multidetector-row computed tomography (MDCT) versus coronary angiography. Eur. J. Radiol. 57, 336–344 (2006).
Sinnaeve, P. R. et al. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS One. 4, e7037 (2009).
Muse, E. D. et al. A whole blood molecular signature for acute myocardial infarction. Sci. Rep. 7, 12268 (2017).
Deng, X., Shen, A. & Jiang, L. Bioinformatics analysis of JUP in patients with acute myocardial infarction and its potential application in clinical prognostic evaluation. Front. Cardiovasc. Med. 12, 1531309 (2025).
Phillips, K. A. Assessing the value of next-generation sequencing technologies: an introduction. Value Health. 21, 1031–1032 (2018).
Zhang, Z. et al. Unraveling the therapeutic potential of sophora flavescens Aiton in myocardial infarction: an integrative approach combining Bioinformatics, network Pharmacology, and experimental validation. Curr. Pharm. Des. 31, 1474–1497 (2025).
Chen, J. et al. Network Analysis-Based approach for exploring the potential diagnostic biomarkers of acute myocardial infarction. Front. Physiol. 7, 615 (2016).
Kiliszek, M. et al. Altered gene expression pattern in peripheral blood mononuclear cells in patients with acute myocardial infarction. PLoS One. 7, e50054 (2012).
Liu, K., Chen, S. & Lu, R. Identification of important genes related to ferroptosis and hypoxia in acute myocardial infarction based on WGCNA. Bioengineered 12, 7950–7963 (2021).
Tsai, A. G., Williamson, D. F. & Glick, H. A. Direct medical cost of overweight and obesity in the USA: a quantitative systematic review. Obes. Rev. 12, 50–61 (2011).
Stępień, M. et al. Serum concentrations of adiponectin, leptin, resistin, Ghrelin and insulin and their association with obesity indices in obese normo- and hypertensive patients - pilot study. Arch. Med. Sci. 8, 431–436 (2012).
Stępień, M. et al. Obesity indices and inflammatory markers in obese non-diabetic normo- and hypertensive patients: a comparative pilot study. Lipids Health Dis. 13, 29 (2014).
Mehta, L. et al. Impact of body mass index on outcomes after percutaneous coronary intervention in patients with acute myocardial infarction. Am. J. Cardiol. 99, 906–910 (2007).
Yusuf, S. et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 364, 937–952 (2004).
Leek, J. T. et al. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Simon, N. et al. Regularization paths for cox’s proportional hazards model via coordinate descent. J. Stat. Softw. 39, 1–13 (2011).
Tibshirani, R. The Lasso method for variable selection in the Cox model. Stat. Med. 16, 385–395 (1997).
Cao, J. et al. A fast gene selection method for multi-cancer classification using multiple support vector data description. J. Biomed. Inf. 53, 381–389 (2015).
Li, H., Lai, L. & Shen, J. Development of a susceptibility gene based novel predictive model for the diagnosis of ulcerative colitis using random forest and artificial neural network. Aging (Albany NY). 12, 20471–20482 (2020).
Wei, D. et al. NR4A2 May be a potential diagnostic biomarker for myocardial infarction: A comprehensive bioinformatics analysis and experimental validation. Front. Immunol. 13, 1061800 (2022).
Luo, W. & Brouwer, C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831 (2013).
Zheng, W. et al. Integrative analysis of a novel signature incorporating metabolism and stemness-related genes for risk stratification and assessing clinical outcomes and therapeutic responses in lung adenocarcinoma. BMC Cancer. 25, 591 (2025).
Desai, A. et al. Chronic pancreatitis and risk of atherosclerotic cardiovascular disease: A US cohort Propensity-Matched study. Pancreas 52, e21–e28 (2023).
Jang, D. K. et al. Risk of cardiovascular disease and mortality in patients with diabetes and acute pancreatitis history: a nationwide cohort study. Sci. Rep. 12, 18730 (2022).
Powell-Wiley, T. M. et al. Obesity and cardiovascular disease: A scientific statement from the American heart association. Circulation 143, e984–e1010 (2021).
Nichols, M. et al. Cardiovascular disease in Europe 2014: epidemiological update. Eur. Heart J. 35, 2950–2959 (2014).
Yeh, R. W. et al. Population trends in the incidence and outcomes of acute myocardial infarction. N Engl. J. Med. 362, 2155–2165 (2010).
Reed, G. W., Rossi, J. E. & Cannon, C. P. Acute myocardial infarction. Lancet 389, 197–210 (2017).
Flegal, K. M. et al. Overweight and obesity in the united states: prevalence and trends, 1960–1994. Int. J. Obes. Relat. Metab. Disord. 22, 39–47 (1998).
Kenchaiah, S. et al. Obesity and the risk of heart failure. N Engl. J. Med. 347, 305–313 (2002).
Ruperez, C. et al. Obesity accelerates cardiovascular ageing. Eur. Heart J. 46, 2161–2185 (2025).
Kooner, J. S. et al. Abdominal obesity, impaired nonesterified fatty acid suppression, and insulin-mediated glucose disposal are early metabolic abnormalities in families with premature myocardial infarction. Arterioscler. Thromb. Vasc Biol. 18, 1021–1026 (1998).
Suwaidi, J. A. et al. Obesity is associated with premature occurrence of acute myocardial infarction. Clin. Cardiol. 24, 542–547 (2001).
Sahu, P. et al. Obesity and heart failure: mechanistic insights and the regulatory role of MicroRNAs. Genes (Basel) ; 16. (2025).
Rabkin, S. W., Mathewson, F. A. & Hsu, P. H. Relation of body weight to development of ischemic heart disease in a cohort of young North American men after a 26 year observation period: the Manitoba study. Am. J. Cardiol. 39, 452–458 (1977).
Rimm, E. B. et al. Body size and fat distribution as predictors of coronary heart disease among middle-aged and older US men. Am. J. Epidemiol. 141, 1117–1127 (1995).
Ridker, P. M. et al. Interleukin-1β Inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory thrombosis outcomes study (CANTOS). Am. Heart J. 162, 597–605 (2011).
Arend, W. P. & Gabay, C. Physiologic role of interleukin-1 receptor antagonist. Arthritis Res. 2, 245–248 (2000).
Shao, G. Integrated RNA gene expression analysis identified potential immune-related biomarkers and RNA regulatory pathways of acute myocardial infarction. PLoS One. 17, e0264362 (2022).
Shishido, T. et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 108, 2905–2910 (2003).
Arslan, F. et al. TLR2 and TLR4 in ischemia reperfusion injury. Mediators Inflamm ; 2010: 704202. (2010).
Hayden, M. S. & Ghosh, S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 (2012).
Kong, P. et al. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal. Transduct. Target. Ther. 7, 131 (2022).
Zhang, Q. et al. Signaling pathways and targeted therapy for myocardial infarction. Signal. Transduct. Target. Ther. 7, 78 (2022).
Li, T. et al. The Role of Matrix Metalloproteinase-9 in Atherosclerotic Plaque Instability. Mediators Inflamm ; 2020: 3872367. (2020).
Yabluchanskiy, A. et al. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiol. (Bethesda). 28, 391–403 (2013).
Wei, X. L. et al. NINJ1 and MMP9: potential biomarkers for intracranial atherosclerosis plaque vulnerability. Front. Neurol. 16, 1552948 (2025).
Deleon-Pennell, K. Y. et al. The circular relationship between matrix metalloproteinase-9 and inflammation following myocardial infarction. IUBMB Life. 67, 611–618 (2015).
Halade, G. V., Jin, Y. F. & Lindsey, M. L. Matrix metalloproteinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol. Ther. 139, 32–40 (2013).
Schäffler, A. et al. Genomic structure of human omentin, a new adipocytokine expressed in omental adipose tissue. Biochim. Biophys. Acta. 1732, 96–102 (2005).
Shibata, R. et al. Omentin as a novel biomarker of metabolic risk factors. Diabetol. Metab. Syndr. 4, 37 (2012).
Lin, X. et al. Omentin-1 modulates macrophage function via integrin receptors αvβ3 and αvβ5 and reverses plaque vulnerability in animal models of atherosclerosis. Front. Cardiovasc. Med. 8, 757926 (2021).
Bai, P. et al. Association between coronary artery disease and plasma Omentin-1 levels. Cureus 13, e17347 (2021).
Huang, D. et al. Machine learning revealed ferroptosis features and a novel ferroptosis-Based classification for diagnosis in acute myocardial infarction. Front. Genet. 13, 813438 (2022).
Yifan, C., Jianfeng, S. & Jun, P. Development and validation of a random forest diagnostic model of acute myocardial infarction based on Ferroptosis-Related genes in Circulating endothelial cells. Front. Cardiovasc. Med. 8, 663509 (2021).
Acknowledgements
We sincerely acknowledge the contributions from the GEO, GeneCards and GSEA databases, and the SangerBox platform (http://sangerbox.com/).
Funding
This work was supported by the National Natural Science Foundation of China (No: 81372147) and Henan University support grant CX3070A0780502.
Author information
Authors and Affiliations
Contributions
Weixia Dong : Data curation, Investigation, Software, Validation. Zixin Xue : Investigation, Formal analysis, Data curation. Min Zhang : Investigation, Data curation. Feng Lu : Conceptualization, Writing-original draft, Writing-review & editing, Project administration, Software, Funding acquisition, Supervision. Shaoping Ji : Conceptualization, Supervision, Resources, Methodology, Writing- review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethic approval and consent to participate
Ethical approval was obtained from the Ethics Committee of Medical School of Henan University, China (HUSOM-2018-282).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, W., Xue, Z., Zhang, M. et al. Deciphering the clinical implication of an obesity-related gene signature as the novel biomarker for acute myocardial infarction diagnosis. Sci Rep 15, 44726 (2025). https://doi.org/10.1038/s41598-025-28461-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28461-0
