
Abstract
Pulmonary hypertension (PH) is a progressive cardiopulmonary disorder. It features elevated pulmonary arterial pressure, which leads to right ventricular failure and increased mortality. PH’s insidious nature, with no specific clinical symptoms, hinders early diagnosis. Recent investigations have implicated vascular cell senescence in the pathogenesis of PH; however, the identification of early diagnostic biomarkers and the development of senescence-targeted therapeutics remain areas of unmet need.In this study, an integrative transcriptomic analysis of multiple datasets was undertaken to delineate differentially expressed senescence-related genes (DESRGs). Feature genes were selected via the implementation of LASSO regression, random forest, and support vector machine algorithms. Subsequently, a diagnostic nomogram was constructed, predicated on six hub genes. The discriminative capacity of the nomogram was rigorously validated utilizing external datasets. Single-gene gene set enrichment analysis (GSEA) was executed to elucidate potential biological functions. The expression profiles of core genes were corroborated through single-cell RNA sequencing and qPCR in an in vitro hypoxia model of pulmonary artery smooth muscle cells. Finally, connectivity map (cMAP) analysis and molecular docking were employed to identify potential therapeutic small molecules.The analysis identified 20 DESRGs. Six feature genes (LCN2, CBS, ABCB1, NQO1, TWIST1, TLR8) were consistently selected by all three machine learning methods. The diagnostic nomogram exhibited an area under the curve (AUC) of 0.974 in the training cohort and demonstrated robust performance in independent validation cohorts. Notably, CBS, TLR8, and NQO1 were significantly downregulated in validation datasets, single-cell sequencing, and the in vitro hypoxia model. GSEA revealed significant enrichment of innate immune responses, the IL-17 pathway, and oxidative stress. cMAP analysis identified TUL-XXI039 as a potential therapeutic compound, with molecular docking studies predicting strong binding affinities (binding energies < -8.0 kcal/mol) to CBS, TLR8, and NQO1.This study introduces a novel, high-precision diagnostic model for PH based on senescence-related genes, underscoring CBS, TLR8, and NQO1 as promising biomarker candidates. TUL-XXI039 emerges as a potential multi-target therapeutic candidate for PH, thus meriting further investigation.
Similar content being viewed by others

Introduction
Pulmonary hypertension (PH) is a severe and irreversible cardiopulmonary disease characterized by a persistent elevation of mean pulmonary arterial pressure (mPAP)1, ultimately resulting in right ventricular dysfunction (RVD) and death2. Globally, the annual incidence of PH is approximately 10 cases per million people3, and the condition is associated with high mortality, with a median survival of only 2–3 years4; delayed diagnosis significantly increases mortality. Numerous pathogenic factors contribute to PH, including genetic predisposition and various acquired factors, such as BMPR2 gene mutations and connective tissue diseases5. In addition to genetic and acquired factors, recent studies have identified vascular cell senescence as a core mechanism driving PH progression6. In its early stage, PH may present with nonspecific symptoms such as exertional dyspnea, fatigue, and reduced exercise tolerance7, often without an obvious murmur of tricuspid regurgitation, which poses substantial challenges for timely diagnosis and clinical decision-making8.
Despite advances in understanding PH pathogenesis, a critical clinical gap persists: current diagnostic methods, primarily right heart catheterization and imaging techniques, are invasive, costly, and ineffective for early detection of aging-driven vascular changes9,10. Previous PH biomarker studies have largely focused on inflammation, genetic mutations, or hemodynamic markers (e.g., BMPR2-related pathways or inflammatory cytokines like IL-6)11,12,13, but these approaches often target late-stage progression rather than the underlying aging mechanisms, limiting their utility for early intervention. Moreover, existing therapies, such as endothelin receptor antagonists, provide symptomatic relief but fail to reverse aging-related vascular remodeling14, including senescence-associated secretory phenotype (SASP)-induced inflammation and endothelial dysfunction15. This highlights the urgent need for novel, non-invasive diagnostic tools based on aging-related biomarkers to bridge this gap and enable early modulation of PH progression.
Cellular senescence, characterized by irreversible cell cycle arrest and cessation of mitotic proliferation, plays a pivotal role in this process16. Aging is a major risk factor for cardiovascular diseases, impairing endothelial function and promoting PH onset and progression17. For instance, every 10-year increase in age correlates with a ~ 30% rise in mortality risk among PH patients18. Senescent endothelium exhibits not only cell cycle arrest but also reduced contractile capacity, mitochondrial dysfunction, telomere shortening, and SASP-mediated chronic inflammation, which further compromises vascular integrity19. Immune cells exacerbate this: senescent T cells secrete proinflammatory SASP factors (e.g., IL-6 and TNF-α), stimulating abnormal proliferation of neighboring pulmonary arterial smooth muscle cells and intensifying pulmonary vascular remodeling20.
In light of the above evidence, we hypothesized that aging-related signature genes may serve as effective biomarkers for the early diagnosis of pulmonary hypertension. Therefore, this study aimed to systematically characterize aging-associated molecular alterations in PH and construct a robust diagnostic model integrating these signatures. Furthermore, candidate small-molecule compounds with potential activity against senescence-driven vascular remodeling were screened to explore possible therapeutic interventions. Collectively, this work provides a novel framework for early, non-invasive detection of PH and offers new insights into targeting aging-related mechanisms for disease modulation.
Methods
Identification of differentially expressed genes (DEGs) and related enrichment analyses
All data sets are download from the Gene Expression Omnibus (GEO) database and listed in Table 1. Raw data were loaded and processed in R. Differential expression analysis was performed using the limma package with thresholds of P < 0.05 and log2FC > 1. We visualized DEG expression profiles as volcano plots (using ggplot2) and clustered heatmaps (using pheatmap). The intersection of DEGs with SRGs yielded aging-related DEGs (DESRG). GO and KEGG enrichment analyses were conducted using the clusterProfiler package21,22,23. Additionally, Gene Set Enrichment Analysis (GSEA) was employed. For GSEA, the correlation between each diagnostic gene and other genes was first calculated, and all genes were ranked in descending order by this correlation. Finally, GSEA analyses were performed using the gseGO and gseKEGG functions in clusterProfiler.
Machine learning
LASSO regression, random forest (RF), and support vector machine (SVM) are three widely used machine learning algorithms for feature selection. LASSO, which incorporates L1 regularization, is a linear model for feature selection and regression analysis. It enhances linear regression by adding an L1 regularization term (an absolute-value penalty), which drives some coefficients toward zero and thereby enables automatic feature selection. LASSO is particularly effective for handling high-dimensional data, mitigating overfitting, and improving the stability of linear models. In this study, key genes corresponding to the optimal λ value were selected using 10-fold cross-validation. RF is an ensemble learning algorithm that constructs multiple decision trees and aggregates their predictions for classification and regression tasks. It performs well with complex data, high-dimensional features, and noisy inputs. SVM is a supervised learning model and associated algorithm commonly applied to classification and regression problems. All machine learning analyses were performed in R using the glmnet, randomForest, and e1071 packages.
Construction of a nomogram
To evaluate the integrated diagnostic performance of the feature genes, the rms package was used to construct a nomogram, and a calibration curve was established to assess the nomogram’s accuracy. In the nomogram, each gene corresponds to a score, and the total score from six genes is used to predict the risk of pulmonary arterial hypertension (PH). Finally, decision curve analysis was performed using the rmda package to evaluate the net benefit of the nomogram predictions.
Evaluation of feature genes and the nomogram
Expression of six feature genes was compared across the training cohort (GSE254617) and validation cohorts (GSE48149 and GSE236251). Receiver operating characteristic (ROC) curves were constructed using the pROC package to assess the diagnostic performance of the feature genes and the nomogram.
Single-cell sequencing analysis
This study performed efficient single-cell data analysis based on the Seurat V5 framework integrated with the BP Cells package. Raw 10x Genomics data were converted to memory-optimized matrices via BPCells, and Ensembl IDs were annotated to gene symbols using the Azimuth package. A dataset combining six samples from Control and PH groups (totaling 69,506 cells) was constructed. Stringent quality control was applied (ribosomal gene proportion < 20%; gene counts 200–5000; UMI > 500). Normalization was performed using LogNormalize, and the top 2000 highly variable genes were used for principal component analysis (PCA; first 30 PCs). Dimensionality reduction was performed with UMAP, and Louvain clustering (resolution = 0.5) identified 29 cell clusters. Cluster markers were detected by the Wilcoxon rank-sum test (min.pct = 0.25, log2FC > 0.25) and manually annotated using canonical marker genes. For the extracted smooth muscle cell (SMC) subpopulation (1234 cells), expression of CBS, TLR8, and NQO1 across groups was visualized with DotPlot and UMAP, and significance was assessed by the Mann-Whitney U test. Data were stored in the BPCells disk format, with the SketchData function (LeverageScore algorithm to extract 50,000 cells) and the future package used to optimize parallel computation.
RNA extraction and real-time quantitative PCR
Pulmonary artery smooth muscle cells (PASMCs) were exposed to normoxic (NX) and hypoxic (HX) conditions for 48 hours. Total RNA was extracted using the RNAeasy™ Animal RNA Extraction Kit (R0026, Beyotime, China) according to the manufacturer’s instructions. Complementary DNA was synthesized from total RNA using HiScript III RT SuperMix (R323-01, Vazyme Biotech, China). Quantitative real-time PCR (qRT-PCR) was performed with TB Green Premix Ex Taq II (RR820Q, TaKaRa, China) to quantify the expression of target genes. Primer sequences (5′→3′) used in this study are: CBS-F: CCAAGCAGATGTACCTGGCA; CBS-R: TTTGGGGGATTTGGTCAGCA; TLR8-F: CCAGCCAGAAAGGGGAAAAC; TLR8-R: GGTTCCAGAGGACAGCAGAC; NQO1-F: CTCTTCAACCCAAGCCCATTG; NQO1-R: CATTCCGGTTAGGGCAAAGC.
Connectivity map (cMAP) analysis
Connectivity Map (cMAP; https://clue.io/) is a database of gene expression profiles based on cellular responses to perturbations or interventions and can be used to identify potential therapeutic compounds relevant to a disease. In this study, PH-altered DEGs were mapped to the cMAP database, and the top 10 compounds with the highest enrichment scores were identified as potential therapeutics for PH.
All statistical analyses were performed using R software (version 4.2.3)
Differences between two groups were assessed with either the Wilcoxon rank-sum test or Student’s t-test. Correlations between variables were evaluated using Pearson’s or Spearman’s correlation coefficients, as appropriate. A two-sided p-value < 0.05 was considered statistically significant.
Molecular docking
Molecular docking was performed using the online docking platform CB-Dock2 (http://clab.labshare.cn/cb-dock2/).
Results
Identification and enrichment analysis of DEGs in PH
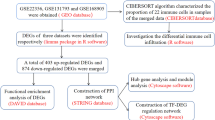
Figure 1 is a flow chart of this study.We first performed principal component analysis (PCA) on the training dataset GSE254617, and the results showed significant differences between the control group and PH group (Fig. 2A). A total of 637 DEG were identified, including 390 up-regulated and 247 down-regulated (Fig. 2B). A hierarchical clustering algorithm was performed to analyze DEG in each group and visualized in the form of a heat map (Fig. 2C). GO and KEGG enrichment analyses were then performed to explore relevant biological functions and signaling pathways. GO enrichment indicates that DEGs are significantly enriched in biological processes (BP) such as humoral immune response, antimicroidal humoral response, and leukocyte migration (Fig. 2D); cellular components (CC) such as the “collagen-containing extractive matrix” and “tertiary granule lumen“ (Fig. 2E); and molecular functions (MF) such as “cytokine activity”, “chemokine receptor binding” and “oxygen carrier activity“ (Fig. 2F). The KEGG enrichment results indicate that DEG is significantly enriched in pathways such as “Cytokine-cytokine receptor interaction”, “Chemokine signaling pathway” and “Neuroactive ligand-receptor interaction“ (Fig. 2G). Enrichment analysis showed that the pathogenesis of PH involves a regulatory network that exacerbates age-related immune deficiencies and ECM remodeling.

Flow diagram of the study.

Screening of differentially expressed genes (DEGs) and senescence-associated DEGs in pulmonary hypertension (PH). (A) Principal component analysis (PCA) of GSE254617 showing distinct clustering between PH and control groups. (B) Volcano plot illustrating upregulated and downregulated DEGs. (C) Heatmap and hierarchical clustering of DEGs between PH and control groups. (D–F) Bubble plots representing Gene Ontology (GO) enrichment analysis of DEGs in biological processes, cellular components, and molecular functions. (G) Circular plot displaying KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis of DEGs. (H) Venn diagram highlighting overlap between DEGs and senescence-associated genes. (I) Correlation dot plot of senescence-associated DEGs. (J–K) GO and KEGG enrichment visualization of differentially expressed senescence-related genes (DESRGs). GO, Gene ontology; KEGG, Kyoto encyclopedia of genes and genomes.
Identification and enrichment analysis of aging-related DEGs (DESRGs).
After intersecting 637 DEG with 866 SRGs, a total of 20 DESRGs were identified (Fig. 2H). Spearman correlation analysis showed significant interactions between DESRGs (Fig. 2I). GO analysis showed that key transcription regulators, including MYCN, TWST1, WT1, VEGFA, IGFBP1, SFN, FGFR2, and GLI1, were significantly associated with multiple important biological processes in the pathogenesis of PH (Fig. 2J). It is worth noting that these genes are mainly enriched in the regulation of cytokine signaling pathways (cytokine receptor binding and cytokine activity), tyrosine kinase activity, molecular isolation processes, and miRNA metabolism processes. The significant enrichment in positive regulation of growth (p < 0.05) suggests a dysfunctional cell proliferation mechanism characteristic of vascular remodeling observed in ages-associated PH. KEGG pathway analysis further clarified the molecular basis of PH in the context of aging (Fig. 2K). The IL-17 signaling pathway became the most significantly enriched pathway (p < 0.001), indicating that the inflammatory process plays an important role. This is consistent with recent evidence that aging-related immune disorders promote chronic inflammation of pulmonary blood vessels. The above results suggest that studying cellular aging in pulmonary arterial hypertension is of great significance for exploring the pathogenesis and treatment methods of pulmonary arterial hypertension.
Machine learning-based identification of diagnostically valuable hub genes
Three machine learning algorithms LASSO, RF and SVM were further used to identify PH and aging-related characteristic genes from 20 DESRGs. In LASSO regression, the parameter family was set to binomial, and gene coefficient plots and binomial deviation plots were generated for cross-verification (Fig. 3A,B). The hub gene is selected from the variables corresponding to the optimal penalty parameter value. Here, the optimal λ value was determined (λ min = 0.00003), and the ROC curve showed that the model demonstrated ideal diagnostic performance (Fig. 3C). Finally, LASSO regression identified 16 hub genes, including NQO1, TLR8, WTL1, ABCB1, WIF1, CBS, LCN2, TWIST1, IGFBP1, GLI1, FGFR2, IFNG, ME2, MUC4, EPHA3, and MYCN (Fig. 3D). For the RF algorithm, diagnostic errors are visualized and candidate genes are arranged in descending order based on the importance of the variables (Fig. 3E,F). Genes with MeanDecreaseGini > 3 were identified as important genes, including LCN2, TLR8, NQO1, ABCB1, ME2, WIF1, GLI1, MUC4, FGFR2 and TWIST1. At the same time, the top 10 genes were identified using SVM algorithm (Fig. 3G). The genes obtained by the three algorithms were overlapped, and six characteristic genes (LCN2, CBS, ABCB1, NQO1, TWIST1 and TLR8) were finally obtained (Fig. 3H). Further comparison of the models found that the three models had excellent overall classification capabilities, and random forest was the best (Fig. 4I).

(A) Identification of hub genes in pulmonary hypertension (PH) using machine learning algorithms. (B) LASSO (Least Absolute Shrinkage and Selection Operator) regression analysis selecting 10 optimal candidate genes for PH at the binomial deviance minimum; coefficients of selected genes shown. (C) ROC (receiver operating characteristic) curve of the LASSO model demonstrating robust diagnostic performance. (D) Coefficients of genes selected by LASSO regression. (E) Diagnostic error visualization for the random forest (RF) algorithm. (F) Ranking of genes by MeanDecreaseGini values in RF. (G) Gene importance analysis based on the support vector machine (SVM) model. (H) Venn diagram identifying six overlapping hub genes shared among LASSO, RF, and SVM as senescence-associated PH signature genes. (I) Comparative performance metrics of machine learning models. LASSO, Least Absolute Shrinkage and Selection Operator; RF, random forest; SVM, support vector machine; ROC, receiver operating characteristic.

Gene set enrichment analysis (GSEA) of individual hub genes. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed via GSEA for six signature genes (LCN2, CBS, TLR8, NQO1, ABCB1, and TWIST1). GO, Gene ontology; KEGG, Kyoto encyclopedia of genes and genomes; GSEA, Gene set enrichment analysis.
Single-gene GSEA of feature genes
To further elucidate the biological functions and signaling pathways associated with the six hub genes (LCN2, CBS, ABCB1, NQO1, TWIST1, and TLR8), we performed single-gene GO and KEGG enrichment analyses. The top 10 GO and KEGG terms for each gene are shown in (Fig. 4A,F). Gene Set Enrichment Analysis (GSEA) revealed that these genes were significantly enriched in multiple important biological processes and signaling pathways. In the biological process (BP) category, several genes (LCN2, CBS, TLR8, NQO1) cooperated in innate immune response and defense response to bacterium (p.adj = 0.0403). In the stress response system, NQO1 was primarily involved in the response to oxidative stress (p.adj = 0.0132). At the core signaling pathway level, within inflammatory signaling, LCN2 and CBS jointly activated the IL-17 signaling pathway (p.adj = 0.0049), with a Ranked Metric peak of 2.5, indicating strong signaling. Collectively, these features suggest that these genes play key roles in innate immunity, pathogen defense, and IL-17 signaling activation, potentially modulating the immune microenvironment and contributing to PH progression.
Construction of a PH diagnostic nomogram
Using the rms package, we constructed a diagnostic nomogram for PH based on the six hub genes. In the nomogram, each gene contributes a score, and the total score from the six genes is used to predict PH risk (Fig. 5A). Calibration curves demonstrated good agreement between predicted probabilities and those of an ideal model (Fig. 5B), and decision curve analysis indicated a net benefit for using the nomogram in clinical decision-making (Fig. 5C). In the training cohort, expression analysis showed downregulation of LCN2, CBS, TLR8, and NQO1 in PH, whereas ABCB1 and TWIST1 were upregulated (Fig. 5D). All six hub genes achieved good diagnostic performance, with ROC AUCs of 0.864 (LCN2), 0.814 (CBS), 0.748 (TLR8), 0.807 (ABCB1), 0.799 (NQO1), and 0.761 (TWIST1) (Fig. 5E,J). The six-gene nomogram further improved diagnostic performance, attaining an AUC of 0.974 (Fig. 5K).

Construction and validation of a diagnostic nomogram for pulmonary hypertension (PH) risk prediction. (A) Diagnostic nomogram integrating six signature genes (LCN2, CBS, TLR8, NQO1, ABCB1, TWIST1), where individual gene scores are summed to estimate PH risk. (B) Calibration curve assessing the nomogram’s predictive accuracy. (C) Decision curve analysis (DCA) evaluating the net clinical benefit of the nomogram for PH diagnosis. (D) Expression levels of the six hub genes in the GSE254617 dataset. (E–K) ROC curves validating the predictive performance of the six-gene nomogram and individual genes (LCN2, CBS, TLR8, NQO1, ABCB1, TWIST1) in PH diagnosis. ROC, receiver operating characteristic; DCA, decision curve analysis.
External validation of feature genes and the nomogram
The expression and diagnostic performance of the six hub genes were further validated in two independent external datasets (GSE48149 and GSE236251). Three hub genes (CBS, TLR8, and NQO1) showed consistent downregulation in both training and validation cohorts (Fig. 6A,C), whereas LCN2, ABCB1, and TWIST1 did not exhibit a clear trend. ROC analyses indicated favorable diagnostic performance for CBS, TLR8, and NQO1: in GSE48149, AUCs were CBS 1.000, TLR8 0.944, and NQO1 0.944 (Fig. 6B); in GSE236251, AUCs were CBS 0.701, TLR8 0.840, and NQO1 0.722 (Fig. 6D). Overall, the consistently trending genes across datasets demonstrate strong diagnostic value and may have potential roles in PH progression.

Validation of six signature genes in external datasets. (A) Comparative expression analysis of six hub genes (LCN2, CBS, TLR8, NQO1, ABCB1, TWIST1) between PH and control groups in the validation dataset GSE48149. (B) ROC curves and nomogram performance for the six-gene panel in GSE48149. (C) Expression validation of the six genes (LCN2, CBS, TLR8, NQO1, ABCBl, TWIST1) in an independent dataset GSE236251. (D) ROC curves and nomogram analysis for the six-gene panel in GSE236251.Abbreviations: ROC, receiver operating characteristic.
Single-cell RNA-seq analysis validates key targets in pulmonary arterial smooth muscle cells
To further validate previously screened key aging-related targets, we re-analyzed published scRNA-seq data from patients with PH, and we successfully identified and annotated multiple lung cell types via UMAP dimensionality reduction clustering, including pulmonary artery smooth muscle cells (PASMC), endothelial cells, fibroblasts, macrophages, neutrophils, T cells, B cells, mast cells, dendritic cells, and natural killer cells (Fig. 7A). Given the central role of PASMC in the pathogenesis of PH, we focused on the expression of CBS, TLR8 and NQO1 in PASMCs. scRNA-seq data showed that TLR8 and NQO1 were significantly down-regulated in PASMC in PH patients compared to controls, while CBS changes were not significant (Fig. 7B). It was then verified that the dot diagram of cell type-specific marker genes supported accurate annotation of major cell subtypes (Fig. 7C). To confirm these findings, we established an in vitro hypoxia (3% O2)-induced pulmonary hypertension model. qPCR analysis showed that CBS, TLR8 and NQO1 mRNA levels were significantly reduced under hypoxia compared to normoxia (Fig. 7D–F).

Validation of signature genes in pulmonary arterial smooth muscle cells (PASMCs) and molecular docking with a small-molecule drug. (A) UMAP visualization of single-cell RNA sequencing (scRNA-seq) data reanalyzed from external datasets GSE135893 and GSE296823. (B) Differential expression of three signature genes (CBS, TLR8, NQO1) in PASMCs between PH and control groups (P < 0.05, unpaired Student’s t-test). (C) Cell type annotation markers for subpopulations in scRNA-seq data. (D–F) mRNA expression changes of CBS, TLR8, and NQO1 in PASMCs. (G–I) Molecular docking simulations of TUL-XXI039 (a small-molecule compound) with CBS, TLR8, and NQO1, demonstrating binding affinities and interactions.
Potential therapeutic drug screening for PH
Based on the above findings, aging-related DEGs were submitted to the Connectivity Map (cMAP) database to identify small-molecule compounds with potential therapeutic value for PH. The screening identified TUL-XXI039 as the top-ranked candidate (Supplementary Table 2), suggesting its potential to reverse PH-associated gene expression signatures.
To evaluate interactions with key targets, molecular docking analyses were performed. The binding energies of TUL-XXI039 with CBS, TLR8, and NQO1 were − 10.8, − 9.3, and − 8.5 kcal/mol, respectively, all below − 8.0 kcal/mol, indicating favorable binding affinity. Docking pattern analyses showed that TUL-XXI039 can stably bind to the active sites of these proteins, forming multiple hydrogen bonds and hydrophobic interactions, implying that the compound may modulate CBS, TLR8, and NQO1 activities concurrently.
Taken together, single-cell transcriptomics and in vitro validation support the roles of CBS, TLR8, and NQO1 in PH, while docking analyses suggest that TUL-XXI039 could function as a multi-target regulator, providing a potential new therapeutic strategy for PH.
Discussion
Early diagnosis of PH remains challenging due to the absence of obvious tricuspid regurgitation murmurs24,25. In this study, we used the training dataset GSE254617 to screen and analyze differentially expressed genes (DEGs) in PH, focusing on aging-related DEGs (DESRGs). Three machine learning algorithms (LASSO, RF, and SVM)26identified six hub genes (LCN2, CBS, ABCB1, NQO1, TWIST1, and TLR8) and built a high-performance diagnostic model (AUC = 0.974). External verification datasets GSE48149 and GSE236251 were used to verify its diagnostic performance. Single-gene GO/KEGG enrichment analysis (GSEA) showed that these genes are involved in innate immune responses, pathogen defense, inflammatory signals, especially the IL-17 pathway, and oxidative stress responses. Single-cell transcriptome data and an in vitro hypoxia induced PASMC model further demonstrated that CBS, TLR8 and NQO1 are down-regulated in PASMC. In addition, connectivity mapping screening and molecular docking identified TUL-XXI039 as the first choice small molecule drug with good binding to CBS, TLR8 and NQO1. In summary, this work provides the first model for aging-related PH diagnosis and points out a promising small molecule drug candidate, providing possibilities for future translational research.
Our GO and KEGG analyses show that the DEGs are enriched in immune responses, cytokine signaling, and extracellular matrix (ECM) remodeling, aligning with previously reported inflammation and vascular remodeling mechanisms in PH27. The identification of aging-related DEGs (DESRGs) reveals that aging regulates PH processes, for example, TWIST1 participates in PASMC proliferation to drive vascular remodeling, and ABCB1 participates in PH-associated immune responses, indicating that aging-related immune reactions and ECM remodeling contribute to PH pathogenesis. Compared to existing PH diagnostic models, which primarily rely on invasive procedures such as right heart catheterization or late-stage hemodynamic biomarkers (e.g., NT-proBNP with AUCs typically ranging from 0.70 to 0.85 in clinical cohorts)28,29, our aging-related model introduces a non-invasive, gene-based approach focused on early senescence mechanisms, achieving a superior AUC of 0.974 and emphasizing preventive detection in aging populations. Compared with Meng et al., who conducted an exploratory study on 18 clinical samples and identified IGF1, HOXB7, and YWHAZ as candidates30, our study leverages a larger cohort (96 PH patients and 52 controls; N = 148), enhancing statistical power and biological reproducibility. Single-gene GSEA showed enrichment of these hub genes in innate immune responses, pathogen defense, and the IL-17 signaling pathway. This implicates them in immune regulation that promotes PH progression. Notably, NQO1 is enriched in oxidative stress pathways, linking vascular aging to oxidative injury. The six hub genes show that IL-17–driven inflammatory signaling and oxidative stress may be central to PH biology.
Furthermore, single-cell and in vitro validations indicate that CBS, TLR8, and NQO1 play critical roles in PASMCs within PH. scRNA-seq analysis shows downregulation of TLR8 and NQO1 in PASMCs from PH patients, with CBS showing a weaker, but trending, decrease; these findings are supported by qPCR in a hypoxia-induced PASMC model, which confirms reduced mRNA levels of CBS, TLR8, and NQO1 under hypoxic conditions. The downregulation of TLR8 may reflect a shift toward an immune-tolerant state under chronic inflammatory stimulation31, while NQO1 downregulation implies suppression of the Nrf2-mediated antioxidant response32, leading to ROS accumulation33, oxidative damage34, and enhanced PASMC proliferation35 and vascular remodeling36. Together, these data underscore aging-related immune mechanisms and PASMC remodeling as core contributors to PH and identify CBS, TLR8, and NQO1 as potential therapeutic targets. However, while these expression changes suggest mechanistic involvement, the absence of functional assays (e.g., gene knockdown or overexpression) limits our ability to establish causal relationships between these genes and PH pathogenesis, warranting future experimental validation.
In clinical significance, the diagnostic nomogram (AUC = 0.974) constructed in this study can be used as an auxiliary diagnostic tool for elderly patients37, providing a new strategy for the diagnosis of elderly cardiovascular patients without obvious tricuspid regurgitation murs in the early stage38. At the same time, TUL-XXI039, a small molecule compound with potential efficacy, was screened through the cMAP database and molecular docking, and showed high affinity for multiple targets, suggesting its potential as a new curative drug, improving PH by regulating CBS, TLR8 and NQO1 expression patterns.This provides a new strategy for precision medicine to achieve personalized intervention for elderly patients with PH, but further empirical evidence is needed to assess safety and efficacy. In summary, these new findings not only deepen the understanding of the underlying mechanisms of PH, but also provide actionable treatment targets for clinical practice.
Although this study has made important progress in clarifying the aging-related mechanisms of PH and providing clinical applications, we must also acknowledge its limitations, which provide direction for future research. This study relies on public databases, and there may be batch effects and sample heterogeneity, resulting in inconsistencies in genes such as LCN2 in the verification set. At the same time, TUL-XXI039, a small molecule compound screened through the cMAP database and molecular docking, showed high affinity for multiple targets (CBS, TLR8, and NQO1), suggesting its potential as a new curative drug to improve PH by regulating their expression patterns; however, these findings remain speculative without in vitro or in vivo validation of its therapeutic efficacy. This provides a new strategy for precision medicine to achieve personalized intervention for elderly patients with PH, but further empirical evidence is needed to assess safety and efficacy. In addition, the small-molecule drug TUL-XXI039 screened by cMAP remains highly speculative, as it lacks any in vitro drug validation (e.g., cell-based assays for efficacy or toxicity) or in vivo studies, which significantly limits its translational potential; needs to be further demonstrated in animal models and clinical trials. At the same time, based on computer simulation of molecular docking, it lacks co-immunoprecipitation verification. Furthermore, while qPCR validation in the hypoxia-induced PASMC model provides valuable correlative evidence, the study does not include functional experiments (e.g., siRNA-mediated knockdown or overexpression of CBS, TLR8, or NQO1) to demonstrate causal roles in PH mechanisms, such as PASMC proliferation or remodeling, highlighting a key area for future mechanistic studies.
Taken together, we report three aging-related hub genes (CBS, TLR8, and NQO1) and present a novel diagnostic model for pulmonary arterial hypertension (PH). We also highlight the expression changes and putative roles of these genes in pulmonary arterial smooth muscle cells (PASMCs). Finally, we identified a small-molecule compound with potential to treat PH—TUL-XXI039—and discuss potential novel mechanisms by which it could exert therapeutic effects in PH.
Conclusions
This study is the first to propose a high-precision early diagnostic model for PH based on senescence-related feature genes and identifies CBS, TLR8, and NQO1 as stable and verifiable core molecular markers. It also screens TUL-XXI039 as a multi-target candidate drug for PH treatment. These findings lay the foundation for early precision diagnosis and novel targeted therapies in PH.
Data availability
The datasets generated and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Ruopp, N. F. & Cockrill, B. A. Diagnosis and treatment of pulmonary arterial hypertension: A review. JAMA 327, 1379–1391 (2022).
Naeije, R., Richter, M. J. & Rubin, L. J. The physiological basis of pulmonary arterial hypertension. Eur. Respir. J. ;59. (2022).
Pi, H. et al. Metabolomic signatures associated with pulmonary arterial hypertension outcomes. Circul. Res. 132, 254–266 (2023).
Bertoletti, L. et al. Association between anticoagulant therapy and survival in pulmonary arterial hypertension: A registry report and updated meta-analysis. J. Am. Coll. Cardiol. 86, 982–995 (2025).
Bisserier, M. et al. Regulation of the methylation and expression levels of the BMPR2 gene by SIN3a as a novel therapeutic mechanism in pulmonary arterial hypertension. Circulation 144, 52–73 (2021).
Piper, B. et al. RAB7 deficiency impairs pulmonary artery endothelial function and promotes pulmonary hypertension. J. Clin. Investig. 134 (2024).
Manders, E. et al. The striated muscles in pulmonary arterial hypertension: adaptations beyond the right ventricle. Eur. Respir. J. 46, 832–842 (2015).
Yasuda, K. et al. Fatal pulmonary arterial hypertension in an infant Girl with incontinentia pigmenti. Pediatr. Int. 58, 394–396 (2016).
Huang, Z. et al. Deep learning-enhanced noninvasive detection of pulmonary hypertension and subtypes via chest radiographs, validated by catheterization. Chest (2025).
Mandras, S. A., Mehta, H. S. & Vaidya, A. Pulmonary hypertension: A brief guide for clinicians. Mayo Clin. Proc. 95, 1978–1988 (2020).
Choudhury, P. et al. Bioinformatics analysis of hypoxia associated genes and inflammatory cytokine profiling in COPD-PH. Respir. Med. 227, 107658 (2024).
Xie, Z. et al. Identification of the key genes in chronic obstructive pulmonary disease by weighted gene co-expression network analysis. Ann. Transl. Med. 10, 665 (2022).
Guan, Q. et al. Identification of potential key genes in the pathogenesis of chronic obstructive pulmonary disease through bioinformatics analysis. Front. Genet. 12, 754569 (2021).
Grünig, E. et al. Randomized trial of Macitentan/Tadalafil Single-Tablet combination therapy for pulmonary arterial hypertension. J. Am. Coll. Cardiol. 83, 473–484 (2024).
Sun, Z. et al. Loss of ALDH2 accelerates the progression of pulmonary arterial hypertension through the 4-HNE/ERK1/2-p16(INK4a) signaling pathway. Biochim. Et Biophys. Acta Mol. Basis Dis. 1871, 167863 (2025).
de Magalhães, J. P. Cellular senescence in normal physiology. Sci. (New York NY). 384, 1300–1301 (2024).
Evangelou, K. et al. Cellular senescence and cardiovascular diseases: moving to the heart of the problem. Physiol. Rev. 103, 609–647 (2023).
Rivas, J. et al. Cardiovascular risk evaluation in a Latin American population with severe mental illness: an observational study. Actas Esp. De Psiquiatria. 53, 742–755 (2025).
Xiao, X. et al. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal. Transduct. Target. Therapy. 6, 354 (2021).
Li, X. et al. Inflammation and aging: signaling pathways and intervention therapies. Signal. Transduct. Target. Therapy. 8, 239 (2023).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. Publ. Protein Soc. 28, 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677 (2025).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Olsson, K. M. et al. Pulmonary hypertension associated with lung disease: new insights into pathomechanisms, diagnosis, and management. Lancet Respir. Med. 11, 820–835 (2023).
DuBrock, H. M. et al. An electrocardiogram-based AI algorithm for early detection of pulmonary hypertension. Eur. Respir. J. ;64. (2024).
Wu, J. et al. Glycosyltransferase-related prognostic and diagnostic biomarkers of uterine corpus endometrial carcinoma. Comput. Biol. Med. 163, 107164 (2023).
Zhang, G. et al. NDRG1 signaling is essential for endothelial inflammation and vascular remodeling. Circul. Res. 132, 306–319 (2023).
Ivy, D. et al. Embracing the challenges of neonatal and paediatric pulmonary hypertension. Eur. Respir. J. ;64. (2024).
Preston, I. R. et al. A long-term follow-up study of Sotatercept for treatment of pulmonary arterial hypertension: interim results of SOTERIA. Eur. Respir. J. 66. (2025).
Meng, Z. Y. et al. Identification and experimental verification of senescence-related gene signatures and molecular subtypes in idiopathic pulmonary arterial hypertension. Sci. Rep. 14, 22157 (2024).
Drobek, A. et al. The TLR7/9 adaptors TASL and TASL2 mediate IRF5-dependent antiviral responses and autoimmunity in mouse. Nat. Commun. 16, 967 (2025).
Wang, S. et al. Iodoacetic acid activates Nrf2-mediated antioxidant response in vitro and in vivo. Environ. Sci. Technol. 48, 13478–13488 (2014).
Hseu, Y. C. et al. The in vitro and in vivo depigmenting activity of pterostilbene through induction of autophagy in melanocytes and inhibition of UVA-irradiated α-MSH in keratinocytes via Nrf2-mediated antioxidant pathways. Redox Biol. 44, 102007 (2021).
Zhao, L. et al. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 15, 284–296 (2018).
Yu, Y. et al. Methyleugenol alleviates pulmonary vascular remodeling in rats with high-altitude pulmonary hypertension by improving pulmonary smooth muscle cell function. Biomed. Pharmacother. 187, 118109 (2025).
Oh, C. J. et al. Dimethylfumarate attenuates restenosis after acute vascular injury by cell-specific and Nrf2-dependent mechanisms. Redox Biol. 2, 855–864 (2014).
Venkataramani, R. et al. Tricuspid regurgitation: A focus on updated interventional anatomy and pathophysiology. J. Cardiothorac. Vasc. Anesth. 37, 2327–2334 (2023).
Gnanenthiran, S. R. et al. Long-term prognostic utility of computed tomography coronary angiography in older populations. Eur. Heart. J. Cardiovasc. Imaging. 20, 1279–1286 (2019).
Funding
This work was partly supported by the Science and Technology Bureau Project of Taizhou (Nos. 23ywa52), and the Zhejiang Provincial Medical and Health Science and Technology Project (Nos. 2024KY540).
Author information
Authors and Affiliations
Contributions
X.J.D. and J.F.L. collected and analyzed the data. L.X.Y. completed the writing of the first draft of the article. H.T.X., J.J.L. and B.W.J. gathered, analyzed and organized data. M.Z.W. and B.W.J. proposed ideas and perfected the article. J.L. and G.Q.P. provided financial support. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, M., Lu, J., Li, X. et al. Construction of an early diagnostic model for pulmonary hypertension based on aging-related signature genes and identification of potential therapeutic targets. Sci Rep 15, 44788 (2025). https://doi.org/10.1038/s41598-025-28921-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28921-7
