
Abstract
Epithelial ovarian cancer is the sixth most common malignant neoplasm in women and the leading cause of death from gynecological malignancies in the Western World. The five-year survival rate is 45–48% because of the high frequency of late-stage diagnoses and the development of chemo-resistant disease. To assess the potential of antibodies and complement to kill platinum (Pt)-resistant and Pt-sensitive cell lines, antibodies S2 and Yth53.1 were tested at varying concentrations using high-grade serous ovarian cancer (HGSOC) cells. Cell lysis was quantified after 60 minutes using the impermeant dyes 7-Aminoactinomycin D and Annexin V. The cytotoxic effect of S2 was demonstrated through the activation of the complement and Yth53.1 monoclonal antibody neutralized the membrane attack complex (MAC) inhibitor CD59 and allowed assembly of the MAC. In contrast to Pt-based therapy, we found no evidence that chemo-resistance plays an important role when considering the antibody based therapy for Pt-resistant HGSOC cell lines. Significant killing efficiencies were achieved using antibodies against breast cancer cells (S2) and antibody against CD59 (Yht53.1) regardless whether the cell lines HGSOC (OVCAR-3, OVCAR-8) or CRISPR-Cas9 BRCA2 deleted OVCAR-4 were homologous recombination (HR) deficient or proficient, Pt sensitive or resistant. This work shows a potential towards antibody-based therapy development for Pt-resistant HGSOC.
Similar content being viewed by others


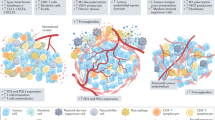
Introduction
High-grade serous ovarian cancer (HGSOC) is the most common and aggressive type of ovarian cancer (OC) and the fifth most common malignancy occurring in women, it is the leading cause of death from gynecological malignancies in the world1. It is estimated that 324,603 new cases of epithelial ovarian cancer (EOC) and 206,956 cancer-related deaths occurred in 2022 worldwide2. This is due to the combination of factors that include delayed diagnosis, the development of chemoresistant disease, recurrent peritoneal metastases, the specific microenvironment of ovarian cancer, and various mechanisms of tumor evasion of host immune response.
The tumors of the HGSOC patient are sensitive to cytoreductive surgery and platinum (Pt)/taxanes-based chemotherapy. Currently, almost all patients receive carboplatin and paclitaxel in the first-line3. Paclitaxel targets tubulin and stops cell division4. Pt (II) drugs such as cisplatin kill cells by crosslinking DNA, which is an effective way to stop aggressively developing tumors. Pt agents induce DNA crosslinks, which removes temporary but potentially toxic double strand-breaks (DSBs) which are repaired by HR5,6. Although first-line chemotherapy with carboplatin and paclitaxel achieves an improvement and most patients are initially highly responsive to platinum chemotherapy; however, recurrence occurs in 25% of patients with early stage disease and more than 80% of patients with advanced disease within 1 to 3 years7.
Approximately 50% of HGSOCs are characterized by inactivation of genes required for HR, which is a major mechanism of double-strand DNA repair, such as BRCA1 and BRCA28,9. Down-regulation of the HR protein BRCA2 significantly promotes apoptosis and enhances carboplatin cytotoxicity; therefore, disruption of functional HR is a potential therapeutic target to prevent resistance to chemotherapy. A key insight into the clinical development of poly (ADP-ribose) polymerase inhibitors (PARPis) has come through the analysis of patients with Pt-sensitive and resistant ovarian cancer9. Despite the efficacy of PARPis in repairing single-stranded DNA breaks, while PARPis showed some response for Pt-sensitive cells, Pt resistant patients still responded poorly2. Maintenance therapy with PARP inhibitors has shown some efficacy in patients following platinum-based treatment10. Patients with germline or somatic BRCA1/2 mutations responded better to PARPis olaparib, niraparib, and rucaparib than patients with wild-type BRCA11,12. Despite their similar mechanisms of action, these three inhibitors had specific toxicity and resulted in dose interruptions and discontinuation of treatments13. Due to survival limits, niraparib, olaparib, and rucaparib used for the treatment of advanced EOC positive for BRCA mutated or HR deficient EOC were withdrawn14,15,16. Therefore, there is an unmet need to identify new treatments for patients with EOC.
T cells play a central role in immunotherapies for solid cancers, as shown by clinical responses (10-35%) achieved in several clinical trials with immune checkpoint inhibitors (ICIs) such as anti-CTLA4 and anti-PD-1/PD-L117. The potential benefit seen in patients with specific genomic markers (like BRCA1/2) mutations or high tumor mutation burden (TMB) resulted in the aim to identify subgroups of ovarian cancer patients who could respond to ICIs in combination with standard therapies18. While some research points to potential benefits for specific patient subsets with homologous recombination deficiency (HRD) or other genetic markers, ICIs have not yet proven effective in unselected ovarian cancer patients and are not in routine use for the treatment18. Combinatorial strategies have become the main focus of research19. Phase III trials like the JAVELIN OVARIAN 100 trial with avelumab (NCT02718417), the DUO-O trial with durvalumab (NCT03737643), IMagyn050 trial with atezolizumab (NCT03038100), ATHENA-COMBO with nivolumab (NCT03522246), and recently KEYLYNK-001 with pembrolizumab (NCT03740165) evaluated the clinical benefit of incorporating ICIs into the first-line treatment regimen for advanced EOC18,20. Although dramatic responses have been observed in some types of non-ovarian tumors to immunotherapies, most clinical trials of ovarian cancer immunotherapy have shown largely negative results with ICIs, none of these trials improved progression free survival in overall ovarian cancer patient population and indicated a lack of clinical benefit of adding ICIs to standard systemic therapy2,18,21,22,23.
Despite advances in targeted therapies and CAR T cell therapies, their efficacy in solid tumors remains limited by antigen heterogeneity, escape, stromal exclusion, and immunosuppressive tumor microenvironments24. Recently, allogenic CAR-NKT cells demonstrated superior anti-ovarian cancer efficacy compared to conventional CAR-T cells25. Tumor heterogeneity leads to variation in immune cell-targeted mutations and also to targeting specific antigens in HGSOC patients and limits the effectiveness of tumor-specific antigen-targeted therapies; therefore, activation of humoral responses and production of antibodies could be an alternative strategy26,27,28,29,30. The promising immunotherapy tumor killer system is to harness the patient’s own effector mechanisms such as antibody-dependent cellular toxicity (ADCC), direct complement-mediated killing (CDC), complement-dependent cellular cytotoxicity (CDCC), or the complement system to destroy malignant cells31,32,33,34,35. In the CDC mechanism, antibodies lyse the unwanted target cancer cells by activating a cascade of complement-related reactions36. CDC mechanism involves the direct formation of the membrane attack complex (MAC) on a targeted cells and their lysis37. When antibody binds to an antigen on the surface of a target cell, it triggers the classical complement pathway where C1q protein binding to the antibody and series of complement proteins assemble to form the MAC causing the cell to die37. While in CDCC, the complement cascade activation leads to the deposition of complement opsonins (like C3b) on the target cell’s surface37. These opsonins can be also recognized by complement receptors on the surface of other cells, such as macrophages, neutrophils, or natural killer cells and by activating them kill the target cancer cells37.
Antigen-antibody interactions enhance the humoral response mounted against the cancer specific antigens34. Antibodies contribute to the immunity in three ways: neutralization, opsonization, and complement activation38. Antibodies once bound to the cancer cells can activate the proteins of the classical pathway of the complement system39. In the absence of infection or disease, complement proteins circulate in the inactive form in our bloodstream40. Complement proteins act as precursor enzymes, effector molecules, they control proteins or receptors. The contact of the first component with an activator i.e. an immunoglobulin leads to a subsequent activation of the second one in a precise order of the complement pathway. The classical complement pathway is activated when IgM or IgG is bound to the tumor cell surface41. Similar to ADCC, antibodies elicit CDC via binding its Fc region to serum complement components, particularly C1q which triggers36 the cascade of the proteolysis zymogens and activates one of the three distinct effector pathways: inflammation, phagocytosis, and membrane attack complex (MAC)42. The complement system recognizes specific features on the cancer cell surfaces: antibody complexes and marks them for destruction by coating them with C3b and iC3b fragments generated on the target cell surface. These fragments can engage complement receptors on cancer cells and induce CDCC by activating the MAC and lysing cells by forming 10 nm pores in ovarian cancer cells membranes43,44,45.
Only a small number of monoclonal antibodies (mAbs) approved by the FDA for anticancer treatment are complement-fixing, highlighting the window of opportunity to increase the efficiency of current immunotherapies36. The efficacy of CDC for tumor targeting antibodies is limited because of production of complement regulatory proteins (CRPs) by cancer cells36. Tumors cells are known to have sophisticated mechanisms to escape the immune attack and usually are not recognized as cancer cells46. Host cells express complement regulators molecules (CD46, CD55 and CD59) on their surfaces to restrict harmful complement activation on the cells to a minimum47,48,49. Previous studies with lymphoma, glioblastoma, prostate, and ovarian cancer cell lines have shown significant differences in complement resistance between cell lines and tumor types34,47,49,50,51.
Our hypothesis is that killing of Pt tumor cells can be achieved, if complement could be targeted to the tumor cells by specific antibodies and complement inhibitors on the surface of the tumor cells could be neutralized by Yth53.1.
In this work we are trying to elucidate possible immune-therapeutic directions with S2 and Yth53.1 for developing humoral immunotherapies using active complement from normal human serum in combination with antibodies targeting Pt resistant HGSOC cells. The purpose of the present study is to evaluate the combined effect of S2 antibodies and Yth53.1 antibody, which targets CD59, to facilitate the assembly of the MAC. In this study the cytotoxic effect of human complement on HGSOC cells was examined in the presence and absence CD59-neutralizing antibodies. Treatment of cells with the Yth53.1 monoclonal antibody together with the complement-activating S2 antibody resulted in increased complement-mediated cytotoxicity. By neutralizing the tumor cell’s ability to exploit CD59 and other resistance mechanisms, this study demonstrate an enhanced complement-dependent killing effect through the combined use of antibody-based therapy in platinum-resistant HGSOC cell lines.
Results
The study included a range of HGSOC cell lines with distinct characteristics. Among them, OVCAR-3, an epithelial and HR-proficient cell line, and OVCAR-8, a mesenchymal and HR-deficient cell line, were examined. Additionally, we studied two single clones derived from the OVCAR-4 cell line (OVCAR-4-262 and OVCAR-4-264). BRCA2 mutated HR-deficient cell lines 262 and 264, and the wild-type OVCAR-4 which is HR-proficient. BRCA2 mutations had been introduced using CRISPR-Cas9 technology, rendering them HR-deficient. In contrast, the original wild-type OVCAR-4 cell line retained its HR-proficient status, offering a comparative basis for the investigation. We observed an increase in cytotoxicity up to 95% upon the addition of S2 antibodies during the assays (Fig. 1 and 2, Supplementary Figures A1–A7, A13—A15). Remarkably, nearly all HGSOC cells (89%) were eliminated, regardless of whether CD59 was simultaneously blocked. The extent of cell death varied depending on the concentration of the S2 antibody, highlighting its pivotal role in driving cytotoxic effects (Fig. 1, 2, Supplementary Figures A1–A7, A13—A15).
In the case of the Pt-resistant OVCAR-8 cell line, the addition of antibodies resulted in enhanced cytotoxicity (Fig. 1 and 2, Supplementary Figure A4). At a constant Yth53.1 concentration of 20 \({\upmu }\)g/ml, the inclusion of 3% (v/v) S2 increased the percentage of cells positive for Annexin V and 7-AAD from 90.2% to 98.8%. Similarly, when Yth53.1 was added to 1% (v/v) S2, the percentage of positive cells for Annexin V and 7-AAD rose from 50% to 96.9%. For OVCAR-8 HR-proficient (Pt-sensitive cell line) the use of 3% (v/v) S2 together with Yth53.1 led to 80% killing of cancer cells, 39% of the cells affected were positive for 7-AAD and PE-Annexin V, in addition 29.4% were early apoptotic cells and 13.8% of the cells were necrotic (Fig. 1, 2, Supplementary Figure A5). In comparison to OVCAR-8, a lower killing efficiency were seen for OVCAR-3 Pt-resistant and sensitive cell lines at 1% (v/v) S2 (Fig. 1 and 2, Supplementary Figures A6 and A7). Overall, very similar killing efficiencies were seen when comparing the Pt-resistant and sensitive OVCAR-3 cells lines. In all OVCAR-4 cells (WT, 264, 262) more than 98% cells were positive for Annexin V and 7-AAD at 3% (v/v) S2 (Supplementary Fig. A1–A3). The addition of YTh53.1 to S2 1% increased the number of positive cells to Annexin V and 7-AAD from 68.3% to 95.4% (WT), from 58.6 to 90.3 % (262), and from 46.9 % to 61% (264). There was no significant difference observed in killing efficiencies between OVCAR-4 262, 264 cell lines and WT cell lines. To sum up all HGSOC cell lines under study demonstrated sensitivity to complement-mediated killing, regardless of their specific subtype (chemo resistant vs. sensitive, HR-deficient vs. HR-proficient).

OVCAR-8 and OVCAR-3 Pt-resistant (R) and sensitive (S) HGSOC cells were treated with S2 0% (A) 1% S2 and 3% S2 (B, D) and 1 % S2 and 3% +20 \({\upmu }\)g/ml Yth53.1 (C, E). HGSOC cells were tracked from PE Annexin V and 7-AAD negative (viable, or no measurable apoptosis), to PE Annexin V positive and 7-AAD negative (early apoptosis, membrane integrity is present) and finally to PE Annexin V and 7-AAD positive (end stage apoptosis and death).
To further assess the differences in the mean killing efficiency between the two replicates at each concentration of S2, both alone and in combination with Yth53.1, all cells were monitored for positivity to PE-Annexin V and 7-AAD (at the end stage apoptosis and cell death) (Fig. 2). With increasing concentration of antibody S2 the killing efficiencies were increased for all cell lines. The end stage of apoptosis was achieved at 3% (v/v) of S2 for most cell lines.

Apoptosis measured after complement killing assay after 60 min incubation with antibodies and normal human serum (NHS). HGSOC cells were tracked from PE-Annexin V and 7-AAD positive (end stage apoptosis and death). (A) OVCAR-8 and OVCAR-3 Pt-resistant and sensitive HGSOC cells treated with S2 antibodies. (B) OVCAR-8 and OVCAR-3 Pt-resistant and sensitive HGSOC cells treated with S2+20 \({\upmu }\)g/ml Yth53.1 antibodies. (C) OVCAR-4 (262, 264, WT) cells treated with S2 alone. (D) OVCAR-4 (262, 264, WT) cells treated with S2 +20 \({\upmu }\)g/ml Yth53.1.
To evaluate differences in cytotoxicity between Pt-resistant and Pt-sensitive HGSOC cell lines, data from 2 replicates at each concentration were pooled for each group and analyzed. Killing efficiency was quantified as the percentage change from each replicate’s mean at specific antibody concentrations for the respective cancer cell lines (Fig. 3). By taking a mean within each concentration we focused on the effect on the effect of the Pt resistivity versus sensitivity. The results shown in Fig. 1 revealed no significant differences in killing efficiencies between Pt-sensitive and Pt-resistant HGSOC cell lines (OVCAR-3, OVCAR-8). Statistical analysis using the Mann-Whitney U-test yielded the p-value of 0.5382 while comparing killing efficiencies of Pt-sensitive and -resistant cells (under S2 alone treatment) and p-value of 0.7930 was obtained under S2+Yth53.1 treatment. The obtained p value indicated that there is no a significant difference between the two groups (Pt resistant and sensitive). The median for Pt-resistant was -2.78 and for Pt sensitive the median was 0.17 (under S2 alone treatment). Under S2+Yth53.1 treatment the median for Pt resistant was 2.95 and for Pt sensitive 2.4.

Killing efficiency after antibodies S2 alone (A) and S2+Yth53.1 treatment (B) for each replicate shown as the percentage change from the mean for each concentration across OVCAR8 and OVCAR3 Pt sensitive and Pt resistant HGSOC cell lines.
To confirm that all the cell lines treated with S2 and Yth53.1 were highly sensitive to complement killing, the formation of MAC on cancer cells was traced by flow cytometry (Fig. 4). Cells were incubated with the antibodies and normal human serum, and prior to further incubation, unbound serum and antibodies were washed away. MAC formation was detected using a monoclonal mouse anti-human C5b-9 antibody (clone aE11) and visualized with a secondary goat anti-mouse IgG antibody conjugated to Alexa Fluor-488 (Fig. 4). The results showed that exposure to 0.1% (v/v) S2 induced significant MAC formation on the cancer cells. In addition, when heat inactivated serum was used in assays no lysis of cancer cells was observed (Supplementary Figure A14).

Membrane attack complex C5b-9 expression quantification after: (A) OVCAR-8 resistant cells were not treated with antibodies; (B) OVCAR-8 were treated with 0.1% S2+NHS; (C) OVCAR-8 were treated with 0.1% S2+20 \({\upmu }\)g/ml Yth53.1+NHS.
To further evaluate the differences in cytotoxicity between Pt-resistant and Pt-sensitive HGSOC cell lines using antibodies and the carboplatin drug, we analyzed the killing efficiency of OVCAR-3, OVCAR-8, and OVCAR-4 cell lines treated with carboplatin alone (Fig. 5 and Supplementary Figures A8–A12). Our results demonstrated that Pt-resistant cell lines exhibited lower sensitivity to carboplatin compared to antibody-mediated killing.

HGSOC cells treated with carboplatin at increasing concentrations: (A) OVCAR-3 Pt sensitive and OVCAR-3 5R Pt resistant (HR-proficient) at concentrations 0, 0.25, 0.5, 1, 2 and 4 \({\upmu }\)M (B) OVCAR-8 Pt sensitive and OVCAR-8 R Pt resistant (HR-deficient) at concentrations 0, 2, 4 and 8 \({\upmu }\)M. IC50 values for OVCAR-8 Pt sensitive: 1.069 \({\upmu }\)M and OVCAR-8 R Pt resistant: 3.215 \({\upmu }\)M and (C) OVCAR-4: WT (HR-proficient), 262, 264 cell lines (HR-deficient) at concentrations 0, 0.5, 1,2, and 4 \({\upmu }\)M. IC50 values for WT: 1.7 \({\upmu }\)M, 262: 0.4 \({\upmu }\)M ja 264: 0.9 \({\upmu }\)M.
Discussion
Drug resistance is one of the most important factors in the failure of chemotherapy in advanced ovarian cancer7. This study demonstrates that the neutralization of CD59, antibody membrane complement regulatory protein (mCRP), combined with the antibody S2, effectively directs complement-mediated cytotoxicity against HGSOC Pt-resistant and sensitive OVCAR-8 and OVCAR-3, as well as the CRISPR-Cas9 BRCA2 deleted OVCAR-4 cell lines 262 and 264. CD59, a key antibody membrane complement regulatory protein that makes tumor cells resistant to complement killing45, has partially lost its protective effect when neutralized by a specific anti-CD59 antibody. Flow cytometry analysis revealed no significant differences in killing efficiencies between HGSOC Pt resistant and Pt sensitive OVCAR-3 and OVCAR-8, as well as CRISPR-Cas9 BRCA2 deleted OVCAR-4 262 and 264 cell lines treated with antibodies S2 and Yth53.1. Incubation with antibodies S2 and Yth53.1 and a single dose of normal human sera resulted in complete elimination of both Pt-resistant and Pt-sensitive HGSOC cells. 85% of the cells were killed within 60 minutes of exposure to normal human sera regardless of HR status or BRCA2 mutation. These findings suggest that complement-activating antibodies could be injected locally into the peritoneal cavity to enhance therapeutic outcomes for ovarian cancer patients. These results underscore the potential of this therapeutic approach for targeting chemotherapy-resistant ovarian cancer.
To date, immunotherapy (alone or in combination) does not improve overall survival in newly diagnosed, Pt-sensitive, and Pt-resistant ovarian cancer patients2. Therefore, activating the B cell response against tumors may be an alternative strategy30,52. Tumor-infiltrating B (TILs-B) cells that infiltrate tissue sites are generally activated and undergo differentiation in tumor microenvironment or migrate from lymphoid organs to produce antibodies30,53,54. B cells can present antigens to T cells either directly or via immune complexes by dendritic cells52. It is particularly effective in poorly immunogenic tumors which are unable to directly activate T cells30,52. The presence of specific B cells antigens matching B cell receptors + T cells help is crucial for B cells to be activated in order to differentiate into antibody secreting cells30,55,56,57,58. Activating T and B cells can also cause the development of tertiary lymphoid organs, which hold significant therapeutic potential59,60. Antibodies once bound to the pathogens can activate the proteins of the classical pathway of the complement system and trigger CDC mechanism, in addition antibodies drive antibody-dependent NK cell activation (ADNKA), complement deposition (ADCD), monocyte phagocytosis (ADCP) and antibody-dependent neutrophil phagocytosis (ADNP)35.
The complement cascade is activated in tumors61,62. CDC mechanism triggers a cascade of activation, where each enzyme cleaves and activates the next component in the pathway, leading to either phagocytosis by immune cells or lysis by the MAC, which is a complex of C5b, C6, C7, C8, and many C9 molecules (C5b9)39,45,63. MAC can cause an influx of calcium ions into the cell, which can open mitochondrial pores and release pro-apoptotic factors, it can also induce apoptosis in cells by membrane disruption and caspase activation64,65,66,67. When a cell undergoes apoptosis, the flippase activity decreases, allowing phosphatidylserine (PS) to be exposed on the outer membrane, which can then be detected by a protein Annexin V, enabling the appearance of 7-ADD-positive cells and the identification of apoptotic cells64,68,69,70. Various antibodies, such as anti-CD20 antibody (rituximab, ofatumumab), anti-HER2 antibody (trastuzumab), anti-CD52 antibody (alemtuzumab), and anti-CD38 antibody (daratumumab) are being used in combination with anti-CD55 and anti-CD59 antibodies to achieve higher CDC efficiency41.
The presence of the tumor cells within the peritoneal cavity containing a functional cytolytic complement system makes this tumor type an attractive target for local antibody therapy for ovarian cancers50. In previous studies, it was observed that in addition to complement, tumor-reactive antibodies of IgG class are present in the ascitic fluids (AF) of most patients with ovarian cancer50. TIL-Bs produce antibodies and likely contribute to tumor elimination by activating tumor immunity in HGSOC tissues71. Further investigation required on the role of B and T cells collaboration and TIL-Bs as a potential therapeutic target to enable the development of a novel cancer immunotherapeutic strategy using antibodies.
In future we aim to produce monoclonal S2 antibodies which are tumor-specific (e.g. EGFR, human epididymis protein 4 (HE4), or CA125 expressed on ovarian cancer cells) and clone antibodies produced by patients B cells in their own ascites, ovarian tumor tissues and plasma to enhance the immune killing of the ovarian cancer cells. To improve further the S2 antibodies, the heat-killed HGSOC cells could be used as immunogen and the S2 antibodies created must be tumor-specific complement-activating mAbs against these proteins. We are extrapolating suggestions from our obtained in-vitro data that will need further confirmations with future in-vivo studies, and later on with clinical evidence. The long-term goal of this study is to combine active complement with monoclonal antibodies (mAbs) and cytotoxic agents to enhance local therapeutic efficacy. In future we suggest using the BCR sequences from ovarian cancer patients to produce anti-tumor monoclonal antibodies that together with complement activating antibodies S2 could be explored in ex vivo and later in-vivo models for their ability to induce the efficacy and specificity of complement mediated tumor killing.
We hypothesize that carboplatin and PARP inhibitors, BMS-754807 (IGF1R inhibitor), Paclitaxel (taxane microtubule stabilizer), and Gemcitabine could be delivered through MAC to enhance tumor cell killing and overcome drug resistance45,72. We will study and evaluate CDC-inducing antibodies synergistically with chemotherapy or checkpoint blockade immunotherapy, and especially cancer mutations inhibitors such as CDK2/CDK9 inhibiotors, dicaciclib, aurothiomalate and other oncogenic targets that contribute to tumour progression and metastasis together with HER2 and PI3K-AKT pathway73,74,75.
While our study offers means to kill Pt-resistant HGSOC cells, there are limitations. Our study is limited to in-vitro analyses and does not directly address the in-vivo immunologic mechanisms underlying platinum sensitivity or resistance. This work highlights the potential translational relevance of the findings and suggests possible future directions for antibody-based therapeutic strategies. In addition, this study does not sufficiently address potential off-target effects or the specificity of the S2 antibody, which was derived from heat-killed breast cancer cells. Since IgG fraction from rabbit serum the concentration of the specific antibody is not known it is important to test whether this antiserum binds to the cells and if the antigen is present in all cell lines and at the same level. In addition in future it would be important to study in more detail binding vs. resistance mechanisms of S2 and to see if the effect of S2 was due to Fc-binding. Moreover, the broader implications of the tumor microenvironment and the potential for synergistic effects with other immunotherapies remain underexplored in this work. Despite these limitations, this research makes a compelling case for further development, suggesting that CDC-based therapies, when optimized and combined with other modalities, could play a transformative role in overcoming platinum resistance in HGSOC as well as other cancers. In summary, antibody-based therapeutics hold great promise in Pt resistant HGSOC treatment.
Methods
HGSOC cell lines and culturing conditions
The human HGSOC cell line OVCAR-3 [OVCAR-3] (ATCC HTB-161) was obtained from the American Type Culture Collection (ATCC) and OVCAR-8 from NCI Frederick Cancer DCTD Tumor/Cell line repository6. Cell lines were tested for mycoplasma contamination using the MycoAlert kit (Lonza). Cell lines were authenticated using the GenePrint\(\text{\textregistered}\) 24 System (Promega) at the Institute for Molecular Medicine Finland (FIMM) Genotyping service. CRISPR-Cas9 genome editing was performed based on Alt-R CRISPR-Cas9 System from Integrated DNA Technologies for OVCAR-4 cell line. OVCAR-4-262 and OVCAR-4-264 are clones from OVCAR-4 after successful BRCA2 mutation by CRISPR-Cas9. OVCAR4 parental cell line was commercially available cell line from Sigma-Aldrich. The target sequence used for BRCA2 is 5’-GGCCTCTCTTTGGATCCAAT-3’. ATTO\(^\textrm{TM}\) 550 modified tracrRNA (catalog number: 1075927) was purchased from Integrated DNA Technologies (USA) for enrichment of transfected cells. Single clones were isolated after genome editing and characterized by Sanger sequencing.
Antibodies and other reagents
The antibody S2 was produced at the Department of Bacteriology and Immunology of the University of Helsinki (Prof. Seppo Meri). Antibodies against human breast cancer cells (S2) were produced by immunizing a rabbit three times with \(10^7\) heat-killed MCF7 (breast adenocarcinoma) cells intramuscularly as described previously34,47,76. Antibody against CD59 Yht53.1 was obtained from Biorad Antibodies, UK.
Ethical declaration
Normal Human Serum (NHS) samples as a complement source were obtained from 7 healthy research lab personnel and the informed consent was obtained from donors before taking normal human serum. The blood was then allowed to clot, and the serum was subsequently harvested, pooled, and stored at − 80 \(^{\circ }\)C degrees until it was further used. The required volume of serum was thawed on ice before each experiment and was used only once. Heat-inactivated serum (HIS) was made by incubating NHS at 56 \(^{\circ }\)C for 1 h. Blood was collected and handled by the licensed nurse of the University of Helsinki Faculty of Medicine according to the protocol which was approved by the University of Helsinki Immunology and Bacteriology Department and all research was performed in accordance with the Declaration of Helsinki in accordance to Helsinki University Hospital (HUS) Regional Committee on Medical Research Ethics.
Generation carboplatin-persistent sub-line
OVCAR-3 and OVCAR-8 cell lines were treated with a pre-determined LC50 concentration of carboplatin (Selleckchem, USA) for 5 days. Thereafter the cultures were allowed to recover in the drug-free medium for 2 weeks. The surviving fraction was expanded and treated with a 2xLC50 concentration of carboplatin. The procedure was repeated 2 more times, the resultant cultures were expanded and cryopreserved. The parental and the persisted-enriched cell lines were tested for carboplatin sensitivity in a viability assay and a clonogenic survival assay to verify the shift in the drug response as described below. The highest concentrations of carboplatin used for the enrichment of the persisted population in the cell lines were treated with 8 \({\upmu }\)M and 5 \({\upmu }\)M of carboplatin, respectively. The persisted-enriched cell lines used in the further experiments are designated OVCAR 8R and OVCAR-3 5R (Pt resistant cell lines).
Viability assay
Viability assay 384-well plates (Corning) were pre-drugged with carboplatin using an acoustic dispenser Echo 550 (Labcyte, USA). 1000 cells were seeded per well in triplicates in 25 \({\upmu }\)L of medium. The plates were incubated for 1 week at 37 5% carbon dioxide. After that, 5 \({\upmu }\)L of staining solution (PBS with 1 mg/mL Hoechst 33342 Invitrogen, USA) and 1/1000 Cello Green (Promega, USA) dead cell-labelling dye was added per well and incubated with cells for 30 min. The wells were imaged at Cytation5 cell imaging multi-mode reader (Biotech, USA), and the number of live cells (visualized by Hoechst-positive, Cello Green-negative nuclei) vs. dead cells (Hoechst-positive, Cello Green-positive) were quantified automatically using Gen5 software (Biotech, USA). The number of live cells in the control was taken for 100
Clonogenic survival assay
For this, 3000 cells were plated per well (6-well plate) in 3 mL of medium with increasing concentrations of carboplatin, in duplicate. Cells were incubated with the carboplatin drug for 5 days before the medium was exchanged with drug-free fresh RPMI (Gibko, UK). The colonies were allowed to grow for 2 weeks before they were fixed with 7:1 MeOH: Acetic acid mix and stained with 0.1% Crystal violet for 1 h. The washed plates were scanned at Cytation5 cell imaging multi-mode reader (Biotech, USA), and the number of colonies was automatically counted from the images using Gen5 software (Biotech, USA) ( Supplementary Materials A12). The number of colonies in the untreated control was used as a reference for 100% clonogenic survival.
Complement-mediated killing of HGSOC cells
Annexin V-FITC Apoptosis detection Kit BMS500FI/300 (eBioscience/Thermo Fisher, USA) was used for apoptosis detection after complement killing assays using Flow Cytometry. Briefly, 500,000 cells per tube were incubated for 20 min. at 22 \(^\circ\)C with appropriate dilutions of antibodies S2, Yth53.1 (20 \({\upmu }\)g/ml) and further with Normal Human Serum (NHS) for 1 hour at 37 \(^\circ\)C in a total volume of 200 \({\upmu }\)L ( Supplementary Table A1). For collecting NHS informed consent was obtained from all subjects and/or their legal guardian(s). In complement killing assays the cytotoxic potential upon incubation with antibodies and normal human serum were studied. The cytotoxicity was measured after 1 hour incubation of HGSOC cell lines with antibodies S2 and Yth53.1 together with NHS. To assess the cytotoxic potential, we tracked the apoptosis from PE Annexin V and 7-AAD negative HGSOC cells (viable, or no measurable apoptosis), PE Annexin V positive and 7-AAD negative (early apoptosis, membrane integrity is present) and finally to PE Annexin V and 7-AAD positive (end stage apoptosis and death). Cells were stained following the manufacturer’s protocol with PE conjugated recombinant Annexin V and 7-AAD69. Cells were washed twice with PBS prior to using the kit. In addition, to confim the killing effect seen in assays, the remaining live cells in assays were quantified using Cell Titer Glo and the dying cells using Cell Tox Green (See Supplementary figures A13, A14). In order to confirm complement activation, the human serum that was heated to 56 \(^\circ\)C to deactivate the complement in the assays (Supplementary Fig. A14).
Complement assays, analysis of C5b-9 deposition on ovarian tumour cells in vitro
The monoclonal mouse anti-human C5b-9 (Abcam, UK) was used to detect the MAC complex (clone aE11), at 5 \({\upmu }\)g/ml in PBS. First cells were incubated with antibodies S2 and Yth53.1 and NHS, then cells were washed twice with PBS. Then the HGSOC cells were incubated in 100 \({\upmu }\)l of the antibody for 45 minutes at RT. Secondary antibody goat anti-mouse IgG Alexa Fluor 488 (Abcam) diluted 1:1000 in PBS was used for detection using the BD Accouri C6 Flow Cytometer.
Statistical methods
Statistical analysis was used to evaluate the difference in killing efficiency between Pt resistant and sensitive HGSOC cells. All replicates were pooled for each Pt resistant and sensitive cell lines and quantified killing efficiency as the percentage change from that replicate’s mean at specific concentrations for OVCAR-3 and OVCAR-8 Pt resistant and sensitive cell lines. The difference between resistant and sensitive cells in this pooled data was tested using a Mann-Whitney U test.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information.
Change history
18 February 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41598-026-40076-7
References
Reid, B. M., Permuth, J. B. & Sellers, T. A. Epidemiology of ovarian cancer: a review. Cancer Biol. Med. 14, 9–32 (2017).
Bogani, G. et al. Incorporating immune checkpoint inhibitors in epithelial ovarian cancer. Gynecol. Oncol. 193, 30–40 (2025).
Luyckx, M., Squifflet, J.-L., Bruger, A. M. & Baurain, J.-F. Recurrent high grade serous ovarian cancer management. Exon Publications 87–103 (2022).
van Zyl, B., Tang, D. & Bowden, N. A. Biomarkers of platinum resistance in ovarian cancer: what can we use to improve treatment. Endocr. Relat. Cancer 25, R303–R318 (2018).
Basourakos, P., S. et al. Combination platinum-based and dna damage response-targeting cancer therapy: evolution and future directions. Curr. Med. Chem. 24, 1586–1606 (2017).
Tumiati, M. et al. A functional homologous recombination assay predicts primary chemotherapy response and long-term survival in ovarian cancer patients. Clin. Cancer Res. 24, 4482–4493 (2018).
Pokhriyal, R., Hariprasad, R., Kumar, L. & Hariprasad, G. Chemotherapy resistance in advanced ovarian cancer patients. Biomark. Cancer 11, 1179299X19860815 (2019).
Konstantinopoulos, P. A., Ceccaldi, R., Shapiro, G. I. & D’Andrea, A. D. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 5, 1137–1154 (2015).
Cleary, J. M. et al. Opportunities for utilization of DNA repair inhibitors in homologous recombination repair-deficient and proficient pancreatic adenocarcinoma. Clin. Cancer Res. 27, 6622–6637 (2021).
Ray-Coquard, I. et al. Olaparib plus bevacizumab first-line maintenance in ovarian cancer: final overall survival results from the PAOLA-1/ENGOT-ov25 trial. Ann. Oncol. 34, 681–692 (2023).
O’Donnell, R. L. et al. Advanced ovarian cancer displays functional intratumor heterogeneity that correlates to ex vivo drug sensitivity. Int. J. Gynecol. Cancer 26, 1004–1011 (2016).
Monk, B. J. et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J. Clin. Oncol. 40, 3952–3964 (2022).
Chelariu-Raicu, A. et al. Parp inhibitors: risk factors for toxicity and matching patients to the proper poly (adp-ribose) polymerase inhibitor (parpi) therapy. Int. J. Gynecol. Cancer 33, 812–822 (2023).
Shah, M. et al. Overall survival and the evolving benefit-risk assessment for poly (adp-ribose) polymerase inhibitors in advanced ovarian cancer. J. Clin. Oncol. 43, 2218–2227 (2025).
Tew, W. P., Lacchetti, C., Kohn, E. C. & in the Management of Ovarian Cancer Guideline Expert Panel, P. I. Poly (adp-ribose) polymerase inhibitors in the management of ovarian cancer: Asco guideline rapid recommendation update. J. Clin. Oncol. 40, 3878–3881 (2022).
Shahzad, M., Naci, H., Esselen, K. M., Dottino, J. A. & Wagner, A. K. Regulatory histories of recently withdrawn ovarian cancer treatment indications of 3 parp inhibitors in the us and europe: lessons for the accelerated approval pathway. J. Pharm. Policy Pract. 17, 2351003 (2024).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
You, B. et al. Impact of adding the immune checkpoint inhibitor atezolizumab to first-line chemotherapy on progression-free survival in poor-prognosis ovarian cancer: A retrospective analysis from the imagyn050 trial. Gynecol. Oncol. 197, 66–73 (2025).
Freyer, G. et al. Bevacizumab, olaparib, and durvalumab in patients with relapsed ovarian cancer: a phase II clinical trial from the gineco group. Nat. Commun. 15, 1985 (2024).
Monk, B. J. et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (javelin ovarian 100): an open-label, randomised, phase 3 trial. Lancet Oncol. 22, 1275–1289. https://doi.org/10.1016/S1470-2045(21)00342-9 (2021).
Zhao, L., Zhai, Y. & Niu, G. Research progress of immune checkpoint inhibitors in ovarian cancer. Explor. Immunol. 4, 853–870 (2024).
Matulonis, U. et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II keynote-100 study. Ann. Oncol. 30, 1080–1087 (2019).
Pujade-Lauraine, E. et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (javelin ovarian 200): an open-label, three-arm, randomised, phase 3 study. Lancet Oncol. 22, 1034–1046 (2021).
Jung, I.-Y. et al. Tissue-resident memory car T cells with stem-like characteristics display enhanced efficacy against solid and liquid tumors. Cell Rep. Med. 4, 101053. https://doi.org/10.1016/j.xcrm.2023.101053 (2023).
Li, Y.-R. et al. Overcoming ovarian cancer resistance and evasion to CAR-T cell therapy by harnessing allogeneic CAR-NKT cells. Med (2025).
Knoche, S. M., Larson, A. C., Sliker, B. H., Poelaert, B. J. & Solheim, J. C. The role of tumor heterogeneity in immune-tumor interactions. Cancer Metastasis Rev. 40, 377–389 (2021).
Conejo-Garcia, J. R., Biswas, S. & Chaurio, R. Humoral immune responses: Unsung heroes of the war on cancer. In Seminars in immunology, vol. 49, 101419 (Elsevier, 2020).
Conejo-Garcia, J. R., Biswas, S., Chaurio, R. & Rodriguez, P. C. Neglected no more: B cell-mediated anti-tumor immunity. In Seminars in immunology, vol. 65, 101707 (Elsevier, 2023).
Mann, D. L. et al. Investigation of cellular and humoral immune responses to tumor-associated antigens and survival of patients with advanced cancers treated with combined radiation and immunotherapy. Med. Res. Arch. 11 (2023).
Sameshima, J. et al. Tumor-infiltrating b cells produce tumor-specific antibodies and may contribute to suppressing tumor in head and neck squamous cell carcinoma. OncoImmunology 14, 2543019 (2025).
Golay, J. & Taylor, R. P. The role of complement in the mechanism of action of therapeutic anti-cancer mAbs. Antibodies 9, 58 (2020).
Macagno, M. et al. Role of adcc, cdc, and cdcc in vaccine-mediated protection against HER2 mammary carcinogenesis. Biomedicines 10, 230 (2022).
Seledtsov, V. I. & Seledtsova, G. V. Attaining threshold antibody cytotoxicity for selective tumor cell destruction: an opinion article. Oncotarget 9, 35790 (2018).
Hakulinen, J. Complement-mediated killing of cancer cells. Phd thesis, University of Helsinki, Helsinki (2003). Available at https://helda.helsinki.fi/bitstream/handle/10138/20483/compleme.pdf?sequence=1.
Irvine, E. B. et al. Fc-engineered antibodies promote neutrophil-dependent control of mycobacterium tuberculosis. Nat. Microbiol. 9, 2369–2382 (2024).
Sonderegger, S. E. et al. The role of the complement system in cancer etiology and management. Clinical Immuno-Oncology-E-Book 41 (2023).
Lee, C.-H. & Delidakis, G. Engineering IgG1 Fc domains that activate the complement system. In Immune Receptors: Methods and Protocols, 187–200 (Springer, 2021).
Bournazos, S., Gupta, A. & Ravetch, J. V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 20, 633–643 (2020).
Goldberg, B. S. & Ackerman, M. E. Antibody-mediated complement activation in pathology and protection. Immunol. Cell Biol. 98, 305–317 (2020).
West, E. E. & Kemper, C. Complosome-the intracellular complement system. Nat. Rev. Nephrol. 19, 426–439 (2023).
Lee, W., Lee, S. M. & Jung, S. T. Unlocking the power of complement-dependent cytotoxicity: engineering strategies for the development of potent therapeutic antibodies for cancer treatments. BioDrugs 37, 637–648 (2023).
Dunkelberger, J. R. & Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 20, 34–50 (2010).
Meri, S. Self-nonself discrimination by the complement system. FEBS Lett. 590, 2418–2434 (2016).
Lkhagvasuren, E. Janeway’s immunobiology. Central Asian J. Med. Sci. 3, 100–101 (2017).
Parsons, E. S. et al. Single-molecule kinetics of pore assembly by the membrane attack complex. Nat. Commun. 10, 2066 (2019).
Gorter, A. & Meri, S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol. Today 20, 576–582 (1999).
Bjørge, L., Hakulinen, J., Wahlström, T., Matre, R. & Meri, S. Complement-regulatory proteins in ovarian malignancies. Int. J. Cancer 70, 14–25 (1997).
Hakulinen, J., Junnikkala, S., Sorsa, T. & Meri, S. Complement inhibitor membrane cofactor protein (MCP; CD46) is constitutively shed from cancer cell membranes in vesicles and converted by a metalloproteinase to a functionally active soluble form. Eur. J. Immunol. 34, 2620–2629 (2004).
Jarvis, G. A. et al. Expression and function of the complement membrane attack complex inhibitor protectin (CD59) in human prostate cancer. Int. J. Cancer 71, 1049–1055 (1997).
Bjørge, L. et al. Ascitic complement system in ovarian cancer. Br. J. Cancer 92, 895–905 (2005).
Junnikkala, S. et al. Exceptional resistance of human H2 glioblastoma cells to complement-mediated killing by expression and utilization of factor H and factor H-like protein 1. J. Immunol. 164, 6075–6081 (2000).
Victor Engelhard, J. R. et al. B cells and cancer. Cancer Cell 39, 1293–1296 (2021).
Laumont, C. M., Banville, A. C., Gilardi, M., Hollern, D. P. & Nelson, B. H. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat. Rev. Cancer 22, 414–430 (2022).
Yang, Y. et al. Pan-cancer single-cell dissection reveals phenotypically distinct B cell subtypes. Cell 187, 4790–4811 (2024).
Janeway Jr, C. A., Travers, P., Walport, M. & Shlomchik, M. J. B-cell activation by armed helper T cells. In Immunobiology: The Immune System in Health and Disease. 5th edition (Garland Science, 2001).
Perugino, C. A. et al. Two distinct durable human class-switched memory B cell populations are induced by vaccination and infection. Cell Rep. 44 (2025).
Rival, C. et al. B cells secrete functional antigen-specific IgG antibodies on extracellular vesicles. Sci. Rep. 14, 16970 (2024).
Barral, P. et al. B cell receptor-mediated uptake of CD1D-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc. Natl. Acad. Sci. 105, 8345–8350 (2008).
Fridman, W. H. et al. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 19, 441–457 (2022).
Langouo Fontsa, M., Padonou, F. & Willard-Gallo, K. Tumor-associated tertiary lymphoid structures in cancer: implications for immunotherapy. Expert Rev. Clin. Immunol. 20, 839–847 (2024).
Markiewski, M. M. et al. Modulation of the antitumor immune response by complement. Nat. Immunol. 9, 1225–1235 (2008).
Kwak, J. W. et al. Complement activation via a C3a receptor pathway alters CD4+ T lymphocytes and mediates lung cancer progression. Can. Res. 78, 143–156 (2018).
Weisser, N. E. et al. An anti-HER2 biparatopic antibody that induces unique HER2 clustering and complement-dependent cytotoxicity. Nat. Commun. 14, 1394 (2023).
Nauta, A. J. et al. The membrane attack complex of complement induces caspase activation and apoptosis. Eur. J. Immunol. 32, 783–792 (2002).
Xie, C. B., Jane-Wit, D. & Pober, J. S. Complement membrane attack complex: new roles, mechanisms of action, and therapeutic targets. Am. J. Pathol. 190, 1138–1150 (2020).
Cragg, M. et al. Complement mediated cell death is associated with DNA fragmentation. Cell Death Differ. 7, 48–58 (2000).
Triantafilou, K., Hughes, T. R., Triantafilou, M. & Morgan, B. P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 126, 2903–2913 (2013).
Wang, J. et al. The role of phosphatidylserine on the membrane in immunity and blood coagulation. Biomarker Res. 10, 4 (2022).
Vermes, I., Haanen, C., Steffens-Nakken, H. & Reutellingsperger, C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184, 39–51 (1995).
Anthony, R. et al. Flow cytometry using annexin V can detect early apoptosis in peripheral blood stem cell harvests from patients with leukaemia and lymphoma. Bone Marrow Transplant. 21, 441–446 (1998).
Banville, A. C. & Nelson, B. H. Breaching B cell tolerance in the tumor microenvironment. Cancer Cell 40, 356–358 (2022).
Garcia, A. A. et al. Phase II study of gemcitabine and weekly paclitaxel in recurrent platinum-resistant ovarian cancer. Gynecol. Oncol. 93, 493–498 (2004).
Talbot, T., Lu, H. & Aboagye, E. O. Amplified therapeutic targets in high-grade serous ovarian carcinoma-a review of the literature with quantitative appraisal. Cancer Gene Ther. 30, 955–963 (2023).
Au-Yeung, G. et al. Selective targeting of cyclin E1-amplified high-grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT inhibition. Clin. Cancer Res. 23, 1862–1874 (2017).
Oh, D.-Y. & Bang, Y.-J. HER2-targeted therapies-a role beyond breast cancer. Nat. Rev. Clin. Oncol. 17, 33–48 (2020).
Hakulinen, J. & Meri, S. Complement-mediated killing of microtumors in vitro. Am. J. Pathol. 153, 845–855 (1998).
Acknowledgements
This research was funded by Finnish Cultural Foundation.

Dr. Daria Bulanova is thanked for providing and generating the Pt resistant and sensitive cell lines OVCAR-3 and OVCAR-8. Dr. Jun Dai is thanked for incorporating a diverse set of HGSOC cell lines of OVCAR-4 with varying homologous recombination statuses and employing CRISPR-Cas9. The flow cytometry analysis was performed at the HiLife Flow Cytometry Unit, University of Helsinki. Biostatistics Unit of the University of Helsinki is thanked for helping to choose the statistical analysis model. Noora Leppänen is thanked for helping to calculate the IC50 values. Open access funded by Helsinki University Library.
Author information
Authors and Affiliations
Contributions
Z.G. and S.M. conceived the experiments; Z.G. and N.A. conducted the experiments, Z.G., N.A., F.A.N.M., S.M. analyzed the results. Conceptualization Z.G, S.M.; methodology Z.G., S.M.; software N.A.; validation Z.G. and N.A.; formal analysis Z.G.; investigation Z.G and F.A.N.M.; resources Z.G, S.M.; data curation Z.G.; writing—original draft preparation Z.G., L.B., F.A.N.M.; writing—review and editing F.A.N.M., L.B., Z.G, S.M.; visualization, Z.G.; supervision S.M., Z.G.; project administration S.M. and Z.G.; funding acquisition Z.G., S.M. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: Line Bjørge and Seppo Meri were omitted from the author list in the original version of this Article. In addition, Seppo Meri was omitted as a corresponding author. Correspondence and requests for materials should also be addressed to seppo.meri@helsinki.fi. As a result, the Author contributions and Acknowledgements have been corrected. Furthermore, Affiliation 1 was incomplete. Full information regarding the corrections made can be found in the correction for this Article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Giedraityte, Z., Aarnio, N., Nait Mohamed, F. et al. Towards antibody-based immunotherapy development for platinum-resistant high-grade serous ovarian cancer. Sci Rep 15, 43733 (2025). https://doi.org/10.1038/s41598-025-30099-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-30099-x
