
Abstract
Osteoarthritis (OA) is a prevalent joint disorder. It is of great importance to identify efficacious therapeutic targets and biomarkers for the treatment of inflammatory joint diseases and the effective management of their condition. Glutamine metabolism is pivotal in regulating chondrocyte function, maintaining bone homeostasis and modulating the inflammatory response. This study aimed to identify glutamine metabolism-related genes that may serve as potential biomarkers for OA. The OA transcriptome dataset and the glutamine metabolism-related genes were obtained. Subsequently, consistency clustering was employed to differentiate between different disease types for the glutamine metabolism-related genes in the OA transcriptome dataset. Furthermore, differential expression analysis was conducted, and functional enrichment analysis was employed to elucidate the underlying mechanisms. A protein-protein interaction (PPI) network was constructed and candidate key genes were identified through Cytoscape’s 7 algorithms. Valuation of the diagnostic ability of OA and prediction of transcription factors (TFs), microRNAs (miRNAs), and long non-coding RNAs (lncRNAs) that were related. Subsequently, drug prediction and molecular docking were conducted. An OA mice model and cell model were established using the DMM and IL-1β methods, respectively, were employed to verify the expression. 1018 differentially expressed genes (DEGs1) were obtained in the OA transcriptome dataset. Subsequently, based on glutamine metabolism-related genes, the transcriptome dataset genes between two subtypes were further 385 differentially expressed genes (DEGs2) were obtained. An intersection of the DEGs1 and the DEGs2 revealed 102 intersecting genes, which were considered candidate genes. LRRFIP1 and MFSD11 as the key genes according to Cytoscape’s 7 algorithms and expression levels verification, and with a better OA diagnostic ability. Subsequently, the OA mice model and cell model were established successfully, LRRFIP1 and MFSD11 both had a lower expression level. Glutamine metabolism-related genes LRRFIP1 and MFSD11 may be potential biomarkers for diagnosing and treating OA.
Similar content being viewed by others


Introduction
Osteoarthritis (OA) is a prevalent joint disorder that affects millions of individuals globally. It is characterized by the deterioration of cartilage in joints, resulting in pain, stiffness, and reduced mobility, which can affect almost any joint1. OA commonly affects weight-bearing joints such as the knees, hips, and spine, but it can also occur in other joints2. Previous research has reported that risk factors for OA include ageing, obesity, joint injury, and genetics, as the obesity and ageing population gradually increases which leads to the incidence rising3. At the same time, overweight persons have a higher risk than nonobese persons4. Treatment options for OA focus on relieving symptoms, improving joint function, and slowing the progression of the disease. These may include medications, physical therapy, lifestyle modifications, and in some cases, surgery5. It’s important for individuals with OA to work closely and long-term with healthcare providers to manage their condition effectively. Significant progress has been achieved in treating inflammatory joint diseases through effective therapeutic targets and biomarkers6. Therefore, it is necessary to find novel biomarkers that are good for OA treatment.
Glutamine is an amino acid-rich in the body and has a wide function. It is capable of being hydrolysed to glutamate by glutaminase and then plays a crucial role in the tricarboxylic acid cycle, heat shock protein responses and immune systems7. Some researchers confirmed that glutamine metabolism serves as a critical regulatory point in various biological processes, such as vessel formation, cancer occurrence, and immune system regulation8,9,10. In the context of osteoarthritis, glutamine metabolism may be relevant in the maintenance of cartilage health and overall joint function, glutamine may be an important substrate for chondrocytes has been reported, and glutamine metabolism has a certain physiological role in chondrocytes11,12. Meanwhile, glutamine metabolism plays a key regulator in chondrocyte proliferation, matrix synthesis and reactive oxygen species (ROS) production, which has been identified as a metabolic regulator13. Inflammation response is a vital driver factor in OA occurrence, glutamine uptake and release have been reported to be involved in the inflammation response of chondrocytes14. Therefore, glutamine could potentially have a positive impact on joint health, but more research is needed to understand its effects on osteoarthritis fully.
Hence, this study aims to screen and identify the potential glutamine metabolism-related biomarkers in OA, analyse the biological pathway these are involved in, and explore the regulatory network and related drugs, provide new theoretical support for OA clinical diagnosis and treatment.
Materials and methods
Data sources
Training set GSE57218 and validation set GSE117999 were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). GSE57218 comprises 40 samples, including 7 normal and 33 OA cartilage tissue samples, the testing platform is Illumina HumanHT-12 V3.0 expression beadchip (Sequencing platform: GPL6947; Species: Homo sapiens) (Supplementary Table 1). GSE117999 comprises 24 samples, including 12 normal and 12 OA cartilage tissue samples, four samples exhibited expression values of not available (NA). As these data were entirely absent from the original dataset, to ensure the quality and reliability of the analysis, this study included the remaining 20 samples with complete data (10 normal and 10 OA cases) for subsequent analysis. The testing platform is Agilent-072363 SurePrint G3 Human GE v3 8 × 60 K Microarray 039494 [Feature Number Version] (Sequencing platform: GPL20844; Species: Homo sapiens) (Supplementary Table 2). Glutamine metabolism-related genes were obtained from previous research15 (Supplementary Table 3).
Differential analysis
The R language’s limma16 (v3.52.4) package was used to perform differential analysis, |log2FC| >0.5 & adj.P.Val < 0.05 as the screening conditions. The volcano map and heatmap are shown using ggplot217 (v3.3.6) and Complexpheatmap18 (v1.0.12) packages, respectively. Consistency clustering analysis identifies subpopulations of samples exhibiting similar glutamine metabolic characteristics, ensuring homogeneity within subtypes and heterogeneity between them. Unsupervised clustering analysis reveals the intrinsic structure of the data, delineating distinct molecular subtypes of the disease. This analysis provides a potential molecular basis for patient stratification and personalised treatment. Therefore, to ensure that the genes targeted for subsequent analysis not only play a role in differentiation across different disease subtypes but also possess universal significance in the broader context of disease onset and progression, thereby avoiding potential biases arising from a single comparative dimension, this study employed cluster analysis. Aimed at identifying sample subpopulations exhibiting similar glutamine metabolic characteristics. First, the expression matrix of glutamine metabolism-related genes15 was extracted from the GSE57218 dataset. Subsequently, the R language’s ConsensusClusterPlus19 package was employed, utilizing the PAM clustering algorithm with Euclidean distance as the metric. Cluster stability was validated through 100 bootstrap resamples. Based on the consensus matrix, the samples were ultimately classified into two stable subtypes. Principal component analysis (PCA) validated the significant separation trend between subtypes. Differential expression analysis using R language’s limma (v3.52.4) package identified subtype-specific genes, which were then intersected with differentially expressed genes between the disease and normal groups. Venn Diagram is shown by ggvenn package20.
Function enrichment analysis
The clusterProfiler21 (v4.4.4) package was utilised for Gene Ontology (GO)22 and Kyoto Encyclopedia of Genes and Genomes (KEGG)23,24,25 enrichment analysis, screening condition was pvalueCutoff < 0.05. Then, visualisation using Enrichplot26 (v1.16.2) package. GO system includes 3 parts Cellular components (CC), Molecular functions (MF), and Biological process (BP).
PPI network establishment and key gene identification
The STRING27 database (https://string-db.org/) was for obtaining the candidate genes’ protein and PPI network information. Cytoscape28 software to construct networks of inter-cooperative relationship genes and screen candidate key genes using 7 algorithms in it. Then, the expression levels of candidate key genes were analysed by ggplot2 packages in GSE57218 and GSE117999 respectively.
Artificial neural network (ANN)
The backpropagation (BP) neural network model is the short title of the error back-propagation (BackPagation) network model, which is established using the R language’s Neuralnet29 (v1.44.2) package. pROC30 (v1.18.0) package was used to map the ROC curve.
Co-expression network establishment and gene set enrichment analysis (GSEA)
Protein-protein interactions can be predicted using the GeneMINIA database (http://genemania.org/). R language’s clusterProfiler and org.Hs.eg.db packages31 respectively performed single gene KEGG enrichment analysis for key genes. Then, further performed GSEA enrichment analysis, with the threshold set to |NES| > 1, NOM P < 0.05 & q < 0.25.
Regulatory network prediction
ChEA3 database (https://maayanlab.cloud/chea3/) was used to search for Transcription factors (TFs), miRDB (https://mirdb.org/), miRWalk (http://mirwalk.umm.uni-heidelberg.de/) and miRtarbase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) databases were used to obtain miRNAs of key genes, Starbase database (https://rnasysu.com/encori/) was used to obtain lncRNAs of miRNAs. SNP sites of miRNA were searched from miRNASNP (https://guolab.wchscu.cn/miRNASNP/#!/).
Drug prediction and molecular Docking
Drugs prediction from Comparative Toxicogenomics Database (CTD, https://ctdbase.org/). PDB database (https://www.rcsb.org/) was used to download the tertiary structure of key genes, PubChem database (https://pubchem.ncbi.nlm.nih.gov/) was used to download the tertiary structure of drugs. Then, performed molecular docking by AutoDockTools32, and visualisation using Pymol33.
Construction of the animal model and experimentation
We obtained C57BL/6J mice (10–12 weeks old) from SPF (Beijing) Biotechnology Co., Ltd. To create the destabilization of the medial meniscus (DMM) mice model, we followed a method described in previous research34. In brief, we induced the DMM model by transecting the meniscal-tibial ligament of C57BL/6J mice, while the ligament was left untouched in the sham control group. The animal experiments were conducted and received approval from the Experimental Animal Ethics Committee of Yunnan Labreal Biotech Co., Ltd. We confirmed that all experiments in this study were performed in accordance with the relevant guidelines and regulations. All the procedure of the study is followed by the ARRIVE guidelines. The animals were euthanized by cervical dislocation under over isoflurane anesthesia. Histological analysis using the Hematoxylin-Eosin (HE) Stain Kit (G1120, Beijing Solebao Biotechnology Co., Ltd.) and the Safranin-O/Fast Green staining Kit (G1371, Beijing Solebao Biotechnology Co., Ltd.), following the manufacturer’s instructions.
Cell model construction and experimentation
Cartilage was obtained from the knee joints of C57BL/6J mice under a microscope in a sterile environment. Then, digested and separated cartilage using 2 mg/mL (0.1%) collagenase II, cartilage was cultured in DMEM/F12 (Gibco, Invitrogen, Shanghai, China) solution supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich, MO, USA) and 1% double-antibody (Sigma-Aldrich, MO, USA). All cells were then incubated at 37 °C with 5% CO2 in a cell incubator. OA cell model using 20 µM mouse recombinant IL-1β (PeproTech) was established. The ELISA Kits of the PGE2 assay kit (ml037542), IL-6 assay kit (ml098430), IL-1β assay kit (ml098416), and TNF-α assay kit (mIC50536-1) were purchased from China Shanghai Enzyme-linked Biotechnology Co., Ltd. Specific operation referenced the instructions of the Kits.
RT-qPCR
Total RNA was extracted using the TRIzol reagent, then subjected to reverse transcription to form cDNA using the One Step Prime Script miRNA cDNA Synthesis Kit (Takara, Kyoto, Japan). For RT-qPCR procedures, we followed the instructions provided by the manufacturer of the SYBR Green PCR Master Mix (Life Technologies, USA). The RT-qPCR primers are shown in Table 1. The expression levels were calculated by the 2-ΔΔCt method, with β-actin as the internal reference.
Western blot
The proteins in tissue samples and cells of each group were extracted using RIPA buffer (Sigma-Aldrich, USA) containing 1% protease inhibitor and phosphatase inhibitor. They were then separated using a 10% SDS-PAGE gel and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, MA, USA). The membrane was sealed with 5% skim milk at room temperature for 2 h. The first antibody was added and allowed to incubate overnight at 4 °C. The second antibody with HRP conjugated (1:2000, ab205718, Abcam, UK) was then incubated at room temperature for 1 h. ECL chemiluminescence solution development (BD Biosciences) was used for exposure and observation. Protein band analysis was done using Image J. Anti-β-actin (1:1000, ab8226, Abcam, UK) as a control. The information of primary antibodies following that: Anti-Col2a1 (1:1000, WH0001280M1, Merck, Germany), Anti-aggrecan (1:1000, ab3778, Abcam, UK), Anti-MMP-3 (1:20000, ab52915, Abcam, UK), Anti-MMP-13 (1:3000, ab39012, Abcam, UK), Anti-LRRFIP1 (sc-398240, Santa Cruz Biotechnology (Shanghai) Co., Ltd.) and Anti-MFSD11 (PA5-54547, Thermo Scientific).
Immunofluorescence experiment
The cells were placed onto a 24-well plate at a density of 2 × 104 cells per well. After 24 h, the cells were washed twice with PBS. Next, they were fixed with 4% paraformaldehyde, permeabilised for 10 min and blocked with bovine serum albumin for 1 h. The cells were then stained with Anti-Col2a1 or Anti-aggrecan and incubated overnight at 4 °C. The following day, the cells were incubated with corresponding secondary antibodies for 1 h, followed by DAPI staining. Finally, the stained cells were observed and photographed under a fluorescence microscope (400857, Nikon, Japan).
Statistical analysis
Data processing and analysis were performed using R software (version 4.2.1). Differential analysis of the dataset was conducted via the limma (v3.52.4) package, with consensus clustering performed using the ConsensusClusterPlus package. Enrichment analysis employed the clusterProfiler package (version 4.4.4), with Cytoscape software utilised for visualisation and candidate gene screening. Expression analysis was conducted using the Wilcoxon rank-sum test via the stat_compare_means function in the ggpubr package. Neural network models were constructed using the Neuralnet package (v1.44.2), with model predictive performance evaluated via ROC curves. GSEA enrichment analysis was performed using the clusterProfiler and org.Hs.eg.db packages. Molecular docking and visualisation were conducted using Autodock and pymol software. The experimental data in this paper are presented as mean ± standard deviation. We used GraphPad Prism 8 to analyse and visualize the data. T-tests were used to compare two groups, One-way ANOVA was used to compare multiple groups, and Two-way ANOVA was used for pairwise comparison between groups.
Results
Identification of the differential genes
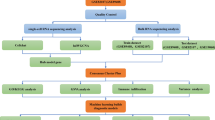
Figure 1 shows a complete analysis flow of this study. First of all, 1018 differentially expressed genes (DEGs1) were obtained in the OA and normal samples (Supplementary Table 4), within 438 were up-regulated (Supplementary Table 5) and 580 were down-regulated (Supplementary Table 6), as shown in Fig. 2A, B. Subsequently, the expression matrix for the glutamine metabolism-related gene set was extracted from the GSE57218 dataset. Consensus clustering analysis was performed using the PAM clustering algorithm with Euclidean distance as the metric. Among the 118 glutamine metabolism genes, 108 were present in the training set. Consensus clustering was conducted based on these 108 genes. Ultimately, the samples were divided into two stable molecular subtypes (Cluster_1 and Cluster_2). The validity of the clustering results was verified through PCA (Fig. 2D) and heatmap visualization (Fig. 2C, Supplementary Table 7). Differential analysis between subtypes was subsequently performed using the screening criteria: |log2FC| > 0.5 and adj.P.Val < 0.05, yielding 385 differentially expressed genes (DEGs2) (Supplementary Table 8). Among these, 161 genes were upregulated and 224 were downregulated (Fig. 2E, F).

A complete analysis flow of this study.

Identification of the differential genes. (A) Volcano map and (B) Heat map of differentially expressed genes in GSE57218. (C) Cluster analysis effect in disease samples. (D) Subtype PCA map. (E) Volcano map and (F) heat map of the differential analysis of genes of two stable molecular subtypes (Cluster_1 and Cluster_2).
Identification of the key genes
This study took an intersection of DEGs1 and DEGs2, 102 intersecting genes obtained as candidate genes and used for subsequent analyses (Fig. 3A, Supplementary Table 9). Subsequently, performed GO and KEGG enriched analysis, 174 GO terms were enriched (Supplementary Table 10), including 118 BP terms mainly artery development, protein localization to the plasma membrane, artery morphogenesis, protein localization to cell periphery and extracellular matrix assembly. 32 CC terms mainly clathrin-coated vesicle membrane, coated vesicle membrane, lysosomal membrane and lytic vacuole membrane. 24 MF terms mainly carbon-oxygen lyase activity, calcium-dependent protein binding and extracellular matrix structural constituent conferring tensile strength (Fig. 3B). 7 KEGG pathways were enriched (Supplementary Table 11), mainly Biosynthesis of amino acids, Arginine and proline metabolism, Ubiquitin mediated proteolysis, Biosynthesis of cofactors, Glycolysis / Gluconeogenesis and Ubiquinone and other terpenoid-quinone biosynthesis pathways (Fig. 3C). Subsequently, we constructed the PPI network of these candidate genes and found only 43 genes had the interaction relationship (Fig. 3D), which had 43 nodes, and 40 edges. Moreover, identified the key genes using Cytoscape’s 7 algorithms (BottleNeck, Closeness, Degree, DMNC, EPC, MCC, and MNC methods, Supplementary Tables 12–18), the Top 10 taken an intersection a total obtained 5 genes, ALDH18A1, LRRFIP1, MFSD11, PGK1 and TPI1 (Fig. 3E). Then, we further verified the expression levels in GSE57218 (Fig. 3F) and GSE117999 (Fig. 3G) and found that LRRFIP1 and MFSD11 both showed a significant difference and had the same trend. Therefore, LRRFIP1 and MFSD11 as the key genes used for subsequent analyses.

Identification of the key genes. (A) Venn of candidate genes. (B) GO and (C) KEGG enriched analysis. (source: www.kegg.jp/kegg/kegg1.html). Cluster analysis effect in disease samples. (D) PPI network of candidate genes. (E) Venn of candidate key genes. The expression levels verification in (F) GSE57218 and (G) GSE117999.
Construction of an ANN model and GSEA
To further evaluate the diagnosability of key genes for OA, we constructed the BP neural network (Fig. 4A) and plotted the ROC curve (Fig. 4B), the AUC = 0.818, which verified that key genes have good predictive ability for OA samples. At the same time, we also constructed the BP neural network for GSE11799 (Fig. 4C), the AUC = 0.75 in the ROC curve (Fig. 4D), which showed a similar result. Subsequently, we constructed the protein-protein interactions and related pathways network of key genes (Fig. 4E). In GSEA, LRRFIP1 enriched 11 KEGG pathways (Fig. 4F), mainly PARKINSONS_DISEASE, OXIDATIVE_PHOSPHORYLATION, ALZHEIMERS_DISEASE, HUNTINGTONS_DISEASE and PROTEASOME (Supplementary Table 19). MFSD11 enriched 15 KEGG pathways (Fig. 4G), mainly OLFACTORY_TRANSDUCTION, TASTE_TRANSDUCTION, SPLICEOSOME, MATURITY_ONSET_DIABETES_OF_THE_YOUNG and UBIQUITIN_MEDIATED_PROTEOLYSIS (Supplementary Table 20).

Construction of an ANN model and GSEA. (A) BP neural network construction for GSE57218 and (B) ROC curve. (C) BP neural network construction for GSE11799 and (D) ROC curve. (E) Co-expression network of key genes. GSEA of (F) LRRFIP1and (G) MFSD11.
Construction of regulatory network
To further verify the mechanism of key genes, a total of 274 TFs were searched (Supplementary Table 21), and we selected the Top 20 TFs based on Rank (Supplementary Table 22) and visualization of the network of TFs and mRNAs (Fig. 5A). Then, further obtained miRNAs of LRRFIP1 from miRDB (Supplementary Table 23), miRWalk (Supplementary Table 24) and miRtarbase (Supplementary Table 25) databases, and an intersection found 6 miRNAs (Fig. 5B, Supplementary Table 26). Also obtained were miRNAs of MFSD11 from miRDB (Supplementary Table 27), miRWalk (Supplementary Table 28) and miRtarbase (Supplementary Table 29) databases, and an intersection found 1 miRNA (Fig. 5C, Supplementary Table 30). Moreover, further predicted the lncRNAs of miRNAs, a total of 272 lncRNAs (Supplementary Table 31), we selected the lncRNAs with clipExpNum ≥ 5 using for lncRNA-miRNA-mRNA network construction (Fig. 5D). SNP sites of miRNA may affect the binding of miRNA and mRNA, so we further searched the SNP (mature), and visualized the network (Fig. 5E, Supplementary Table 32), which had 56 nodes and 54 edges.

Construction of regulatory network. (A) TF-mRNA network construction. (B) Venn of LRRFIP1 miRNA. (C) Venn of MFSD11 miRNA. (D) Construction of ceRNA network. (E) SNP-miRNA-mRNA network construction.
Drug prediction and molecular docking
Based on key genes MFSD11 and LRRFIP1 were downregulated in OA samples, so this study searched the CTD database for drugs that can promote their expression (Supplementary Tables 33–34), and obtained 14 drugs after taking an intersection (Fig. 6A, Supplementary Table 35), and visualized the key genes-drugs network (Fig. 6B), which had 16 nodes and 28 edges. Subsequently, we selected D014635, which was the Reference Count maximum (Supplementary Table 36) performed molecular docking (1KCal = 4.184KJ). Figure 6C shows the result of LRRFIP1 and Valproic Acid, and Fig. 6D shows the result of MFSD11 and Valproic Acid.

Drug prediction and molecular docking. (A) Venn of drug prediction. (B) Drug prediction network construction. (C) Molecular docking of LRRFIP1 and Valproic Acid. (D) Molecular docking of MFSD11 and Valproic Acid.
DMM mice model validation
We constructed the DMM mice model to further verify key genes. Compared with Sham group, erosion and hypocellularity of the superficial articular cartilage, as well as proteoglycan loss, were observed in the DMM group, as indicated in HE staining and Safranin O staining (Fig. 7A). Similarly, the OARSI score was higher level in the DMM group (Fig. 7B). These results are enough to demonstrate successful of the DMM mice model. Subsequently, we verified the expression levels of key genes using IHC (Fig. 7C), RT-qPCR (Fig. 7D) and Western blot (Fig. 7E) and found that LRRFIP1 and MFSD11 both had a lower expression level in the DMM group, compared with Sham group.

DMM mice model validation. (A) HE staining and Safranin O staining. (B) OARIS scores of mice cartilage. (C) IHC, (D) RT-qPCR and (E) Western blot detected the expression of key genes.
Cellular experimental validation
We constructed an OA cell model using IL-1β-induced cartilage, ELISA assay showed that compared with the NC group, IL-1β-induced significantly promoted the levels of prostaglandin E2 (PGE2), IL-6, IL-1β, and TNF-α (Fig. 8A). Moreover, Matrix metalloproteinase 3 (MMP-3) and Matrix metalloproteinase 13 (MMP-13) played a crucial role in OA occurrence for proteoglycan synthesis and matrix degradation in chondrocytes35. After IL-1β-induced the expression of MMP-3 and MMP-13 was increased significantly by Western blot (Fig. 8B). Also, Col2a1 (type II collagen alpha 1) and aggrecan as extracellular matrix proteins which in cartilage are essential for function36. Similarly, we found that reduced the expression of Col2a1 and aggrecan in IL-1β-induced cartilage by Western blot (Fig. 8B) and IF (Fig. 8C). Interestingly, the expression levels of LRRFIP1 and MFSD11 also down-regulated after IL-1β-induced (Fig. 8D-E).

Cellular experimental validation. (A) The content of IL-6, IL-1β, TNF-α and PGE2 using ELISA. (B) Western blot detected the expression of MMP-3, MMP-13, Col2a1 and aggrecan. (C) IF detected the expression of Col2a1 and aggrecan. (D) RT-qPCR and (E) Western blot detected key gene expression.
Discussion
Glutamine is a type of non-essential amino acid, and more and more research is focusing on its biological functions in various diseases and cellular energy37. Glutamine plays a vital role in chondrocytes which can maintain glycosaminoglycan, protein synthesis and extracellular matrix metabolism, also glutamine metabolism has crucial regulatory roles in sustaining bone homeostasis38. Moreover, glutamine metabolism is involved in chondrocytes’ inflammatory response, which is important for Osteoarthritis (OA), as reported in previous research11. However, this regulation of biological processes is complex, the molecular mechanism deserves further to be elucidated. This study aimed to find glutamine metabolism-related genes as potential biomarkers in OA, transcriptome datasets for bioinformatics analysis of OA and normal samples based on the GEO database.
Articular cartilage injury and degeneration are the major characteristics of OA development, chondrocytes regulating the function and structure of cartilage are of utmost importance39. However, previous studies confirmed that glutamine is vital for maintaining bone homeostasis at the cellular level, such as glutamine is essential in promoting cell cycle and proliferation in vitro40,41, and glutamine metabolism can provide ATP for BMSC differentiation42. Moreover, glutamine is closely related to glutathione, which can protect chondrocytes by upregulating glutathione concentration, also which can play a crucial effect in inhibiting chondrocyte apoptosis and cartilage degeneration43,44. This study obtained 102 candidate genes based on combining differential genes in OA samples with glutamine metabolism-related differential genes and found that mainly related to extracellular matrix assembly, calcium-dependent protein binding and extracellular matrix structural constituent conferring tensile strength, Arginine and proline metabolism, Ubiquitin mediated proteolysis, and Glycolysis/Gluconeogenesis pathways in function enrichment. Extracellular matrix (ECM) is an important part of chondrocytes’ maintaining tissue homeostasis, the ECM will lost after OA tissue damage, new ECM is secreted by chondrocytes can repair the damaged tissue39. Moreover, the calcium-dependent enzyme transglutaminase 2 (TG2) has been reported to have high expression in OA tissue, which induces the expression of MMP-13 increase and further affects the OA progression45. Functional amino acids (AA) such as arginine and proline are highly beneficial for OA inflammation response46. Also, arginine and proline metabolism pathways have been verified significant effects on OA47. Ubiquitin mediated proteolysis has also been confirmed important pathway for participation in OA multiple times48. Similarly, Glycolysis/Gluconeogenesis pathways have also been confirmed49.
Subsequently, we identified the key candidate genes using the PPI network and interaction relationship and performed the expression levels verification, finally finding LRRFIP1 and MFSD11 as the key genes, which had a significantly low expression in OA samples. And showed a better diagnostic validity for OA. Leucine Rich Repeat of Flightless-1 Interacting Protein 1 (LRRFIP1) has been found as one part of the artificial chimeric cDNA in the early, which played broad roles in various biological processes and multiple human diseases50. Interestingly, LRRFIP1 is a dysregulation, which coincides with our results. LRRFIP1/GCF2 has been reported to affect TNF-α overexpression and multiple inflammation diseases by decreasing the binding with TNF-α promotor51, TNF-α is one of the vital inflammatory cytokines in OA52. The research on major facilitator superfamily domain 11 (MFSD11) is deficient, and mechanisms are unclear. Previous studies have reported that MFSD11 is a key cancer prognostic gene with a significant correlation with ovarian cancer (OA)53 and esophageal squamous cell carcinoma (ESCC)54. Moreover, MFSD11 has wide expression in mice, such as the nervous system (NS)55. Meanwhile, in this study, we detected the expression of MFSD11 in mice tissue and cells and found that had a low expression after constructing the OA model.
In summary, this study conducted initial research into the potential mechanisms of glutamine metabolism-related genes in OA tissue and further verified the role of glutamine metabolism-related genes in OA occurrence. Further found that LRRFIP1 and MFSD11 as the key genes with better diagnostic capabilities in OA and which may be potential biomarkers for the diagnosis and treatment of OA. However, this study has certain limitations: key genes were selected through differential analysis and clustering strategies, and gene selection may have been influenced by the initial clustering approach. Nevertheless, independent GEO datasets, DMM mouse models, and IL-1β-induced cellular models all confirm the downregulation of these key genes in OA, demonstrating the validity of the screening strategy. However, the precise mechanisms by which these two key genes regulate OA require further validation through functional experiments. Subsequent research will delve deeper into the specific molecular mechanisms of LRRFIP1 and MFSD11 in OA.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Katz, J. N., Arant, K. R. & Loeser, R. F. Diagnosis and treatment of hip and knee osteoarthritis: a review. Jama 325, 568–578. https://doi.org/10.1001/jama.2020.22171 (2021).
Hong, H. et al. Associations of Homocysteine, Folate, and vitamin B12 with osteoarthritis: a Mendelian randomization study. Nutrients 15, 2563. https://doi.org/10.3390/nu15071636 (2023).
Johnson, V. L. & Hunter, D. J. The epidemiology of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 28, 5–15. https://doi.org/10.1016/j.berh.2014.01.004 (2014).
Losina, E. et al. Lifetime risk and age at diagnosis of symptomatic knee osteoarthritis in the US. Arthritis Care Res. 65, 703–711. https://doi.org/10.1002/acr.21898 (2013).
Vargas Negrín, F., Abellán, M., Hermosa, M. D., Hernán, J. C. & de Felipe Medina, R. Treatment of patients with osteoarthritis. Aten. Primaria. 46 (Suppl 1), 39–61. https://doi.org/10.1016/s0212-6567(14)70043-5 (2014).
Shams, S. et al. The therapeutic landscape of rheumatoid arthritis: current state and future directions. Front. Pharmacol. 12, 680043. https://doi.org/10.3389/fphar.2021.680043 (2021).
Newsholme, P., Diniz, V. L. S., Dodd, G. T. & Cruzat, V. Glutamine metabolism and optimal immune and CNS function. Proc. Nutr. Soc. 82, 22–31. https://doi.org/10.1017/s0029665122002749 (2023).
Biancur, D. E. et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 8, 15965. https://doi.org/10.1038/ncomms15965 (2017).
Prideaux, M., Findlay, D. M. & Atkins, G. J. Osteocytes: The master cells in bone remodelling. Curr. Opin. Pharmacol. 28, 24–30. https://doi.org/10.1016/j.coph.2016.02.003 (2016).
Yao, S., Li, C., Beckley, M. & Liu, D. Expression of odontogenic ameloblast-associated protein in the dental follicle and its role in osteogenic differentiation of dental follicle stem cells. Arch. Oral Biol. 78, 6–12. https://doi.org/10.1016/j.archoralbio.2017.02.001 (2017).
Arra, M. et al. Glutamine metabolism modulates chondrocyte inflammatory response. eLife 11, 256. https://doi.org/10.7554/eLife.80725 (2022).
Yang, R. et al. Functional gelatin hydrogel scaffold with degraded-release of glutamine to enhance cellular energy metabolism for cartilage repair. Int. J. Biol. Macromol. 221, 923–933. https://doi.org/10.1016/j.ijbiomac.2022.09.039 (2022).
Stegen, S. et al. Glutamine metabolism controls chondrocyte identity and function. Dev. Cell. 53, 530–544e538. https://doi.org/10.1016/j.devcel.2020.05.001 (2020).
Piepoli, T. et al. Glutamate signaling in chondrocytes and the potential involvement of NMDA receptors in cell proliferation and inflammatory gene expression. Osteoarthr. Cartil. 17, 1076–1083. https://doi.org/10.1016/j.joca.2009.02.002 (2009).
He, S. et al. Turbulence of glutamine metabolism in pan-cancer prognosis and immune microenvironment. Front. Oncol. 12, 1064127. https://doi.org/10.3389/fonc.2022.1064127 (2022).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Wang, J. et al. PROS1 shapes the immune-suppressive tumor microenvironment and predicts poor prognosis in glioma. Front. Immunol. 13, 253. https://doi.org/10.3389/fimmu.2022.1052692 (2022).
Gu, Z. & Hübschmann, D. Make interactive complex heatmaps. Bioinf. (Oxford England). 38, 1460–1462. https://doi.org/10.1093/bioinformatics/btab806 (2022).
Qiu, C. et al. Identification of molecular subtypes and a prognostic signature based on Inflammation-Related genes in colon adenocarcinoma. Front. Immunol. 12, 769685. https://doi.org/10.3389/fimmu.2021.769685 (2021).
Mao, W., Ding, J., Li, Y., Huang, R. & Wang, B. Inhibition of cell survival and invasion by Tanshinone IIA via FTH1: a key therapeutic target and biomarker in head and neck squamous cell carcinoma. Exp.Therapeut. Med. 24, 521. https://doi.org/10.3892/etm.2022.11449 (2022).
Wu, T. et al. ClusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. (Cambridge (Mass)). 2, 100141. https://doi.org/10.1016/j.xinn.2021.100141 (2021).
The Gene Ontology Resource. 20 years and still going strong. Nucleic Acids Res. 47, D330–d338. https://doi.org/10.1093/nar/gky1055 (2019).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462. https://doi.org/10.1093/nar/gkv1070 (2016).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Science: Publication Protein Soc. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Xu, Q. et al. Multi-omics analysis reveals prognostic value of tumor mutation burden in hepatocellular carcinoma. Cancer Cell Int. 21, 253. https://doi.org/10.1186/s12935-021-02049-w (2021).
Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–452. https://doi.org/10.1093/nar/gku1003 (2015).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Gutman, R., Aronson, D., Caspi, O. & Shalit, U. What drives performance in machine learning models for predicting heart failure outcome? Eur. Heart J. Digit. Health. 4, 175–187. https://doi.org/10.1093/ehjdh/ztac054 (2023).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77. https://doi.org/10.1186/1471-2105-12-77 (2011).
Lian, Y. et al. Network Pharmacology assessment of Qingkailing injection treatment of cholestatic hepatitis. J. Traditional Chin. Med. = Chung I Tsa Chih Ying Wen Pan. 41, 167–180. https://doi.org/10.19852/j.cnki.jtcm.20201208.001 (2021).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. https://doi.org/10.1002/jcc.21256 (2009).
Seeliger, D. & de Groot, B. L. Ligand Docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 24, 417–422. https://doi.org/10.1007/s10822-010-9352-6 (2010).
Glasson, S. S., Blanchet, T. J. & Morris, E. A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 15, 1061–1069. https://doi.org/10.1016/j.joca.2007.03.006 (2007).
Li, C., Li, W., Pu, G., Wu, J. & Qin, F. Exosomes derived from miR-338-3p-modified adipose stem cells inhibited inflammation injury of chondrocytes via targeting RUNX2 in osteoarthritis. J. Orthop. Surg, Res. 17, 567. https://doi.org/10.1186/s13018-022-03437-2 (2022).
Hodax, J. K. et al. Aggrecan is required for chondrocyte differentiation in ATDC5 chondroprogenitor cells. PloS One. 14, e0218399. https://doi.org/10.1371/journal.pone.0218399 (2019).
El-Lekawy, A. M., Abdallah, D. M. & El-Abhar, H. S. Alanyl-glutamine heals Indomethacin-induced gastric ulceration in rats via antisecretory and Anti-apoptotic mechanisms. J. Pediatr. Gastroenterol. Nutr. 69, 710–718. https://doi.org/10.1097/mpg.0000000000002474 (2019).
Zhou, T., Yang, Y., Chen, Q. & Xie, L. Glutamine metabolism is essential for stemness of bone marrow mesenchymal stem cells and bone homeostasis. Stem Cells Int. 2019, 8928934. https://doi.org/10.1155/2019/8928934 (2019).
Akkiraju, H. & Nohe, A. Role of chondrocytes in cartilage Formation, progression of osteoarthritis and cartilage regeneration. J. Dev. Biol. 3, 177–192. https://doi.org/10.3390/jdb3040177 (2015).
Yuan, L. et al. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. Relat. Cancer. 22, 577–591. https://doi.org/10.1530/erc-15-0192 (2015).
Lenz, P. et al. Cell-cycle control in urothelial carcinoma: large-scale tissue array analysis of tumor tissue from Maine and Vermont. Cancer Epidemiol. Biomark. Prevent. 21, 1555–1564. https://doi.org/10.1158/1055-9965.Epi-12-0261 (2012).
Yu, Y. et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell metabolism 29, 966–978.e964. https://doi.org/10.1016/j.cmet.2019.01.016 (2019).
Issa, R., Boeving, M., Kinter, M. & Griffin, T. M. Effect of Biomechanical stress on endogenous antioxidant networks in bovine articular cartilage. J. Orthop. Res. 36, 760–769. https://doi.org/10.1002/jor.23728 (2018).
Tonomura, H. et al. Glutamine protects articular chondrocytes from heat stress and NO-induced apoptosis with HSP70 expression. Osteoarthr. Cartil. 14, 545–553. https://doi.org/10.1016/j.joca.2005.12.008 (2006).
Han, M. S., Jung, Y. K., Kim, G. W. & Han, S. Transglutaminase-2 regulates Wnt and FoxO3a signaling to determine the severity of osteoarthritis. Sci. Rep. 10, 13228. https://doi.org/10.1038/s41598-020-70115-w (2020).
Li, Y. et al. Alterations of amino acid metabolism in osteoarthritis: its implications for nutrition and health. Amino Acids. 48, 907–914. https://doi.org/10.1007/s00726-015-2168-x (2016).
Chen, R. et al. Perturbations in amino acids and metabolic pathways in osteoarthritis patients determined by targeted metabolomics analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1085, 54–62. https://doi.org/10.1016/j.jchromb.2018.03.047 (2018).
Ge, Y. et al. Identification and validation of hub genes of synovial tissue for patients with osteoarthritis and rheumatoid arthritis. Hereditas 158, 37. https://doi.org/10.1186/s41065-021-00201-0 (2021).
Ma, Z. et al. GCTOF-MS combined LC-QTRAP-MS/MS reveals metabolic difference between osteoarthritis and osteoporotic osteoarthritis and the intervention effect of erxian Decoction. Front. Endocrinol. 13, 905507. https://doi.org/10.3389/fendo.2022.905507 (2022).
Takimoto, M. Multidisciplinary roles of LRRFIP1/GCF2 in human biological systems and diseases. Cells 8, 253. https://doi.org/10.3390/cells8020108 (2019).
Suriano, A. R. et al. GCF2/LRRFIP1 represses tumor necrosis factor alpha expression. Mol. Cell. Biol. 25, 9073–9081. https://doi.org/10.1128/mcb.25.20.9073-9081.2005 (2005).
Wojdasiewicz, P., Poniatowski, Ł. & Szukiewicz, D. A. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014, 561459. https://doi.org/10.1155/2014/561459 (2014).
Yao, S. et al. Alternative splicing: a new therapeutic target for ovarian cancer. Technol. Cancer Res. Treat. 21, 15330338211067911. https://doi.org/10.1177/15330338211067911 (2022).
Liu, H. et al. JMJD6 functions as an oncogene and is associated with poor prognosis in esophageal squamous cell carcinoma. BMC Cancer. 23, 253. https://doi.org/10.1186/s12885-023-11171-z (2023).
Perland, E. et al. The putative SLC transporters Mfsd5 and Mfsd11 are abundantly expressed in the mouse brain and have a potential role in energy homeostasis. PloS One. 11, e0156912. https://doi.org/10.1371/journal.pone.0156912 (2016).
Acknowledgements
We acknowledge GEO, GSEA, GO, KEGG, STRING, DGIdb, miRDB, miRWalk, miRtarbase, GeneMINIA, ChEA3, Starbase, PDB, PubChem and CTD databases for providing their platforms and contributors for uploading their meaningful datasets. And thank our colleagues for their helpful suggestions.
Funding
Yunnan Provincial Department of Science and Technology Basic Research Program (202301AZ070001-091).Yunnan Provincial Department of Science and Technology Basic Research Program (202401AZ070001-067).
Author information
Authors and Affiliations
Contributions
Dongyun Li: Formal analysis, Validation, Writing-original draft. ChenWang: Formal analysis, Data curation, Validation. Yuelei Qing: Data curation, Validation. Xitong Bao: Data curation, Validation. Jingxiao Xu: Data curation, Formal analysis. Xinyu Wang: Resources. Wenping Bao: Resources. Xiaoying Wang: Conceptualization, Writing-review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The animal experiments with the assistance of Yunnan Labreal Biotech Co., Ltd., the animal experiment was conducted and received approval from the Experimental Animal Ethics Committee of Yunnan Labreal Biotech Co., Ltd. (IACUC Issue No. PZ20240111).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, D., Wang, C., Qing, Y. et al. Potential glutamine metabolism-related biomarkers were identified in osteoarthritis by bioinformatics. Sci Rep 15, 45497 (2025). https://doi.org/10.1038/s41598-025-29541-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-29541-x
