
Abstract
Traumatic brain injury (TBI) remains a leading cause of morbidity and mortality, with no effective treatments available. Biperiden, a muscarinic receptor antagonist, has demonstrated beneficial effects by modulating neural plasticity and reducing excitotoxicity. This study investigated the acute impact of cellular and structural changes in a moderate TBI model using lateral fluid percussion injury (LFPI) in male Wistar rats. Biperiden (8 mg/kg) was administered intraperitoneally at 6 h post-injury, followed by two additional doses at 8-hour intervals. Brain and blood were collected twenty-four hours after TBI for, histological, immunohistochemical, and Single Molecule Array (SIMOA) analyses. Results revealed that biperiden-treated (TBI-BIP) rats exhibited diminished degenerating neurons, lower T-tau and NfL levels, decreased astrocyte activation, and restored microglia morphology compared to saline-treated (TBI-SAL) rats, attenuating TBI-induced damage. Motor deficits were most pronounced on Day 1 post-trauma, with non-significant statistical difference in neuroscore performance between the TBI groups, however the biperiden treatment modulates astrocyte reactivity, diminish gliosis, and promote a neuroprotective phenotype. Additionally, biperiden preserves microglial branching following injury. These findings demonstrate that biperiden exerts beneficial effects during the acute phase of TBI by decreasing neuronal degeneration, excitotoxicity, and inflammation, highlighting its therapeutic potential for post-TBI intervention.
Similar content being viewed by others


Introduction
Traumatic brain injury (TBI) is a neurophysiological brain dysfunction caused by an external force (i.e., rapid acceleration or deceleration, blast, penetrating injury, or impact) that causes transient amnesia, alteration of consciousness, mortality, and morbidity1. TBI is the leading cause of both disability and mortality in individuals below the age of 45 years2.
Traditionally, TBI is classified based on the degree of impairment per the Glasgow Coma Scale (GCS) as mild, moderate, or severe3. Moderate TBI is diagnosed by confusion lasting from days to weeks, loss of consciousness, and cognitive, physical, and behavioral impairment lasting for months or sustained permanently, with a GCS score between 9 and 124. Approximately 20% of TBI cases are estimated to be moderate and severe5.
The mechanism behind TBI involves a complex process of primary and secondary injuries. The primary injury results from the initial injury forces applied to the brain, causing immediate tissue distortion and destruction6. Secondary injury is a cascade of pathophysiological responses that occur after the initial insult to the brain, leading to alterations in cell function and propagation of injury through excitotoxicity, edema formation, blood-brain barrier disruption, inflammation, ischemic injury, disruption of calcium homeostasis, and intracranial hypertension6,7. Secondary injury plays a critical role in propagating damage, making the focus of research to identify the molecular mechanisms involved in avoiding or reducing the long-term damage following the initial brain injury8.
To provide better insight into TBI questions, animal models facilitate the translation of preclinical findings to humans9. Among TBI models, lateral fluid percussion injury (LFPI) produces brain injury by rapidly injecting a small volume of fluid directly into the cranium, resulting in transient displacement and deformation of the ipsilateral hemisphere10. This model is widely used to induce a mixed focal-diffuse brain injury pattern that closely mimics human closed-head TBI.
Despite decades of research11, no pharmacological treatments have been approved for TBI. Current management primarily relies on surgical interventions, supportive care, and rehabilitation strategies, which aim to minimize secondary complications rather than reverse neuronal damage. Several experimental therapies are under investigation5,12,13,14. Given the lack of approved pharmacological options, investigating early drug-induced modulation of injury mechanisms may provide critical insights for the development of future therapeutic strategies.
Biperiden, a cholinergic antagonist that acts on the muscarinic receptor, has been shown to influence neuronal excitability and plasticity after brain injury. It has also been reported to delay the onset of seizures and reduce their severity15, raising clinical interest in its potential as a therapeutic intervention following TBI13.
In this study, we aim to investigate the effects of biperiden in a moderate TBI animal model during the acute phase of injury (24 h) focusing on cellular and structural changes in the brain. The null hypothesis was that biperiden treatment would not alter neuronal or glial responses in the acute phase after TBI. The alternative hypothesis was that biperiden could attenuate acute neuronal degeneration and inflammation, suggesting an early modulatory effect on injury progression.
Methods
Animals
A total of 129 male Wistar/EPM rats (8 weeks old, weighing between 220 and 350 g) from the CEDEME—Center for the Development of Animal Models at the Universidade Federal de São Paulo (UNIFESP) were housed in cages measuring 41 cm x 34 cm x 64 cm. The animals were maintained at room temperature (22 ± 2 °C), under a 12:12 light/dark cycle (lights on at 6:00 a.m.), and had free access to food and water. The exclusive use of male rats in this study is justified by epidemiological data showing that young adult males are disproportionately affected by TBI, with an incidence approximately five times higher than that of females17.
All animal handling and experimental procedures complied with the ARRIVE 2.0 guidelines and the EU Directive 2010/63/EU for animal care and use of laboratory animals and were approved by the Board for Ethics in the Use of Animals (CEUA, Comissão de Ética no Uso de Animais), an institutional ethics committee of the UNIFESP, protocol no. 7,641,161,222. All efforts were made to minimize the suffering and the number of animals.
Experimental design and groups
The group size was determined according to previous publications18,19, which employed 20 animals per group. Animals were assigned randomly to experimental groups. The investigators were blinded to group allocation throughout the experiments and during outcome assessments. The sample size employed between 19 and 21 animals per group (81 total)15,16. Animals were assigned randomly to experimental groups. The investigators were blinded to group allocation throughout the experiments and during outcome assessments.
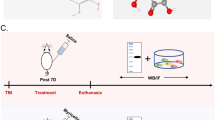
The experimental design proceeded, as it is shown in Fig. 1.

Experimental design. Experimental design overview. Schematic figures adapted from the rat brain atlas by Paxinos and Watson24 represent the regions (in pink and orange) that were analyzed in the ipsilateral cortex and hippocampus, ranging from − 2.76 to − 4.08 mm in the anterior–posterior axis relative to bregma.
All animals were distributed into four groups:
-
SHAM-SAL: Animals that had a craniotomy but did not receive the TBI and received saline solution injection via intraperitoneal (i.p.) (n = 20).
-
SHAM-BIP: Animals that had a craniotomy but did not receive the TBI and received biperiden injections (i.p.) (n = 21).
-
TBI-SAL: Animals that had a craniotomy received the TBI and saline solution injections (i.p.) (n = 19).
-
TBI-BIP: Animals with craniotomy receive the TBI and biperiden injections (i.p.) (n = 21).
Due to variability in tissue integrity following LFPI, final sample sizes for histological and biochemical analyses ranged from 5 to 7 per group for histology and immunohistochemistry, and 9–12 for SIMOA, as only samples meeting predefined quality criteria were included.
Model of traumatic brain injury: lateral fluid percussion injury (LFPI)
Animals were anesthetized through an intraperitoneal (i.p.) injection of a cocktail containing ketamine 10% (95 mg/kg) and xylazine 2% (12 mg/kg) as a single dose. After anesthetic induction, the depth was assessed, and two drops of 2% lidocaine were instilled into the animal’s right and left ear canals, where the bars of the stereotaxic apparatus would be introduced to secure the animal. Then, a dose of lidocaine (20 mg/kg) as a local anesthetic was administered intradermally into the surgical site, and a 10 mm midline incision was made. The skull was exposed, and a burr hole (diameter: 3 mm) was made using a dental drill at -2.76 mm anterior-posterior (AP) to bregma and − 3 mm medial-lateral (ML) taking extreme care to keep the dura mater intact. At the craniotomy site, a cannula (female Luer-lock) was fixed with dental cement and afterward filled with sterile 0.9% NaCl. The cannula remained in place throughout the experimental period and was removed only after perfusion. After surgery, animals were administered ibuprofen (10 mg/kg, oral), pentabiotic (24.000 ul/kg, intramuscular), and two hours after surgery, buprenorphine (0.1 mg/kg, i.p.) was administered after surgery.
Twenty-four hours after surgery, animals received inhalation anesthesia (4% isoflurane with maintenance in 2%, at a flow rate of 1–1.5 L/min in freely breathing oxygen), and the succeeding anesthetic depth, the fluid-percussion device (AmScien Instruments, model FP302, Richmond, VA, USA) was connected via the male Luer-lock fitting to the rat. The LFPI model was applied at a 17–18° angle with a brief (10– 15 ms) transient fluid pressure pulse impact against the exposed dura. The pressures of the pulses were measured by a transducer extracranially and recorded on a storage oscilloscope. The impact force ranged from 1.78 to 2.61 atm against the exposed dura mater, affecting the regions of the primary motor cortex (M1), secondary motor cortex (M2), and primary somatosensory cortex (S1).
Additionally, the righting reflex test was performed to assess neurological function following TBI. This test measures the time required for an animal to regain consciousness or recover the righting reflex after injury. Immediately after the trauma, animals were placed in a supine position, and the time taken to return to a quadrupedal stance was recorded, indicating the recovery of the righting reflex. Moreover, mortality rate and trauma severity (measured in atmosphere (atm)) were recorded after TBI induction. Sham animals underwent the same procedures but did not receive the transient fluid percussion pulse.
Animal exclusion
Animals with dura mater rupture during surgery were excluded and euthanized according to the Guide for the Implementation of a Humane Endpoint20. Following TBI, only moderate models (1.78–2.61 atm) were included. Pain monitoring adhered to the same exclusion and intervention criteria, with humane endpoints applied when necessary.
Biperiden treatment
Animals received intraperitoneal injections of biperiden hydrochloride (8 mg/kg, Cristália, Brazil) or saline solution for 24 h. Treatment consisted of three doses: the first dose administered 6 h after the trauma, followed by two additional doses at 8-hour intervals (see Fig. 1). The dose of 8 mg/kg was selected based on previous rodent studies demonstrating the safety and efficacy of biperiden in modulating excitotoxicity and epileptogenesis15,21. Importantly, this dose reliably induced memory impairments in rodents, serving as an indirect indicator of effective muscarinic receptor blockade22. Moreover, the dose-dependent effects of a similar treatment with anticholinergics were also investigated by Benassi and colleagues21 and can be safely administered after TBI.
Composite neuroscore
Alterations in the somatomotor and vestibular functions were subjected to the composite neuroscore test, described previously by McIntosh, 198910 and Andrade et al., 202223. The test was conducted at three time points: before surgery and TBI (baseline; D-1), after surgery but before TBI (D0), and after both surgery and TBI (D1). The measurement taken on D0 allowed us to distinguish the effects of the surgical procedure from those of the TBI.
All tests were performed from 9 to 10 a.m by two researchers. Rats were scored on a 0 to 4 ordinal scale (0 = severely impaired, 4 = normal) for the following parameters:
(1) Left and right forelimb flexion during tail suspension (4 points each), (2) Left and right hindlimb flexion when the animal was gently pulled backward by the tail along a flat surface (4 points each), (3) Resistance to left and right lateral pulsion (4 points each), and (4) Ability to maintain posture on a 45° inclined board (4 points). The maximum possible score was 28, indicating normal motor and vestibular function.
Blood and brain tissue preparation
Twenty-five hours after the trauma, animals designated for biomarker analysis were decapitated without anesthesia, precisely two hours after the third biperiden administration. Immediately after decapitation, blood was collected in 4 mL EDTA tubes, gently inverted, and immediately processed in a Baby® centrifuge (FANEM, model 206) at 3.000 rpm for 10 min at room temperature. After processing, the plasma was aliquoted into 200 µL volumes and stored at -80 °C for subsequent analysis using the Single Molecule Array (SIMOA) technique.
At the same timepoint (25 h post-trauma and 2 h after the final biperiden dose), animals intended for histological and immunohistochemical analysis were deeply anesthetized. A sternotomy was made to access the heart. Then, the animals were perfused through the heart. Perfusion started with 250 mL of 0.1 M PBS solution (phosphate buffer solution; 5.52 g of monobasic sodium phosphate plus 21.88 g of dibasic sodium phosphate), then 250 mL of 4% paraformaldehyde (diluted in PBS solution). The brains were removed and post-fixed in a 4% formaldehyde solution, followed by a 30% sucrose solution (diluted in PBS) for cryoprotection. The brains remained in this solution until they showed signs of dehydration, a procedure usually occurring within 24 h. Immediately after this period, the brains were dried and frozen at -80 °C. Subsequently, the brains were sectioned in a cryostat (Leica CM1850) in coronal brain Sect. (40 μm thick). Then, the sections were stored in an anti-freezing solution (300 g of sucrose, 500 mL of PBS solution, and 300 mL of ethylene glycol) at -20 °C until they were processed for histological and immunohistochemical analysis. Brain sections were selected according to the stereotaxic atlas24, ranging from − 2.76 to − 4.08 mm in the anterior–posterior axis relative to bregma (dorsal hippocampus), ensuring consistent sampling of both cortical and hippocampal ipsilateral regions for the assessment of histological.
Histological Nissl staining
Brain slices were mounted on gelatin-coated glass slides and stained for 5 min with 1% cresyl violet dissolved in distilled water and filtered. Stained slides were dehydrated for 1 min using 100%, 96%, and 70% ethanol, cleared in xylene for 2 min, covered with Entellan ® mounting medium, and coverslipped. The percentage of the area of the lesion was calculated according to the following formula25,26:
Lesion area (%)=(Ipsilateral hemisphere area / Contralateral hemisphere area) * 100.
Slides containing the hippocampus and the cortex among the groups SHAM-SAL, SHAM-BIP, TBI-SAL, and TBI-BIP (6 brain sections per animal per group) were delineated to include remaining tissue on the contralateral and ipsilateral hemispheres motor cortex (M1 and M2), the somatosensory cortex (S1), and the hippocampal region (CA1). We used the program ImageJ (National Institute of Health- NIH Image https://imagej.net/ij/, and the polygon section plugin to delineate the brain slices. The mean values for each animal were calculated.
Single molecule array (SIMOA)®
The SIMOA (Quanterix Corporation, Lexington, MA) was used to detect brain biomarkers in the blood related to TBI and epileptogenesis, such as total tau (T-tau; a marker of neuronal function)27, Neurofilament Light Chain (NfL, an indicator of neuronal damage), Acid Fibrid Glial Protein (GFAP; a marker of astrocytic damage), and Ubiquitin C-Terminal Hydrolase L1 (UCH-L1; an early marker of neuronal injury)28. These biomarkers were selected for their sensitivity to TBI-related damage29,30. Moreover, this technology has previously been applied in rodent models, including rats31,32,33.
Plasma biomarker levels were quantified using the Neurology 4-Plex B (N4PB) assay, a specific immunoassay kit that detects NFL, total tau, GFAP, and UCH-L1. The samples were thawed, vortexed, and centrifuged for the analysis. The analysis was conducted at Richet/IDOR in Rio de Janeiro using the SIMOA HD-1 device.
Plasma samples from animal models were thawed, and 60 µL of each sample was pipetted into each well of a 96-well plate, along with the validated calibrators for the assay. The device was loaded with the necessary reagents and supplies, including pipettes, commercial kit beads, discs, and the sample plate. After adjusting the parameters, the assay was initiated and lasted approximately 4–5 h. The automated calibration cycles, pipetting, washing, incubation, matrix transfer, and signal reading recorded the intensity of the fluorescent signals emitted by the microparticles, expressed in pg/mL. The calibration curve converted the signals obtained from the samples into quantifiable concentrations of the biomarkers, a conversion typically performed automatically by the SIMOA software.
Fluoro-Jade® B
Fluoro-Jade® B (FJ-B, AG310 Merck-Millipore) was used to stain degenerating neurons. Fixed sections were mounted on glass slides, and FJ-B staining was performed as follows: The slides were mounted and let dry at room temperature for 3 to 5 days. Then, the slices were placed in an oven at 37 °C for 20 min.
The slices were then immersed in 100% ethanol for 3 min, followed by 1 min in 70% ethanol and 1 min in distilled water. Slides were then incubated in 0.06% potassium permanganate (KMnO₄) in distilled water for 15 min. Next, slides were rinsed in distilled water and transferred to a 0.0001% FJ-B staining solution in 0.1% acetic acid for 30 min. The FJ-B working solution was prepared from a stock FJ-B solution (0.01% in distilled water). After rinsing, the slides were coverslipped with DPX.
The slides were analyzed using a 20X objective and a computer-based digitizing image system (Zeiss Axiovert; Carl Zeiss, Germany) connected to an Axiocam 208 color camera. Image analysis was conducted with ImageJ, employing the multi-point tool for cell counting in the areas of interest (cortex (M1, M2, S1) and the hippocampus (CA1).
Immunofluorescence
Immunofluorescence was performed to identify astrocytes, water channels, and their respective nuclei. Free-floating sections were washed five times with PBS solution to remove all anti-freezing solution and incubated in a blocking solution (90 mL of 0.1 M PBS, 250 µL of Triton X-100, 40 µL of bovine serum albumin) for 30 min at room temperature under constant stirring. After blocking, cells were incubated for 2 h with the following primary antibodies: polyclonal chicken anti-glial fibrillary acidic protein (GFAP) antibody (1:1000, Merck, AB5541) for astrocytes, and a polyclonal primary antibody anti-Iba1 made in rabbit (1:1000, Wako, 019-19741) diluted in the blocking solution. After washing in PBS, sections were incubated with secondary antibodies (Alexa 555 anti-chicken, Alexa 594 anti-rabbit, all 1:600, Invitrogen) for 2 h. Nuclei were stained with DAPI (1:10.000; Thermo Fisher Scientific, Carlsbad, CA, USA).
In both immunofluorescence procedures, DAPI was used for nuclear staining for 10 min, followed by three washes with PBS (each for 10 min). The slides were then mounted and covered with dibutylphthalate polystyrene xylene (DPX).
The slides were analyzed in a blind manner using a 20X objective using a computer-based digitizing image system (Zeiss Axiovert 100 M; Carl Zeiss, Germany) connected to an LSM 810 Confocal Laser Scanning System. The laser and detector were maintained at constant settings during the acquisition of each staining set.
For each section, cells were qualitatively evaluated for their localization in the brain areas of interest (cortex (M1, M2, S1) and hippocampus (CA1)) and their morphology. Slides and resultant images were coded, and the quantification of fluorescence signals from GFAP and Iba-1 cells was quantified by integrated density and ImageJ’s plugin Skeletonize as a semiquantitative methodology according to Marques et al. 202316 to analyze the total number, the number of branches, and the length of the branches of astrocytes and microglia in order to have a clearer morphology. The analysis used ImageJ (National Institute of Health-NIH Image).
Statistical analysis
Sample size calculation was performed a priori using G*Power version 3.1.9.734 software. The calculation is based on an F-test for one-way ANOVA (fixed effects), considering four groups, a medium-sized effect (f = 0.35), a significance level of 5% (α = 0.05), and a statistical power of 90%. Effect size was calculated with Cohen´s d formula35, randomization and group allocation were performed by an investigator blinded to the experimental conditions.
Data were analyzed using GraphPad Prism (version 10.1.1; GraphPad Software Inc., San Diego, CA). Parametric tests were applied to variables with a normal distribution, including Student’s t-test, one-way ANOVA, and two-way ANOVA for repeated measures with Tukey’s post hoc test. For non-parametric variables, the Mann-Whitney U and Kruskal-Wallis tests were used, followed by Holm-Sidak post hoc analysis. Results are expressed as mean ± SD, with a P-value < 0.05 considered statistically significant.
Results
Mortality and the righting reflex are affected by LFPI
After standardizing the parameters for moderate TBI using our equipment, we determined that the optimal pressure range is between 1.78 and 2.86 atm, with a mean of 2.20 ± 0.2 atm and impact angle is between 17° and 18°, which were established during pilot experiments with a separate set of animals, ensuring consistent induction of moderate TBI, according to the Kabadi et al., 2010 classification36.
Among the 129 animals, 11 died during anesthesia, 7 during surgery, and 21 due to trauma. Additionally, 9 animals were excluded due to complications, including dura mater rupture, insufficient atm (mean 0.62 ± 0.47), pre-trauma bleeding following surgery, unsecured male Luer-lock, and other technical difficulties.
We conducted the righting reflex test to assess neurological function immediately following the impact, comparing the SHAM groups and the TBI groups, and analyzed the results using the one-way ANOVA test (F [3, 63] = 11.78; p < 0.0001). The results showed that animals subjected to TBI exhibited significantly impaired neurological function, taking longer to return to the four-paw position in the TBI-SAL (379.6 ± 198.5 s) and TBI-BIP (372.9 ± 196.8 s) groups compared to the SHAM-SAL (164.2 ± 65.91 s) and SHAM-BIP (119.8 ± 76.06 s) groups (Fig. 2A).

Validation of moderate traumatic brain injury. (A) Comparison of the righting reflex between animals of SHAM and TBI groups. (B) Composite neuroscore test (in points) conducted pre-surgery (Day − 1), before the impact (Day 0), and during the recovery phase (Days 1). (C) Comparison of the neuroscore points on Day 1 among groups. Data are presented as mean ± SEM. Final sample size per group: n = 19–21. (D) Percentage of lesion area between SHAM and TBI animals, and images of Nissl representation per SHAM and TBI animals. Statistical significance: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.001 (n = 6 per group). Data are shown as the mean ± standard deviation of the mean.
Neuroscore
To assess motor deficits, we performed the compost neuroscore test. We evaluated the effects of craniotomy, LFPI impact, and the biperiden treatment, which were followed up over three days (Day-1, Day 0, and Day 1). The data were analyzed using a two-way ANOVA of repetitive measurements, considering both time variation and experimental groups.
The results showed significant effects for the interaction between time and group (F [6, 154] = 5.51; p < 0.001); as well as for time (F [1.15, 89] = 29.09; p < 0.0001), and group (F [3, 77] = 5.77; p = 0.0013. These findings indicate that the motor performance changed over time, independent of group differences.
Tukey´s post-hoc analysis revealed significant differences between days. On Day 0 neuromotor deficits were significantly greater than on Day − 1 (p = 0.0002), with a decrease in the neuromotor performance following surgery [Day 0: 27.45 ± 1.18 points vs. Day − 1: 28 ± 00 points]. Similarly, performance declined further from Day 0 to Day 1(p = 0.0001) following trauma [Day 1: 25.16 ± 4.66 points vs. Day 0: 27.45 ± 1.18 points). As expected, a significant difference was also observed between Day − 1 and Day 1(p < 0.0001), highlighting the progressive motor impairment over time (Fig. 2B).
Regarding inter-group differences, they were detected only on Day 1, there was a significant difference between the SHAM (SHAM-SAL and SHAM-BIP) and the TBI-SAL group. Showing that TBI-SAL (21.97 ± 6.40; p = 0.027) had the worst motor performance than the SHAM-SAL group (26.7 ± 1.91 points). This poor performance of the TBI-SAL group (p = 0.016) was also compared with the SHAM-BIP group (27 ± 1.76 points) (Fig. 2C). Additionally, it was observed that the TBI-BIP group (25.28 ± 4.6 points) had better motor performance than the TBI-SAL; however, this difference was not significant.
Evaluation of the lesion area and the effects of biperiden
The percentage of lesion area resulting from TBI was evaluated using one-way ANOVA, which revealed statistically significant differences among the experimental groups (F [3, 20] = 13.17; p < 0.0001). Tukey’s post hoc test indicated that the TBI-SAL group (15.86 ± 3.18%) exhibited a significantly higher lesion volume loss compared to the SHAM-SAL (4.97 ± 2.03%; p < 0.0001) and SHAM-BIP (6.93 ± 1.64%; p = 0.0007) groups. Similarly, the TBI-BIP group (11.32 ± 5.09%) demonstrated a significantly increased lesion volume loss relative to the SHAM-SAL group (p = 0.015) (Fig. 2D).
Biperiden reduces blood biomarkers of neuronal damage after TBI
To evaluate blood biomarkers for TBI, the ultra-sensitive SIMOA technique was employed to measure T-tau, NfL, GFAP, and UCH-L1 blood levels. Significant variations in some of these biomarkers were observed among groups.
For T-tau levels, one-way ANOVA revealed a statistically significant difference among groups (F [3, 34] = 5.11; p = 0.005). Tukey’s post-hoc test showed higher levels of T-tau in the TBI-SAL group (1.20 ± 0.85 pg/mL) than SHAM-SAL (0.37 ± 0.09 pg/mL; p = 0.019), SHAM-BIP (0.54 ± 0.51 pg/mL; p = 0.041), and TBI-BIP (0.37 ± 0.23 pg/mL; p = 0.0108) (Fig. 3A).

Levels of plasma biomarkers among the groups in pg/mL. (A) Total tau (T-tau) level in pg/mL. (B) Neurofilament Light Chain (NfL) level in pg/mL. (C) Acid Fibrid Glial Protein (GFAP) level in pg/mL. (D) Ubiquitin C-Terminal Hydrolase L1 (UCHL-1) level in pg/mL. Statistical significance: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.001. Data are shown as the mean ± standard deviation of the mean. Data are presented as mean ± SEM. Sample size per group: n = 9–12. Only samples meeting predefined biochemical quality criteria were included in the analysis.
For NfL levels, one-way ANOVA indicated a significant difference among the groups (F [3, 32] = 3.8; p = 0.019). Tukey’s post-hoc test revealed also higher levels of NfL TBI-SAL (871.4 ± 920.4 pg/mL) compared to TBI-Bip (229 ± 142.4 pg/mL; p = 0.043) (Fig. 3B).
Regarding GFAP and UCH-L1 levels, one-way ANOVA did not reveal significant differences among the groups (GFAP, F [3, 31] = 2.34; p = 0.0927; UCH-L1, F [3, 39] = 1.44; p = 0.25) (Figs. 3C-D). These findings indicate that while T-tau and NfL levels are reduced by biperiden after TBI, GFAP and UCH-L1 levels do not show statistically significant differences between TBI and treatment groups.
Biperiden decreases neuronal degeneration in the cortex after TBI
FJ-B measured neuronal degeneration in the cortex. The two-way ANOVA showed a significant difference among groups (F [3, 19] = 8.44; p = 0.0009). Tukey’s post-hoc test determined that the TBI-SAL group has a significantly higher level of degenerated neurons (48.27 ± 22.27) compared to SHAM-BIP (3.98 ± 4.95, p = 0.014), SHAM-SAL (8.69 ± 7.29, p = 0.027), and TBI-BIP (19.68 ± 21.38, p = 0.033). These findings show that TBI may increase degeneration of neurons in the cortex, while treatment with biperiden could mitigate these effects (Figs. 4A-B).

Fluorojade B (FJ-B) positive cells in cortex and CA1. (A) Images of FJ-B in the cortex for each group I-SHAM-SAL, II-SHAM-BIP, III- TBI-SAL, IV- TBI-BIP. (B) Graphic representation of the number of FJ-B positive cells in the cortex for each group. (C) Images representative of FJ-B in the CA1(dorsal hippocampus) for each group: I-SHAM-SAL, II-SHAM-BIP, III-TBI-SAL, IV-TBI-BIP. (D) A graphic representation of the number of FJ-B positive cells in the cortex for each group (n = 5–7 per group).
In CA1, the Kruskal-Wallis test did not find statistically significant differences among groups (H = 5; d.f.=3; p = 0.17) (Figs. 4C-D).
Biperiden reduces astrocyte activation and resets cellular morphology in the cortex and CA1 after TBI
The quantity and integrated density of astrocytes (GFAP) were evaluated in the cortex and CA1. In the cortex (H = 2.75; d.f.=3; p = 0.43) and in CA1 (F [3, 20] = 1.28; p = 0.31), the integrated density analysis did not show any significant differences between the groups (Figs. 5 and 6A-B).

GFAP positive cells in the cortex. (A) Images representative of GFAP in the cortex for each group: I-SHAM-SAL, II-SHAM-BIP, III-TBI-SAL, IV-TBI-BIP. (B) Graphic representation of the integrated density of GFAP corrected with DAPI for each group. (C) Number of astrocytes for each group. (D) Number of branches for each group. (E) Average branch length measured in µm for each group. Statistical significance: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.001. (n = 5–7 per group. Only samples meeting predefined histological quality criteria were included in the analysis). Data are shown as the mean ± standard deviation of the mean.

GFAP-positive cells in CA1. (A) Images representative of GFAP in CA1 for each group: I-SHAM-SAL, II-SHAM-BIP, III-TBI-SAL, IV-TBI-BIP. (B) Graphic representation of the integrated density of GFAP corrected with DAPI for each group. (C) Number of astrocytes for each group. (D) Number of branches for each group. (E) Average branch length measured in um for each group. Statistical significance: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.001. (n = 5–7 per group. Only samples meeting predefined histological quality criteria were included in the analysis). Data are shown as the mean ± standard deviation of the mean.
Regarding the number of astrocytes in the cortex, the one-way ANOVA test showed significant differences among groups (F [3, 20] = 28.24; p < 0.0001). Tukey’s post-hoc test showed that TBI-SAL group (463.2 ± 161.9) had more astrocytes compared to SHAM-SAL (94.43 ± 18.87 astrocytes; p < 0.0001), SHAM-BIP (107.1 ± 38.96 astrocytes; p < 0.0001), and TBI-BIP (97 ± 6.58 astrocytes; p < 0.0001) (Fig. 5C).
To evaluate the astrocytes’ morphology, the number of astrocyte branches in the cortex was analyzed, and the one-way ANOVA test showed significant differences among groups (F [3,20] = 54.79; p < 0.0001). Tukey’s post-hoc test showed that the TBI-SAL group (16.91 ± 3.4 branches/cell) had more branches compared to SHAM-SAL (5.81 ± 077 branches/cell; p < 0.0001), SHAM-BIP (6.70 ± 0.76 branches/cell; p < 0.0001), and TBI-BIP (5.67 ± 0.68 branches/cell; p < 0.0001). About the branch farther, a one-way ANOVA showed significant differences between groups (F [3,20] = 13.25; p < 0.0001). Tukey’s post-hoc test showed that TBI-SAL (1.71 ± 0,16 μm) had shorter branches than SHAM-SAL (2.18 ± 0.18 μm/branch; p = 0.0012) and SHAM-BIP (2.35 ± 0.21 μm/branch; p < 0.0001) (Fig. 5D-E).
Regarding the number of astrocytes in CA1, a significant difference was found among groups (F[3,20] = 92.26; p < 0.0001). Tukey’s post-hoc test revealed that the TBI-SAL group had a higher number of astrocytes (491.7 ± 85.37 astrocytes) than TBI-BIP (97.14 ± 21.28 astrocytes; p < 0.0001), SHAM-SAL (131.7 ± 14.64 astrocytes; p < 0.0001), and SHAM-BIP (139.9 ± 31.34 astrocytes; p < 0.0001) (Fig. 6C).
To analyze the morphology of astrocytes in CA1, the number of branches was measured using one-way ANOVA, which shows statistical differences among the groups (F [3,20] = 162.4; p < 0.0001). Tukey’s post-hoc test showed that the TBI-SAL (16.71 ± 1.92 branches/cell) group had more branches compared to the SHAM-SAL (5.81 ± 0.22 branches /cell; p < 0.0001), SHAM-BIP (6 ± 0.6 branches/cell; p < 0.0001), and TBI-BIP (5.65 ± 0.53 branches/cell; p < 0.0001) (Fig. 6D).
Finally, the branch length in CA1 was analyzed by a one-way ANOVA test that showed significant differences among groups (F [3,20] = 32.26; p < 0.0001). Tukey’s post-hoc analysis showed that the TBI-SAL group (1.95 ± 0.12 μm/branch) had shorter branches compared to SHAM-SAL (2.7 ± 0.18 μm/branch; p < 0.0001), SHAM-BIP (2.59 ± 0.15 μm/branch; p < 0.0001), and TBI-BIP (2.55 ± 0.14 μm/branch; p < 0.0001) (Fig. 6E).
Biperiden restores microglial morphology in the cortex and CA1 after TBI
We employed the skeleton analysis method to assess microglial quantity and morphology, with a focus on the number and length of branches in the cortex and CA1 regions (Figs. 7A-B and 8A-B). No significant differences were found in the integrated density among groups in the cortex and CA1 (Figs. 7B and 8B).

Iba-1 positive cells in the cortex. (A) Images representative of Iba-1 in the cortex for each group: I-SHAM-SAL, II-SHAM-BIP, III-TBI-SAL, IV-TBI-BIP. (B) Graphic representation of the integrated density of Iba-1 corrected with DAPI for each group. (C) Number of microglia for each group. (D) Number of branches for each group. (E) Average branch length measured in µm for each group. Statistical significance: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.001. (n = 5–7 per group. Only samples meeting predefined histological quality criteria were included in the analysis). Data are shown as the mean ± standard deviation of the mean.

Iba-1 positive cells in CA1. (A) Images representative of Iba-1 in CA1 for each group I-SHAM-SAL, II-SHAM-BIP, III- TBI-SAL, IV- TBI-BIP. (B) Graphic representation of the integrated density of Iba-1 corrected with DAPI for each group. (C) Number of microglia for each group. (D) Number of branches for each group. (E) Average branch length measured in µm for each group. Statistical significance: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.001. (n = 5–7 per group. Only samples meeting predefined histological quality criteria were included in the analysis). Data are shown as the mean ± standard deviation of the mean.
In the cortex, one-way ANOVA revealed significant differences in microglial cell numbers among groups (F[3,18] = 6.34, p = 0.004). Tukey’s post hoc test showed that the SHAM-BIP group had significantly more cells (201 ± 77.31 microglia) than both the SHAM-SAL (121.4 ± 6.9; p = 0.036) and TBI-SAL (92.64 ± 24.15; p = 0.0025) groups (Fig. 7C).
Morphological analysis also showed significant differences in the number of branches per cell (F[3,19] = 20.86, p < 0.0001). The TBI-SAL group had significantly fewer branches (10.58 ± 2.1 branches/cell) than SHAM-SAL (26.94 ± 4.8; p < 0.0001), SHAM-BIP (26.14 ± 5.79; p < 0.0001), and TBI-BIP (22.39 ± 2.59; p = 0.0003) groups (Fig. 7D). However, the average branch length did not differ significantly across groups (F[3,19] = 1.214, p = 0.3317) (Fig. 7E).
In the CA1 region, one-way ANOVA showed significant differences in microglial numbers (F[3,18] = 14.16, p < 0.0001; Fig. 8C). Tukey’s test indicated that the TBI-SAL group had significantly reduced number of microglia (87.85 ± 21.99) than both SHAM-SAL (188.5 ± 57.8; p = 0.0211) and SHAM-BIP (299.3 ± 80.9; p < 0.0001). Additionally, SHAM-BIP showed a higher number of microglia than both SHAM-SAL (p = 0.015) and TBI-BIP (178.6 ± 40.34; p = 0.011).
Branches number analysis also revealed significant group differences (F[3,18] = 39.59, p < 0.0001). The TBI-SAL group had significantly diminished branches (8.28 ± 1.94 branches/cell) than SHAM-SAL (36.12 ± 5.37), SHAM-BIP (27.44 ± 6.16), and TBI-BIP (29.1 ± 4.16) groups (p < 0.0001 for all comparisons). Notably, the SHAM-BIP also had a reduced number of branches than SHAM-SAL (Fig. 8D).
Finally, the average branch length in CA1 also varied significantly among groups (F[3,18] = 6.049, p = 0.0049). The TBI-SAL group exhibited longer branches (2.49 ± 0.06 μm) compared to SHAM-SAL (2.09 ± 0.23 μm; p = 0.0259) and SHAM-BIP (1.97 ± 0.13 μm; p = 0.0052) (Fig. 8E).
Discussion
This study evaluated the effects of biperiden administration during the acute phase following moderate TBI induced by the LFPI model in male rats. Our experiments revealed that the TBI-BIP group exhibited decreased degenerating neurons, lower biomarkers such as T-tau and NfL levels, and diminished astrocyte activation, as well as decreased microglia activation, compared to the TBI-SAL group. This study, demonstrates that biperiden may mitigate TBI-induced damage within 24 h. In contrast, the TBI-SAL group exhibited an elevated number of degenerating neurons, higher levels of T-tau and NfL in plasma, as well as astrocyte and microglial activation, highlighting the detrimental impact of trauma on neuronal integrity.
At day 1 post-trauma, consistent with previous reports9,40,41, LFPI-induced impairments in motor performance were evident and are known to persist for several weeks. The decline observed in the TBI groups likely reflects cytoskeletal disruption, neurochemical imbalance, and acute excitotoxicity associated with the injury9. Although the TBI-BIP group showed a numerically higher neuroscore (approximately 3.4 points above TBI-SAL), this difference did not reach statistical significance. Given the short evaluation window (24 h) and the severity of the injury, robust behavioral recovery would not be expected at this early stage. Nevertheless, the trend toward better performance may reflect the biperiden’s anticholinergic action, which may reduce excitotoxicity and cytotoxic neuronal injury by blocking muscarinic receptors37,38. Experimental evidence suggests that excessive acetylcholine release after brain trauma contributes to neuronal hyperexcitability and glutamate-mediated cytotoxicity37,38. By competitively blocking M1 receptors, biperiden may reduce postsynaptic excitability and attenuate early neuronal stress, which could influence long-term functional outcomes16,39. However, confirming such effects will require longer follow-up and comprehensive behavioral testing at subacute and chronic stages.
It is well established that neuronal loss and inflammation persist for days to weeks following LFPI, and assessing longer-term outcomes is essential for translational relevance9,40,41. Nevertheless, we deliberately restricted our analyses to the 24-hour time point in order to specifically characterize the acute phase of injury, which is marked by peak excitotoxicity, neuronal degeneration, and glial activation. This approach provides critical insights into the immediate effects of biperiden on early injury processes, prior to the onset of endogenous repair and plasticity mechanisms that could confound the interpretation of drug-induced changes.
By focusing on this acute window, we aimed to isolate and characterize the early mechanisms underlying secondary injury and to identify potential initial modulatory effects of biperiden, rather than to assess its long-term therapeutic efficacy. Moreover, according to PubChem42,43, biperiden exhibits good tissue penetration, with a distribution half-life of 0.6 h and a terminal plasma half-life of 18 h. Thus, 24 h seems to a good point to analyze the acute effects of biperiden. Future studies including subacute and chronic time points will be required to determine whether these early effects translate into sustained neuroprotection and functional recovery.
Although the present study did not include targeted experiments to directly assess muscarinic receptor expression or signaling, previous evidence supports the hypothesis that biperiden’s effects are mediated, at least in part, through M1 muscarinic receptor blockade. In fact, biperiden’s ability to block muscarinic receptors, particularly the M1 subtype, was already demonstrated22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46. These studies, mostly related to cholinergic cognitive impairment, proposed that M1 receptors are abundantly expressed in forebrain regions such as the neostriatum, amygdala, hippocampus, and cerebral cortex, where they play critical roles in synaptic plasticity and information storage. These findings provide indirect support for the involvement of M1 receptor pathways in biperiden’s effects.
Neuronal damage biomarkers in the TBI-SAL group may be evidence of a more severely injured brain. Tau, a microtubule-associated protein that supports axonal transport and microtubule dynamics, becomes hyperphosphorylated after TBI, leading to neurofibrillary tangles and impaired neuronal stability47. Studies have reported increased plasma T-tau levels post-TBI, with peaks immediately after injury and between 12 and 36 h, which aligns with our plasma sampling period48,49. This second peak likely reflects secondary neurodegenerative processes50.
Likewise, NfL, an intermediate filament primarily expressed in the soma and axons of long myelinated neurons. Following neuroaxonal damage, NfL is released into the circulation and identified as a potential biomarker for various forms of neurotrauma51. Clinical studies investigating this protein in mild TBI indicate that circulating NfL levels stay for prolonged periods post-trauma and are also predictors of symptom recovery and long-term outcomes29. In this context, the lower NfL levels observed in the TBI-BIP group indicate reduced axonal damage, which may translate into better neurological outcomes.
In contrast, the plasma biomarkers GFAP, associated with astrocytic damage, and UCH-L1, indicative of early neuronal injury, did not show statistically significant differences between experimental groups. These findings indicate that the impact of TBI on these specific cellular responses may be more limited or undetectable at the systemic level under the conditions evaluated. In particular, UCH-L1 is known to peak in plasma within the first hours after injury (approximately 8 h) and to decline rapidly thereafter, which may explain the absence of changes at the 24 h time point analyzed here52,53. Accordingly, biperiden did not exert a modulatory effect on circulating levels of GFAP and UCH-L1. Nevertheless, our histological data demonstrated an increase in cerebral GFAP expression after TBI, supporting the notion of enhanced astrocytic activation despite the absence of detectable changes in the plasma compartment. This discrepancy may reflect the kinetics of GFAP release: following acute injury, GFAP is upregulated in the brain and has been reported to peak in blood around 20 h post-TBI53, followed by a rapid decline. Thus, our sampling may have missed this peak. In addition, sensitivity limitations of plasma assays could have masked subtle increases in circulating GFAP.
Regarding the number of degenerated neurons, Hallam and collaborators (2004)9 reported that the LFPI model induces damage and degeneration of pyramidal neurons throughout Ammon’s horn, which aligns with our observation. Furthermore, Anderson and collaborators (2005)54 found that the CA3 region and the dentate gyrus exhibited the greatest number of FJB-positive neurons following TBI, while area CA1 had the lowest. This aligns with our findings, as we observed no significant differences in the CA1 region. However, this contrasts with Sato and collaborators (2001)55, who reported that CA1 had the highest density of FJB-positive neurons. Although our results in the CA1 region were not statistically significant, it should be noted that there was an increase in the TBI-SAL and a slight increase in the TBI-BIP group. The discrepancies between works may be attributed to variations in the position of the craniotomy and the application of the fluid pulse, both of which can significantly influence the severity of LFPI54.
Interestingly, although both the cortex and hippocampus exhibited astrocytic and microglial activation, neuronal degeneration was observed only in the cortex and not in the CA1 subfield of the hippocampus. This apparent dissociation may reflect region-specific thresholds of vulnerability and distinct temporal dynamics of injury responses. After a focal injury in the central nervous system, microglia rapidly migrate to sites of tissue damage and blood–brain barrier disruption56. Astrocytes, in contrast, remain in situ but become reactive, hypertrophic, and in some cases proliferative in response to injury severity or ischemia53. Neuronal degeneration may follow a different time course and may be more pronounced in cortical than hippocampal neurons in the acute phase. Thus, the early glial response in CA1 could represent an anticipatory reaction to injury-related signals, preceding or occurring independently of detectable neuronal death.
Vulnerability to injury is region-specific. Studies have shown that the CA1 subfield is structurally more rigid, whereas the adjacent CA3 region is mechanically softer and experiences higher tensile strain during trauma54,57. This biomechanical property provides a plausible explanation for the relative preservation of CA1 neurons, despite their anatomical proximity to the cortex57.
Is important to emphasize that in this study, we focused our histological assessment on the CA1 subfield, given its well-established vulnerability to excitotoxic and ischemic injury58, as well as its anatomical proximity to the cortical regions affected by LFPI59,60. This proximity to the lesion site and its established vulnerability makes CA1 a relevant initial target for assessing neurodegeneration after TBI. As our design aimed to characterize the acute-phase response, we prioritized the cortical and CA1 regions, where neuronal degeneration is most prominent at this time point. Nevertheless, we recognize the value of assessing other hippocampal regions, such as CA3 and DG, in subacute and chronic phases, as it could provide a more comprehensive picture of hippocampal vulnerability and the potential region-specific effects of biperiden.
Concerning the decrease of degenerative neurons in the TBI-BIP vs. TBI-SAL group, we speculate that it might be related to biperiden, as it blocks muscarinic ACh receptors, leading to reduced neuronal degeneration by decreasing presynaptic cholinergic hyperactivity, which attenuates excitotoxicity and prevents excessive calcium influx into nerve cells, protecting them from glutamate-induced cell death61.
Regarding astrocytes, after the injury, they are rapidly activated and undergo a series of biological variations, including significant hypertrophy of protrusions, proliferation, and migration62. In our experiment, the TBI-SAL group displayed increased astrocytic activity in the hippocampus’ cortex and CA1 region. Notably, the astrocytes in this group had shorter branches than the other groups, indicating a distinct pattern of glial reactivity that may contribute to impaired neural repair or inflammation. The astrocytic response observed in TBI can be explained by the fact that TBI triggers an intense inflammatory environment, which stimulates astrocyte proliferation and the remodeling of their processes63. The observed reduction in proliferation, lower number of processes, and increased process length may indicate a partial recovery of astrocytic homeostasis, possibly mediated by biperiden-induced cholinergic or anti-inflammatory mechanisms.
Importantly, our results showed that administering biperiden within the first 24 h after TBI reduced the number of processes to five per astrocyte, a value similar to that of the control groups. These morphological changes may be functionally relevant, as astrocytic shape is closely related to their ability to interact with synapses. Astrocytes are the primary cells affected by edema, which compromises their neuroprotective and supportive functions64.
Regarding microglia, we observed similar responses in both the cortex and the CA1 region. In the cortex, TBI decreased microglial cell numbers (compared to the SHAM BIP group) and reduced morphological complexity, mainly a lower number of branches, though branch length remained unchanged. In the CA1, however, TBI also decreased microglial density, but with fewer and longer branches, indicating a mixed reactive state65. Notably, treatment with biperiden for 24 h post-TBI partially restored branching levels to baseline in both regions. Surprisingly, the number of microglial cells was reduced instead of an expected increase, likely due to excessive activation, debris clearance, and subsequent cell death. Biperiden is likely to modulate to modulate this response by possibly blocking M1 muscarinic receptors on immune cells, reducing intracellular calcium, and dampening microglial activation66. This may help limit inflammation and protect neural tissue after injury66.
Conclusion
In summary, our findings show that biperiden administration within the first 24 h after TBI reduces astrocyte reactivity, and diminishes microglia activation, and potentially improves neurological outcomes. The observed reduction in degenerating neurons, lower T-tau and NfL levels, and astrocytic modulation indicate that biperiden may exert early modulatory effects, likely by attenuating inflammation and glutamate excitotoxicity during the first 24 h post-trauma and consequently improving inflammatory outcomes and reducing neuronal degeneration. While these results are promising, further studies are necessary to investigate the long-term effects of biperiden on TBI recovery. Would the results remain the same if biperiden were administered with a 24-hour delay post-trauma or with a bigger gap between applications?
Study limitations
A limitation of the present study is that all assessments were confined to the acute phase (24 h post-injury). While this timeframe provides insight into early excitotoxic, inflammatory, and neurodegenerative processes, it does not capture the persistence of neuronal loss and glial activation, which are known to extend for days to weeks after TBI9,40,41. Given the severity of the injury and the short evaluation window, it is unlikely that robust functional improvements would be detectable at this acute stage, as behavioral recovery typically requires more extensive structural and synaptic reorganization over time. Consequently, the absence of statistically significant differences in neuroscore does not necessarily preclude the possibility of later functional benefits. Ongoing studies in our laboratory are evaluating whether the acute effects of biperiden observed here translate into sustained structural and functional improvements over time.
Moreover, only male rats were examined, leaving potential sex-specific effects unresolved; future experiments will address this by including both sexes to improve translational relevance. Finally, histological analyses were restricted to the CA1 subfield due to its known vulnerability to excitotoxic and ischemic injury, and its proximity to cortical regions affected by LFPI. While CA3 and the dentate gyrus may also be impacted, their assessment was beyond the scope of this acute-phase study. Future work will extend analyses to these subregions to better characterize hippocampal involvement and the neuroprotective potential of biperiden.
Data availability
The datasets used and/or analyzed during the current study are available at the following link: https://docs.google.com/spreadsheets/d/109Y8Cu1QkPRAMoGiyRsYDMu6Sa-t_iao/edit?usp=sharing&ouid=103508239238716550597&rtpof=true&sd=true upon reasonable request.
Abbreviations
- ACh:
-
Acetylcholine
- GCS:
-
Glasgow Coma Scale
- GFAP:
-
Acid Fibrid Glial Protein
- LFPI:
-
Lateral fluid percussion injury
- NfL:
-
Neurofilament Light Chain
- SIMOA:
-
Single Molecule Array
- TBI:
-
Traumatic brain injury
- UCH-L1:
-
Ubiquitin C-terminal hydrolase L1
References
Katz, D. I., Cohen, S. I. & Alexander, M. P. Mild Traumatic Brain Injury. Handbook of Clinical Neurology vol. 127Elsevier B.V., (2015).
World Health Organization. Neurological disorders. in Public Health Challenges (Geneve, Switzerland, (2006).
Teasdale, G. & Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet 37, 52–56 (1974).
Dixon, K. J. Pathophysiology of traumatic B rain injury. Phys. Med. Rehabilitation Clin. NA. 1–11. https://doi.org/10.1016/j.pmr.2016.12.001 (2017).
Abdelmalik, P. A., Draghic, N. & Ling, G. S. F. Management of moderate and severe traumatic brain injury. Transfusion (Paris) 59, (2019).
Katz, P. S. & Molina, P. E. A lateral fluid percussion injury model for studying traumatic brain injury in rats. in Traumatic and Ischemic Injury: Methods and Protocols (ed Tharakan, B.) vol. 1717 27–36 (Springer, New York, (2019).
Awasthi, A., Bhaskar, S., Panda, S. & Roy, S. Brain multiphysics review Article A review of brain injury at multiple time scales and its clinicopathological correlation through in Silico modeling. Brain Multiphys. 6, 100090 (2024).
Greve, M. W., Zink, B. J. & Greve, M. W. Pathophysiology of traumatic brain injury address correspondence to. Mt Sinai J. Med. 76, 97–104 (2009).
Hallam, T. M. et al. Comparison of behavioral deficits and acute neuronal degeneration in rat lateral fluid percussion and Weight- drop brain injury models. J. Neurotrauma. 21, 521–539 (2004).
McIntosh, T. K. et al. Traumatic brain injury in the rat: characterization of lateral fluid-percussion model. Neuroscience 28, 233–244 (1989).
Monsour, M., Ebedes, D. & Borlongan, C. V. A review of the pathology and treatment of TBI and PTSD. Exp. Neurol. 351, 114009 (2022).
Abdelmalik, P. A., Draghic, N. & Ling, G. S. F. Management of moderate and severe traumatic brain injury. Transfus. (Paris). 59, 1529–1538 (2019).
Diaz-Arrastia, R. et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the department of defense neurotrauma Pharmacology workgroup. J. Neurotrauma. 31, 135–158 (2014).
Meyfroidt, G. et al. Correction to: Management of moderate to severe traumatic brain injury: an update for the intensivist. Intensive Care Med. 48, 989–991 (2022).
Bittencourt, A. S. et al. Modification of the natural progression of epileptogenesis by means of biperiden in the pilocarpine model of epilepsy. Epilepsy Res (2017).
Foresti, M. L. et al. Biperiden for prevention of post-traumatic epilepsy: A protocol of a double-blinded placebo-controlled randomized clinical trial (Biperiden trial). PLoS One. 17, 1–11 (2022).
Tamás, V., Kocsor, F., Gyuris, P., Kovács, N. & Czeiter, E. The young male Syndrome — An analysis of Sex, Age, risk taking and mortality in patients with severe traumatic brain injuries. Front Neurol 10, (2019).
Schwarzbold, M. L. et al. Effects of traumatic brain injury of different severities on emotional, cognitive, and oxidative stress-related parameters in mice. J. Neurotrauma. 27, 1883–1893 (2010).
van Hameren, G. et al. Mitochondrial dysfunction underlies impaired neurovascular coupling following traumatic brain injury. Neurobiol. Dis. 186, 106269 (2023).
Cavalcanti, P. V. Guia para a implementação de Ponto Final Humanitário. UNIVERSIDADE FEDERAL DE SÃO PAULO COMISSÃO DE ÉTICA NO USO DE ANIMAIS 1–24 (2020).
Benassi, S. K. et al. Two decades of research towards a potential first anti-epileptic drug. Seizure 90, 99–109 (2021).
Roldán, G., Bolaños-Badillo, E., González-Sánchez, H. & Quirarte, G. L. Prado-Alcalá, R. A. Selective M1 muscarinic receptor antagonists disrupt memory consolidation of inhibitory avoidance in rats. Neurosci. Lett. 230, 93–96 (1997).
Andrade, P. et al. Seizure Susceptibility and Sleep Disturbance as Biomarkers of Epileptogenesis after Experimental TBI. Biomedicines 10, (2022).
Paxinos, G. & Watson, C. The Rat Brain in Stereotaxic Coordinates - The New Coronal Set. English 5th editio, 209 (2004).
Wang, Y. et al. An activated protein C analog protects from ischemic stroke and extends the therapeutic window of tPA in aged female mice and hypertensive rats. Stroke 44, 3529–3536 (2013).
Romariz, S. A. A., Main, B. S., Harvey, A. C., Longo, B. M. & Burns, P. Delayed treatment with ceftriaxone reverses the enhanced sensitivity of TBI mice to chemically-induced seizures. PLoS ONE. 18, 1–17 (2023).
Pase, M. P. et al. Plasma total-tau as a biomarker of stroke risk in the community. Ann. Neurol. 86, 463–467 (2019).
Spitz, G. et al. Plasma biomarkers in chronic single moderate – severe traumatic brain injury. Brain 147, 3690–3701 (2024).
Shahim, P. et al. Serum NfL and GFAP as biomarkers of progressive neurodegeneration in TBI. Alzheimer´s Dement. 20, 4663–4676 (2024).
Castaño-Leon, A. M. & Carabias, S. Serum assessment of traumatic axonal injury: the correlation of GFAP, t-Tau, UCH-L1, and NfL levels with diffusion tensor imaging metrics and its prognosis utility. J. Neurosurg. 138, 454–464 (2023).
Wong, K. R. et al. Serum neurofilament light as a biomarker of traumatic brain injury in the presence of concomitant peripheral injury. Biomark. Insights. 26, 15–18 (2021).
Heiskanen, M. et al. Plasma neurofilament light chain (NF-L) is a prognostic biomarker for cortical damage evolution but not for cognitive impairment or epileptogenesis following experimental TBI. Int. J. Mol. Sci. 23, 15208 (2022).
Sillau, S. H. et al. Plasma biomarkers of neurodegeneration in the brain (UCH-L1 and NfL) become exponentially higher with age from early childhood, and treatment of alzheimer ’ s disease participants with granulocyte-macrophage colony-stimulating factor (GM-CSF) lowers. Alzheimer´s Dement. 20, 1–2 (2024).
Faul, F., Erdfelder, E., Lang, A. G. & Buchner, A. G * Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods Res. Methods. 39, 175–191 (2007).
Cohen, J. Statistical Power Analysis for the Behavioral Sciences (Routledge, 1988).
Kabadi, S. V., Hilton, G. D., Stoica, B. A., Zapple, D. N. & Faden, A. I. Fluid-percussion-induced traumatic brain injury model in rats. Nat. Protoc. 5, 1552–1563 (2010).
Dixon, C. E. et al. Time course of increased vulnerability of cholinergic neurotransmission following traumatic brain injury in the rat. Behav. Brain. Res. 70, 125–131 (1995).
Shin, S. S. & Dixon, C. E. Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain injury. J. Neurotrauma. 32, 1429–1440 (2015).
Sanabria, V. et al. Anticholinergics: A potential option for preventing posttraumatic epilepsy. Front. Neurosci. 1–8 https://doi.org/10.3389/fnins.2022.1100256 (2023).
Sharma, R. et al. Neuroinflammation in Post-Traumatic epilepsy: pathophysiology and tractable therapeutic targets. Brain Sci 9, (2019).
Leung, W. L. et al. Chronic outcomes after mild-moderate traumatic brain injury in adult seizure-prone (FAST) and seizure-resistant (SLOW) rats: A model for Understanding genetic contributions to acquired epileptogenesis? Epilepsy Behav 166, (2025).
Biperiden | C21H29NO | CID. 2381 - PubChem. https://pubchem.ncbi.nlm.nih.gov/compound/Biperiden
Hines, K. M., Ross, D. H., Davidson, K. L., Bush, M. F. & Xu, L. Large-Scale structural characterization of drug and drug-Like compounds by High-Throughput ion Mobility-Mass spectrometry. Anal. Chem. 89, 9023–9030 (2017).
Gieling, E. et al. Performance of conventional pigs and Göttingen miniature pigs in a Spatial holeboard task: effects of the putative muscarinic cognition impairer biperiden. Behav Brain Funct 9, (2013).
Sambeth, A., Riedel, W. J., Klinkenberg, I., Kähkönen, S. & Blokland, A. Biperiden selectively induces memory impairment in healthy volunteers: no interaction with Citalopram. Psychopharmacol. (Berl). 232, 1887–1897 (2015).
Svoboda, J., Popelikova, A. & Stuchlik, A. Drugs interfering with muscarinic acetylcholine receptors and their effects on place navigation. Front Psychiatry 8, (2017).
Hernandez, F., Avila, J. & Tauopathies Cell. Mol. Life Sci. 64, 2219–2233 (2007).
Rubenstein, R. et al. Comparing plasma phospho Tau, total Tau, and phospho Tau–Total Tau ratio as acute and chronic traumatic brain injury biomarkers. JAMA Neurol. 74, 1063–1072 (2017).
Shahim, P. et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 71, 684–692 (2014).
Hossain, I., Blennow, K., Posti, J. P. & Zetterberg, H. Tau as a fluid biomarker of concussion and neurodegeneration. Concussion 1, 2056–3299 (2022).
Zetterberg, H. & Blennow, K. Fluid biomarkers for mild traumatic. Nat. Rev. Neurol. 12, 563–574 (2016).
Bazarian, J. J. et al. Accuracy of a rapid glial fibrillary acidic protein/ubiquitin carboxyl-terminal hydrolase L1 test for the prediction of intracranial injuries on head computed tomography after mild traumatic brain injury. Acad. Emerg. Med. 28, 1308–1317 (2021).
Papa, L. et al. Time course and diagnostic accuracy of glial and neuronal blood biomarkers GFAP and UCH-L1 in a large cohort of trauma patients with and without mild traumatic brain injury. JAMA Neurol. 73, 551–560 (2016).
Anderson, K. J., Miller, K. M., Fugaccia, I. & Scheff, S. W. Regional distribution of Fluoro-Jade B staining in the hippocampus following traumatic brain injury. Exp. Neurol. 193, 125–130 (2005).
Sato, M., Chang, E., Igarashi, T. & Noble, L. J. Neuronal injury and loss after traumatic brain injury: time course and regional variability. Brain Res. 917, 45–54 (2001).
da Fonseca, A. C. C. et al. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 8, 1–13 (2014).
Mao, H., Elkin, B. S., Genthikatti, V. V., Morrison, B. & Yang, K. H. Why is CA3 more vulnerable than CA1 in experimental models of controlled cortical impact-induced brain injury? J. Neurotrauma. 30, 1521–1530 (2013).
Lana, D., Ugolini, F. & Giovannini, M. G. 1 An overview on the differential interplay among Neurons–Astrocytes–Microglia in CA1 and CA3 hippocampus in Hypoxia/Ischemia. Front Cell. Neurosci 14, (2020).
Bramlett, H. M., Green, E. J. & Dietrich, W. D. Exacerbation of cortical and hippocampal CA1 damage due to posttraumatic hypoxia following moderate fluid-percussion brain injury in rats. J. Neurosurg. 91, 653–659 (1999).
Akasu, T., Muraoka, N. & Hasuo, H. Hyperexcitability of hippocampal CA1 neurons after fluid percussion injury of the rat cerebral cortex. Neurosci. Lett. 329, 305–308 (2002).
Baracaldo-santamar, D., Ariza-salamanca, D. F. & Jos, M. Hernandez-duarte, I. Revisiting excitotoxicity in traumatic brain injury: from bench to bedside. Pharmaceutics 14, 1–26 (2022).
Sofroniew, M. V. & Astrogliosis Cold Spring Harb Perspect. Biol. 7, 1–16 (2015).
Lawrence, J. M., Schardien, K., Wigdahl, B. & Nonnemacher, M. R. Roles of neuropathology - associated reactive astrocytes: a systematic review. Acta Neuropathol. Commun. 11, 1–28 (2023).
Shenaq, M. et al. Neuronal damage and functional deficits are ameliorated by Inhibition of Aquaporin and HIF1 α after traumatic brain injury (TBI). J. Neurol. Sci. 323, 134–140 (2012).
Hsieh, C. L., Kim, C. C., Ryba, B. E. & Niemi, E. C. Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 43, 2010–2022 (2013).
Henry, R. J. et al. Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J. Neurosci. 40, 2960–2974 (2020).
Acknowledgements
We thank Professor Dr. Fabio Cruz for letting us use his microscope.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP grants 2018/24561-5 and 2022/00249-8), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (CAPES; Finance Code 001), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grants 311619/2019-3 and 312904/2021-5). VS and SR (2017/05242-3) receive post-doctoral fellowships from the São Paulo Research Foundation (FAPESP).
Author information
Authors and Affiliations
Contributions
Matheus B. Braga: Conceptualization, Methodology, Formal Analysis, Validation, and Data Curation . Viviam Sanabria: Methodology, Validation, Writing- Original Draft, Review, and Editing. Simone Romariz: Methodology and Validation, Review and Editing. Michele Longoni Calió: Methodology, Validation, Review, and Editing . Maria Licia Foresti : Conceptualization, Validation, Review, and Editing. Christiane Gimenes : Methodology, Review, and Editing. Luiz Eugênio Mello: Conceptualization, Review, and Editing . Beatriz M. Long o: Conceptualization, Methodology, Writing- Original Draft, Review, Editing, and Supervision.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was carried out in animals in accordance with approved guidelines (protocol no: 7641161222) by the Board for Ethics in the Use of Animals (CEUA, Comissão de Ética no Uso de Animais), an institutional ethics committee of the Universidad Federal de São Paulo, Brazil.
Consent for publication
All authors have agreed to the published version of this manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Braga, M.B., Sanabria, V., Romariz, S. et al. Effects of acute biperiden treatment following traumatic brain injury in male rats. Sci Rep 15, 44775 (2025). https://doi.org/10.1038/s41598-025-28575-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28575-5
