
Abstract
JMJD6, a Jumonji C domain-containing protein located in the cancer-amplified region 17q25, has emerged as a putative oncogene in several malignancies. Only a few reports identified its function in GC (gastric cancer). In this study, we analyzed the detail oncogenic and immunosuppressive roles of JMJD6 and clinical relevance in GC. We analyzed six GC cell lines and 174 primary tumors. Overexpression of JMJD6 was frequently detected in GC cell lines (4/6 cell lines, 66.7%) and 69 primary GC specimens. Overexpression of JMJD6 was significantly associated with advanced tumor stage and was an independent poor prognostic factor (P < 0.001, Hazard ratio 4.15). Knockdown of JMJD6 suppressed GC cell proliferation, migration and invasion in a TP53 mutation-independent manner, and reduced PD-L1 (Programmed cell death ligand 1) expression via BRD4 (Bromodomain-containing protein 4) and IRF1 (Interferon regulatory factor 1) down-regulation. Ectopic overexpression of JMJD6 induced PD-L1 expression via BRD4 and IRF1 upregulation in low-PD-L1-expressing cells. In co-culture assays with activated T cells, JMJD6 knockdown enhanced tumor immunogenicity and T cell-mediated cytotoxicity. In primary GC samples, JMJD6 expression correlated positively with PD-L1 expression. These findings suggest that JMJD6 functions as a dual regulator of tumor aggressiveness and immune escape, highlighting its utility as a prognostic factor and a promising therapeutic target in GC.
Similar content being viewed by others

Introduction
Gastric cancer (GC) remains a major contributor to cancer mortality worldwide, despite continual improvements in diagnostic methods, surgical interventions, perioperative chemotherapy, and patient management strategies1,2,3. Numerous studies have elucidated the molecular mechanisms underlying gastric tumorigenesis4,5, highlighting genetic alterations such as amplifications of MET and ERBB2, mutations in E-cadherin, Allophycocyanin (APC), and TP536,7,8, hypermethylation of p169,10, and oncogenic activation of K-ras and β-catenin11. Additionally, microsatellite instability resulting from inactivation of mismatch repair genes like hMLH1 has been implicated12. Moreover, considerable attention has been directed toward cancer immunotherapy in recent years. Immune checkpoint blockade (ICB), especially PD-1/PD-L1 targeting, has become a promising therapy for GC13,14,15,16,17,18,19,20. However, ICB efficacy remains limited because of low response rates and adverse events in over 50% of patients21. Therefore, identifying novel molecular drivers contributing to tumor progression and immune evasion is crucial for improving therapeutic strategies in GC.
Jumonji domain-containing protein 6 (JMJD6), a member of the Jumonji C (JmjC) domain-containing family, is an epigenetic regulator involved in histone demethylation and protein hydroxylation. Located in the cancer-amplified region 17q25, JMJD6 has crucial roles in diverse biological processes, including embryonic development, cell differentiation, and transcriptional regulation22,23,24, and recent evidence highlights its significant involvement in cancer progression as an oncogene. Overexpression of JMJD6 occurs in various malignancies, such as breast cancer25, colorectal cancer26, lung cancer27, and hepatocellular carcinoma28, and correlates with advanced tumor stage and poor prognosis. Additionally, JMJD6 contributes to tumor progression by promoting epithelial-mesenchymal transition (EMT)29, enhancing cell proliferation, invasion, and metastasis26,28,30. Notably, in vitro analyses found that JMJD6 also regulates PD-L1 expression31, thus facilitating tumor immune evasion mechanisms and implicating it as a novel potential therapeutic target for cancer immunotherapy. However, the detailed molecular mechanisms of JMJD6 in gastric carcinogenesis, cancer immunosuppression, and its clinical and prognostic significance in patients with GC remain to be fully elucidated.
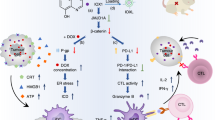
In this study, we investigated the clinical relevance and molecular function of JMJD6 in GC. We evaluated JMJD6 expression in GC cell lines and primary tumor samples, assessed its impact on malignant phenotypes and PD-L1 regulation via BRD4 and IRF1, and explored its effects on tumor immune evasion using co-culture systems with activated T cells (Fig. 1). These findings suggest a hypothetical model of the JMJD6–BRD4–IRF1–PD-L1 regulatory mechanism. Our findings not only reveal JMJD6 as a key regulator of GC malignancy but also suggest its potential as a dual biomarker for prognosis and immunotherapeutic targeting.

Graphical abstract. Graphical abstract illustrating the hypothetical mechanism by which JMJD6 promotes tumor progression and immune evasion in GC. JMJD6 is overexpressed in gastric cancer cells and promotes BRD4 expression, which upregulates IRF1 and consequently increases PD-L1 expression. Elevated PD-L1 expression on tumor cells inhibits T cell–mediated antitumor immunity, thereby facilitating immune escape.
Results
Therapeutic target gene selection
Among the group of 245 genes, possessing oncogenic functions and located in gene-amplified regions of various human cancers (listed in the NCBI database), we identified 33 genes not previously reported in GC. Following the application of exclusion criteria, we selected 11 genes associated with poor prognosis that have high mRNA expression; these were identified in a publicly available database (KM-plotter)28. Finally, we focused on JMJD6, selected from the pool of 11 candidate genes. JMJD6 has been extensively investigated in various cancers and is gaining attention in non-cancer contexts. We plan to extend our research to other related genes in future studies (Supplementary Figure S1).
Overexpression of JMJD6 in GC cell lines
We assessed JMJD6 mRNA and protein abundance in GC cell lines and in the fibroblast strain WI-38 by conducting quantitative RT–PCR and western blotting with JMJD6-specific antibodies. JMJD6 protein expression was shown with mRNA expression in GC cell lines (Fig. 2a). Four of the six GC cell lines (66.7%) —KATOIII, NUGC4, MKN45, and MKN74—displayed elevated JMJD6 expression, suggesting that this molecule may act as an activation target in these cells. In addition, quantitative RT–PCR analysis comparing GC cell lines with normal gastric mucosal tissues obtained from non-cancer patients (Normal-1–3) and a commercial normal gastric sample (Normal-C) demonstrated markedly higher JMJD6 mRNA expression in the cancer cell lines (Supplementary Figure S2), further supporting the overexpression of JMJD6 in GC.

Overexpression of JMJD6 in gastric cancer. (a) The expression of JMJD6 in six GC cell lines compared with the fibroblast cell strain WI-38. The level of JMJD6 mRNA was determined by quantitative RT-PCR in a panel of GC cell lines. The results shown are means ± SD (bars). Black bars represent cell lines where up-regulation of JMJD6 mRNA expression was observed, compared with that in WI-38. The status of a TP53 mutation was the reported status of a TP53 mutation in the database (http://p53.free.fr/index.html, W: wild-type TP53, M: mutant TP53). Note that among TP53-mutated GC cell lines, KATO-III had a p53 gene deletion and a frameshift mutation. (b) Specific JMJD6 immunostaining in a representative primary tumor sample. Based on this result, the intensity scores for JMJD6 staining were determined as follows: 0 = negative, 1 = weak, 2 = moderate, 3 = strong. Kaplan–Meier plots depending on the intensity (c and proportion (d) scores of specific immunostainings of JMJD6. The log-rank test was used for statistical analysis; P < 0.05 was considered to be statistically significant. e. For scoring JMJD6 expression by immunohistochemistry (IHC), the intensity and the percentage of the total cell population (0% ≤ 0 ≤ 20%, 20% < 1 ≤ 40%, 40% < 2 ≤ 60%, 60% < 3 ≤ 100%) that expressed JMJD6 were evaluated for each case. Expression of JMJD6 was graded as high expression (intensity plus proportion scores ≥ 4 for tumor cells showing immunopositivity) or low expression (intensity plus proportion scores ≤ 3 for tumor cells showing immunopositivity). f. Cancer-specific survival rates of patients with GC (as determined by Kaplan–Meier plots), depending on JMJD6 expression.
Immunohistochemical analysis of JMJD6 expression in primary tumors of GC
We investigated the clinicopathological significance of JMJD6 expression in primary tumor samples of GC based on its immunohistochemical staining pattern. A total of 174 GC cases were categorized into JMJD6-positive (n = 69, 40%) and JMJD6-negative (n = 105, 60%) groups based on staining intensity and the proportion of tumor cells exhibiting JMJD6 positivity. In primary cases, JMJD6 protein expression was negative in most of the non-tumorous gastric mucosal cell population (Fig. 2b). Kaplan–Meier analysis revealed that increased JMJD6 staining intensity (Fig. 2c) and proportion (Fig. 2d) were both linked to poorer cancer-specific survival outcomes. JMJD6 expression was scored and categorized as described in Methods (Fig. 2e). Patients in the high JMJD6 expression group exhibited a significantly worse prognosis for cancer-specific survival than those in the low-expression group (P = 0.0027, log-rank test; Fig. 2f). These data suggest that high JMJD6 expression contributes to aggressive tumor behavior and poor prognosis in GC.
Clinicopathological significance of JMJD6 expression in GC
To further evaluate its clinicopathological relevance, we next examined whether JMJD6 protein expression was linked to malignant clinicopathological features in GC by evaluating primary tumor samples via immunohistochemistry. The relationship between JMJD6 status and clinicopathological variables is shown in Table 1. Elevated JMJD6 expression was correlated significantly with higher pT classification. Furthermore, multivariate Cox proportional hazards analysis (Table 2) demonstrated that JMJD6 positivity in tumor cells was an independent factor predicting worse cancer-specific survival rates (P < 0.001, Hazard ratio 4.15; 95% confidence interval: 2.14–8.07), alongside advanced pT and pN stages.
Suppression of cell proliferation by JMJD6 knockdown and its effect according to TP53 mutation status in GC cell lines
Cell proliferation assays were conducted to determine whether elevated JMJD6 expression plays a role in promoting GC development. Silencing JMJD6 with specific siRNAs, we examined whether JMJD6 knockdown suppresses the proliferation of GC cells that overexpress JMJD6. In both the TP53 wild-type MKN45 and TP53 mutant MKN74 cell lines, JMJD6 protein levels were efficiently reduced by transfection with a JMJD6-targeting siRNA (siRNA-JMJD6), compared with a luciferase-specific siRNA (siRNA-Luc) as a negative control. The proliferation of these cell lines was notably suppressed in these cell lines after silencing endogenous JMJD6 (Fig. 3a).

Suppression of malignant activities in GC cells by JMJD6 knockdown. (a) The effects of JMJD6 knockdown on cell proliferation at the indicated times by siRNA (siRNA-JMJD6) compared with control siRNA in MKN45 (wild-type TP53) and MKN74 (mutant TP53) cell lines. (b) Representative population results in each phase of the cell cycle in each cell, as assessed by FACS at 72 h post-treatment with siRNA. (c) Suppression of migration, invasion, and epithelial-mesenchymal transition in GC cells by JMJD6 knockdown. Knocking down JMJD6 by siRNA-JMJD6 suppressed migration and invasion in MKN45 (wild-type TP53) (A) and MKN74 (mutant TP53) cell lines. The bar graphs show the means ± SD (n = 4). The Mann–Whitney U-test was used for statistical analysis. P < 0.05 was considered statistically significant.
Cell cycle analyses by JMJD6 knockdown using fluorescence-activated cell sorting
To explore the molecular basis underlying the suppression of cell proliferation by JMJD6 knockdown, we conducted a cell cycle analysis. FACS analysis revealed that introducing siRNA-JMJD6 into TP53 wild-type MKN45 and TP53 mutant MKN74 cells led to cell cycle arrest at G1 when compared with control siRNA-treated cells (Fig. 3b).
JMJD6 knockdown suppresses proliferation, migration, and invasion of GC cells
To verify the association between JMJD6 and cell migration and invasion in vitro, we performed trans-well migration and invasion assays. We assessed the motility of MKN45 and MKN74 cells transfected with siRNA-JMJD6 by testing their ability to traverse membrane pores under varying conditions. Migration assays employed uncoated membranes, while invasion assays utilized membranes coated with Matrigel. As shown in Fig. 3c, under both experimental conditions, siRNA-JMJD6–transfected MKN45 and MKN74 cells exhibited a markedly reduced number of migrated cells into the lower chamber compared with those transfected with siRNA-Luc. These findings suggest that JMJD6 overexpression contributes to the enhanced migration and invasion of GC cells, regardless of TP53 mutation status.
JMJD6 regulates PD-L1 expression via BRD4 and IRF1
Based on previous reports32,33,34, we focused on the relationship between JMJD6 and BRD4. In MKN74 cells, which have overexpression of both JMJD6 and BRD4, JMJD6 knockdown downregulated both BRD4 and PD-L1 at the protein level, whereas BRD4 knockdown suppressed PD-L1 without affecting JMJD6 expression (Fig. 4a). Immunofluorescence also confirmed that JMJD6 suppression reduced BRD4 and PD-L1 localization (Fig. 4b). Additionally, knockdown of JMJD6 or BRD4 decreased IRF1 protein levels, suggesting that IRF1 acts downstream of the JMJD6–BRD4 axis (Fig. 4a). These results suggest that JMJD6 functions upstream of BRD4 in this axis.

JMJD6 regulates BRD4, IRF1 and PD-L1 expression. (a) The knockdown of JMJD6 by transfection with siRNA-JMJD6 suppressed BRD4, IRF1 and PD-L1 in MKN74. In addition, the knockdown of BRD4 by transfection with siRNA-BRD4 suppressed IRF1 and PD-L1 in MKN74. In contrast, the knockdown of BRD4 did not suppress JMJD6 in MKN74. (b) Knockdown of JMJD6 suppressed PD-L1 and BRD4 expression in MKN74 gastric cancer (GC) cells. White dotted lines indicate nuclear boundaries. (c) Co-culture assay of GC cells and T cells. Under JMJD6 knockdown, T cells had more potent anti-tumor activity against GC cells compared with NC, and the proliferation ratio of GC cells was significantly decreased (mean ± SD, n = 3; error bars indicate SD, n = 3). (d) An impedance-based tumor-cell killing assay. The knockdown of JMJD6 increased the anti-tumor activity of T cells and inhibited the proliferation of GC cells. (e) JMJD6 overexpression using plasmid transfection promotes BRD4, IRF1 and PD-L1 expression. (f) A hypothetical model of the overexpression or activation of JMJD6 in GC cells.
JMJD6 modulates T cell–mediated cytotoxicity by regulating PD-L1
To evaluate the impact of JMJD6 knockdown on T cell–mediated immunity, we co-cultured GC cells with activated T cells isolated from a healthy volunteer and stimulated using anti-CD3/CD28 beads, as previously reported35. An in vitro tumor-cell killing assay using counting beads and flow cytometry revealed that JMJD6 knockdown enhanced T cell cytotoxicity and reduced GC cell proliferation (Fig. 4c). We further assessed the effect of JMJD6 silencing on the real-time cytotoxic activity of T cells using the xCELLigence system, which monitors the impedance generated by viable cells attached to the well surface. To account for potential effects of T cell adhesion to GC cells in the xCELLigence assay, we evaluated tumor-only and co-culture conditions independently. This analysis demonstrated that JMJD6 knockdown enhanced T cell–mediated anti-tumor activity and suppressed the growth of GC tumor cells (Fig. 4d).
Ectopic JMJD6 expression induces PD-L1
To examine whether overexpression of JMJD6 promotes the expression of PD-L1 in GC cells, transient expression assays were performed by transfection of JMJD6 expression constructs into MKN45 cells, a weakly expressing PD-L1 cell line. Expression of HaloTag-JMJD6 was verified by western blotting. Moreover, western blotting showed that overexpression of JMJD6 induced the expression of BRD4 and PD-L1. Consistently, overexpression of JMJD6 also upregulated IRF1 expression (Fig. 4e).
A hypothetical model of the overexpression or activation of JMJD6 in GC cells
Overexpression of JMJD6 upregulates BRD4 expression, which subsequently enhances IRF1 and PD-L1 expression. This signaling cascade is proposed to contribute to the development of adaptive immune resistance in the tumor microenvironment. Additionally, JMJD6 may regulate other unidentified substrates that promote tumor cell proliferation, migration, and invasion.
In this model, solid arrows represent pathways experimentally supported in this study, including the JMJD6–BRD4–IRF1–PD-L1 axis, whereas dashed arrows indicate other hypothetical or unverified mechanisms. However, as this figure represents an overall hypothetical framework, both solid and dashed arrows should be interpreted as part of a hypothetical model rather than definitive evidence (Fig. 4f).
JMJD6 expression positively correlates with PD-L1 in GC tissues
Among 40 selected cases (20 high-JMJD6 and 20 low-JMJD6), immunohistochemistry demonstrated a significant association between JMJD6 and PD-L1 positivity using CPS scoring (P = 0.0281; Fig. 5), reinforcing a potential biomarker role for JMJD6 in predicting immune checkpoint ligand expression in vivo.

JMJD6 expression is positively correlated with PD-L1 in GC tissues. Immunohistochemical staining of JMJD6 and PD-L1 was performed on GC tissues from 174 patients. Twenty high JMJD6-expressing cases and twenty low JMJD6-expressing cases were selected for PD-L1 staining and CPS evaluation. Representative images from four cases are shown. High JMJD6 expression was significantly associated with CPS ≥ 1 (P = 0.0281, Fisher’s exact test).
Discussion
In the present study, we provided novel evidence that JMJD6 is not only a molecular driver of GC progression but also a key modulator of immune evasion through the BRD4-IRF1-PD-L1 signaling axis. JMJD6 knockdown led to marked inhibition of GC cell proliferation, migration, and invasion independent of TP53 status. Additionally, JMJD6 regulated PD-L1 expression through BRD4 and IRF1, and JMJD6 suppression downregulated PD-L1 and significantly enhanced T cell–mediated cytotoxicity in vitro. These dual effects highlight JMJD6 as both a driver of tumor aggressiveness and a suppressor of anti-tumor immunity. While prior studies have implicated JMJD6 in the oncogenesis of various cancers25,27,28,36, our findings uniquely establish JMJD6 as an upstream regulator of PD-L1 via BRD4 and IRF1 in GC. Our hypothesis that IRF1 mediates BRD4-driven PD-L1 expression is supported by several previous reports demonstrating the interplay between BRD4 and IRF1 in the regulation of PD-L1 expression37,38,39,40. Although a recent study showed that JMJD6 enhances PD-L1 expression31, our study is the first report demonstrating that JMJD6 positively regulates BRD4 and IRF1 expression and consequently enhances PD-L1 expression in GC, validating its impact on T cell anti-tumor activity in vitro and clinical specimens.
Regarding the molecular mechanisms by which JMJD6 regulates PD-L1, previous studies have shown that JMJD6 and BRD4 form transcriptional complexes that facilitate pause-release of RNA polymerase II32,34. Our results extend this understanding by establishing a causative relationship whereby JMJD6 drives BRD4 and IRF1 upregulation. BRD4 functions as a co-activator of these transcription factors, contributing to PD-L1 expression41. BRD4 also regulates MYC expression in various cancers42. In contrast, JMJD6 deficiency suppresses M2-like macrophage polarization and enhances the efficacy of ICB therapy, potentially through modulation of the STAT3/IL-10 signaling pathway43. Although MYC, STAT3 and PI3K/Akt have been implicated in PD-L1 regulation44,45,46,47,48, in our study, JMJD6 or BRD4 knockdown did not significantly affect MYC, STAT3 and PI3K/Akt levels in GC cells (Supplementary Figure S3b). This suggests that the JMJD6–BRD4–IRF1–PD-L1 axis may operate independently of canonical oncogenic pathways in GC. Given that IRF1 directly regulates PD-L1 transcription and is modulated by both JMJD6 and BRD4, our findings position IRF1 as a key functional mediator in this axis. This notion is further supported by findings demonstrating that JAK2/STAT1/IRF-1 signaling regulates PD-L1 expression in GC49. Notably, Ebine et al. have also reported that BRD4 enhances PD-L1 expression through IRF1 independently of the MYC pathway40, further supporting the results of the present study.
Collectively, this study highlights JMJD6 as a key regulator of the tumor immune landscape in GC. While ICB using anti-PD-1/PD-L1 antibodies has revolutionized cancer therapy, its clinical efficacy in GC remains limited18,19,50,51,52. Recently, some reports have highlighted the relationship between oncogenes and PD-L1 expression53,54. Our findings also bridge a critical knowledge gap in the immune-oncology field, suggesting that JMJD6 is a key molecular link between oncogenic signaling and immune checkpoint regulation. JMJD6 expression may explain variability in PD-L1 levels and ICB responses among GC patients, offering a new stratification biomarker and combinatorial target.
Our immunohistochemical data demonstrated a significant correlation between JMJD6 and PD-L1 expression in primary GC specimens, validating our in vitro findings and suggesting that JMJD6 may serve as a surrogate biomarker for immune checkpoint molecule expression. Given the growing interest in epigenetic modulation to enhance immunotherapy55, JMJD6 inhibition—alone or combined with ICB—may represent a rational therapeutic approach in GC. Notably, JMJD6 regulation of PD-L1 in GC31 supports JMJD6 or BET inhibition as promising future combinatorial strategies with ICB. While our study includes comprehensive clinicopathological data (Table 1), molecular subtypes such as MSI and EBV status were not assessed, which may influence PD-L1 expression and immune contexture. These subtypes should be considered in future patient stratification.
In conclusion, JMJD6 promotes tumor progression and immune evasion via BRD4-mediated PD-L1 induction. Our findings advocate for further exploration of JMJD6-targeted therapies and suggest that JMJD6 may serve as both a prognostic biomarker and a predictive marker of the response to immunotherapy. In vivo models and clinical cohorts are needed to assess whether pharmacological JMJD6 inhibition can synergize with ICB and improve outcomes in refractory GC.
Methods
Cell lines and primary tissue samples
Six GC cell lines (KATO-III, NUGC4, MKN7, HGC27, MKN45, and MKN74) and the fibroblast strain WI-38 were employed in this study. All cell lines and the cell strain were obtained from RIKEN BioResource Center Cell Bank (Tsukuba, Japan) and were authenticated by short tandem repeat profiling before distribution. The HGC27 cells were cultured in Dulbecco’s Modified Eagle Medium; all other cells were cultured in Roswell Park Memorial Institute 1640 medium. All media were purchased from Nacalai Tesque (Japan) and added to 100 mL/L fetal bovine serum (Corning, USA). Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Formalin-fixed, paraffin-embedded primary GC tissue specimens were obtained from 174 consecutive patients who underwent curative gastrectomy at the Division of Digestive Surgery, Kyoto Prefectural University of Medicine (Kyoto, Japan) between January 2012 and December 2019. Paraffin blocks were stored at room temperature in the dark, and the sliced specimens were stained within two weeks. Relevant clinical and survival data were obtained for all patients. All experiments complied with relevant institutional and national regulations, and written informed consent was obtained from each patient. The study protocol conformed to the Declaration of Helsinki and was approved by the institutional review board of Kyoto Prefectural University of Medicine. All enrolled patients had no history of endoscopic mucosal resection, palliative surgery, neoadjuvant chemotherapy, or radiotherapy. In addition, there were no cases with other organ malignancies occurring either simultaneously or at a different time point. Tumor staging and nodal/metastatic status were assessed based on the TNM classification of the International Union Against Cancer (UICC)56.
Quantitative RT–PCR
Quantitative RT–PCR was performed as previously described57,58,59,60 with minor modifications. Total RNA was isolated from the cell lines using a RNeasy Mini Kit (Qiagen, Valencia, CA, USA). For normal gastric tissues, frozen specimens were homogenized using a Power Masher II homogenizer (Nippi, Tokyo, Japan) before RNA extraction, and commercial total RNA from human stomach tissue (Invitrogen, CA, USA) was also used as a non-malignant gastric control. The reverse transcription reaction was performed using a TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). Quantification of mRNA levels was performed by qPCR on a StepOnePlus PCR System (Applied Biosystems), and Ct (cycle threshold) values were determined via StepOne Software v2.0 (Applied Biosystems) employing TaqMan Gene Expression Assays (Hs00397095_m1 for JMJD6; Hs00204257_m1 for PD-L1; Applied Biosystems) in accordance with the manufacturer’s instructions. The gene expression results were calculated as the ratio between JMJD6 and an internal reference gene (Hs01060665_g1 for β-actin; Applied Biosystems) which served as a normalization factor for RNA isolated from a specimen. All reactions were conducted in triplicate for every sample.
Western blotting
Anti-JMJD6 mouse monoclonal antibody (sc-28348; Santa Cruz Biotechnology, TX, USA), anti-PD-L1 rabbit monoclonal antibody (13684; Cell Signaling Technology, MA, USA), anti-ACTB rabbit monoclonal antibody (3700; Cell Signaling Technology), anti-BRD4 rabbit polyclonal antibody (A301-985A50; Bethyl Laboratories, TX, USA), and anti-IRF1 rabbit monoclonal antibody (8478; Cell Signaling Technology) were used. Cells were lysed, and their proteins were extracted using M-PER® Mammalian Protein Extraction Reagent (Thermo Scientific, USA).
Immunofluorescence staining
Immunofluorescence staining was conducted based on the method reported by Takashima et al.58, with slight modifications. For immunofluorescence staining, cells were treated with 4% paraformaldehyde for fixation at room temperature for 20 min, then permeabilized using 0.25% Triton X-100 in phosphate-buffered saline (PBS), and subsequently incubated in a blocking solution containing 1% bovine serum albumin. The cells were next exposed to anti-JMJD6 and anti-PD-L1 antibodies (ab3373) and kept overnight at 4 °C. Following three washes with PBS, cells were incubated for 1 h at room temperature with Alexa Fluor 488–labeled goat anti-mouse and Alexa Fluor 594–labeled goat anti-rabbit secondary antibodies. After another set of three PBS washes, the samples were treated with rhodamine phalloidin and 4′,6-diamidino-2-phenylindole for 30 min. Subsequently, DAPI staining was completed, and slides were mounted using Vectashield Mounting Medium (Vector Laboratories, Burlingame, CA, USA). The subcellular distribution of JMJD6 and PD-L1 was visualized with a BZ-X700 microscope (Keyence, Tokyo, Japan). The nuclear regions were manually outlined with white dotted lines in the merged images to indicate nuclear boundaries.
Knockdown by SiRNA and cell growth analysis
For analyzing loss-of-function because of the knockdown of endogenous gene expression, the siRNA #1 targeting JMJD6 (siRNA, 5’- CAGGAGGCTGGTGGCATGTTGTCCT − 3’ for siRNA-JMJD6; Invitrogen, CA, USA) or Luciferase (Luc) (5’ -CGUACGCGGAAUACUUCGA- 3’; Sigma) were transfected into cells (10 nmol/L) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Knockdown of the target gene was confirmed by western blot analysis. We also investigated the knockdown effects using the other siRNA #2 targeting JMJD6 (siRNA,5’- CAGCAGCTATGGTGAACACCCTAAA − 3’ for siRNA- JMJD6; Invitrogen) and siRNA #3 targeting JMJD6 (siRNA, 5’-CCAGTGCCTGGAATGCCTTAGTTCA − 3’ for siRNA- JMJD6; Invitrogen) to mitigate the potential for off-target effects. A predesigned siRNA targeting BRD4 (Silencer Select siRNA, s23901; Thermo Fisher Scientific) was also used for mechanistic analysis by western blotting.
Proliferation assays and cell cycle analysis
For cell growth measurements, the number of viable cells at various post-transfection time points was assessed by a colorimetric water-soluble tetrazolium salt assay (Cell Count Reagent SF; Nacalai Tesque). Cell cycle position was evaluated 72 h post-transfection by FACS, following previously reported procedures57,58,59,61.
Transwell migration and invasion assays
Transwell migration and invasion assays were performed following the protocol reported in previous studies62,63,64,65. Assays were set up in a 24-well Transwell chamber containing cell culture inserts with 8-µm pore membranes. The upper surface of the 6.4-mm-diameter membranes was either coated with Matrigel (Corning BioCoat Matrigel Invasion Chamber, CORNING, USA) or left uncoated (Falcon Cell Culture Inserts, CORNING, USA). siRNA-transfected cells (1.0 × 10⁶ per well) were plated in the upper chamber with serum-free medium, while complete medium was placed in the lower compartment. After 22 h incubation, cells that migrated or invaded to the underside of the membranes were fixed and stained using Diff-Quik (Sysmex, Kobe, Japan). Nuclei of stained cells were enumerated in triplicate.
Plasmid construction and overexpression
For transient overexpression experiments, we followed the method described previously with slight modifications58. A Halo-tagged JMJD6 construct (FHC00568 / pFN21ASDA0585) was obtained from PROMEGA KK (Tokyo, Japan). Overexpression assays were performed using MKN45 cells, which were transfected with either an empty vector (pCI-neo Mammalian Expression Vector; E1841; PROMEGA KK) or the HaloTag-JMJD6 plasmid via Lipofectamine 3000 (Invitrogen). JMJD6 protein expression in transfected cells was validated by western blotting.
Immunohistochemistry
A primary anti-JMJD6 mouse monoclonal antibody (sc-28348; Santa Cruz Biotechnology) and an anti-PD-L1 rabbit monoclonal antibody (Cell Signaling Technology) were used. Tumor specimens were fixed in 10% formaldehyde prepared in PBS, paraffin-embedded, and stored at room temperature in the dark. Sections were cut and stained within two weeks using a horseradish peroxidase method. After deparaffinization, antigen retrieval was achieved by heating in 10 mmol/L citrate buffer (pH 9.0) at 95 °C for 60 min. Endogenous peroxidase activity was blocked by incubation in 3% H₂O₂ for 20 min. Sections were then treated with Block Ace (Dainippon Sumitomo Pharmaceutical, Osaka, Japan) for 30 min at room temperature, followed by a 60-min incubation with anti-JMJD6 antibodies (1:2000) at the same temperature. All washing and dilution steps were carried out in PBS. Bound antibodies were visualized with the EnVision™+ HRP System (EnVision + Dual Link System-HRP; Dako North America, Inc., Carpinteria, CA, USA), and color development was performed using diaminobenzidine tetrahydrochloride. Slides were finally counterstained with Mayer’s hematoxylin.
For the evaluation of JMJD6 expression, both the staining intensity (0 = negative, 1 = weak, 2 = moderate, 3 = strong) and the proportion of the total cell population positive for JMJD6 (0% ≤ 0 ≤ 20%, 20% < 1 ≤ 40%, 40% < 2 ≤ 60%, 60% < 3 ≤ 100%) were assessed in each case. JMJD6 expression was categorized as high (sum of intensity score and proportion score ≥ 4 in tumor cells exhibiting immunopositivity) or low (sum ≤ 3 in tumor cells exhibiting immunopositivity) under high-power (×200) microscopy66. In addition, PD-L1 expression was evaluated using a combined positive score (CPS). Cases with CPS < 1 were classified as low expression, and cases with CPS ≥ 1 were classified as high expression.
Statistical analysis
Categorical clinicopathological variables were analyzed between the high and low JMJD6 expression groups using a Chi-squared or Fisher’s exact test. Comparisons of non-categorical variables between subgroups were performed using the non–parametric Mann–Whitney U test. For the survival analysis, Kaplan–Meier curves were generated for each group according to univariate variables, and differences between groups were evaluated using the log-rank method. Both univariate and multivariate survival analyses were carried out via the likelihood ratio approach within a stratified Cox proportional hazards model. All comparisons were conducted using two-tailed statistical tests, and P-values less than 0.05 were regarded as statistically significant.
Flow cytometry
To evaluate PD-L1 expression in GC cells, we used a PE-conjugated anti-human PD-L1 antibody (BioLegend) for staining. EpCAM surface expression on GC cells was determined by PE-conjugated anti-human EpCAM antibody (BioLegend). For detecting CD3 on T cells, an APC-conjugated anti-human CD3 antibody (BioLegend) was employed. Flow cytometric measurements were performed as described previously35, using a BD FACS Accuri C6 Plus system (BD Biosciences), and the acquired data were analyzed with FlowJo software (Tree Star).
In vitro tumor-cell killing assay
The tumor-cell killing assay was conducted following the protocol of Kamiya et al.35. GC cells (6 × 10⁴ cells/well) were plated in six-well dishes and cultured for 24 h, followed by transfection with negative control (Luc) or JMJD6-targeting siRNA. After an additional 24 h, activated T cells—stimulated via anti-CD3/anti-CD28-coated beads—were added to each well at a density of 9 × 10⁴ cells/well. Following 72 h of co-culture, cells were harvested, stained with PE-conjugated anti-EpCAM and APC-conjugated anti-CD3 antibodies to distinguish GC cells from T cells, and mixed with 10,000 Count Bright Absolute Counting Beads (Thermo Fisher Scientific). The population of viable tumor cells (EpCAM⁺/CD3⁻ fraction) was quantified by flow cytometry until 1,000 counting beads were recorded. Relative proliferation was calculated by setting the value to 1.0 for tumor cells cultured alone.
Impedance-based tumor-cell killing assay
The influence of JMJD6 knockdown on tumor immunity was further examined using an impedance-based cytotoxicity assay with xCELLigence Real-Time Cell Analysis (RTCA; ACEA Biosciences). GC cells (7 × 10³ cells/well) were seeded into an E-Plate 16 (ACEA Biosciences) after background impedance measurement. Twenty-four hours later, cells were transfected with either NC (Luc) or siRNA-JMJD6 (final concentration 12 nM). After another 24 h, activated T cells stimulated with anti-CD3/anti-CD28 beads were introduced at 1.4 × 10⁴ cells/well. Once tumor growth reached a plateau, impedance was continuously recorded as a cell index using the xCELLigence RTCA DP system, and data were processed with RTCA Software v2.0 (ACEA Biosciences) according to the previously described protocol35.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available due to the personal information protection law in Japan, but are available after the permission from the institutional review board and the corresponding author on reasonable request.
Abbreviations
- GC:
-
Gastric cancer
- JMJD6:
-
Jumonji domain containing 6
- PD-L1:
-
Programmed cell death ligand 1
- BRD4:
-
Bromodomain-containing protein 4
- IRF1:
-
Interferon regulatory factor 1
- ICB:
-
Immune checkpoint blockade
References
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 63, 11–30 (2013).
Komatsu, S. & Otsuji, E. Essential updates 2017/2018: recent topics in the treatment and research of gastric cancer in Japan. Ann. Gastroenterol. Surg. 3, 581–591 (2019).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Smyth, E. C. et al. Gastric cancer. Lancet 396, 635–648 (2020).
Ushijima, T. & Sasako, M. Focus on gastric cancer. Cancer Cell. 5, 121–125 (2004).
Becker, K. F. et al. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 54, 3845–3852 (1994).
Tahara, E. & Yokozaki, H. The sequential accumulation of genetic alterations characteristic of the colorectal adenoma-carcinoma sequence does not occur between gastric adenoma and adenocarcinoma. J. Pathol. 178, 475–476 (1996).
Lee, J. H. et al. Inverse relationship between APC gene mutation in gastric adenomas and development of adenocarcinoma. Am. J. Pathol. 161, 611–618 (2002).
Oue, N. et al. Distinct promoter hypermethylation of p16INK4a, CDH1, and RAR-beta in intestinal, diffuse-adherent, and diffuse-scattered type gastric carcinomas. J. Pathol. 198, 55–59 (2002).
Ding, Y. et al. Methylation and mutation analysis of p16 gene in gastric cancer. World J. Gastroenterol. 9, 423–426 (2003).
Park, W. S. et al. Frequent somatic mutations of the beta-catenin gene in intestinal-type gastric cancer. Cancer Res. 59, 4257–4260 (1999).
Vissers, J. H. et al. The many faces of ubiquitinated histone H2A: insights from the dubs. Cell. Div. 3, 8 (2008).
Bachy, E. & Coiffier, B. Anti-PD1 antibody: a new approach to treatment of lymphomas. Lancet Oncol. 15, 7–8 (2014).
Lipson, E. J. et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin. Cancer Res. 19, 462–468 (2013).
Hamid, O. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl. J. Med. 369, 134–144 (2013).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1-Positive Non-Small-Cell lung cancer. N Engl. J. Med. 375, 1823–1833 (2016).
Bang, Y. J. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697 (2010).
Kang, Y. K. et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 2461–2471 (2017).
Muro, K. et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 17, 717–726 (2016).
Janjigian, Y. Y. et al. CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J. Clin. Oncol. 36, 2836–2844 (2018).
Chen, C. et al. Efficacy and safety of immune checkpoint inhibitors in advanced gastric or gastroesophageal junction cancer: a systematic review and meta-analysis. Oncoimmunology 8, e1581547 (2019).
Bastian, B. C. et al. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 58, 2170–2175 (1998).
Lin, M. et al. DNA copy number gains in head and neck squamous cell carcinoma. Oncogene 25, 1424–1433 (2006).
Aprelikova, O., Chen, K. & Touny, E. The epigenetic modifier JMJD6 is amplified in mammary tumors and cooperates with c-Myc to enhance cellular transformation, tumor progression, and metastasis. Clin. Epigenetics. 8, 38 (2016).
Biswas, A. et al. Both EZH2 and JMJD6 regulate cell cycle genes in breast cancer. BMC Cancer. 20, 1159 (2020).
Wang, F. et al. JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS Biol. 12, e1001819 (2014).
Zhang, J. et al. High expression of JMJD6 predicts unfavorable survival in lung adenocarcinoma. Tumour Biol. 34, 2397–2401 (2013).
Wan, J. et al. JMJD6 promotes hepatocellular carcinoma carcinogenesis by targeting CDK4. Int. J. Cancer. 144, 2489–2500 (2019).
Liu, X. et al. JMJD6 promotes melanoma carcinogenesis through regulation of the alternative splicing of PAK1, a key MAPK signaling component. Mol. Cancer. 16, 175 (2017).
Liu, H. et al. JMJD6 functions as an oncogene and is associated with poor prognosis in esophageal squamous cell carcinoma. BMC Cancer. 23, 696 (2023).
Zhang, X. & Na, D. Jumonji domain-containing protein 6 promotes gastric cancer progression: modulating immune evasion through autophagy and oxidative stress pathways. Cytojournal 22, 6 (2025).
Liu, W. et al. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155, 1581–1595 (2013).
Wong, M. et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 10, 3319 (2019).
Donati, B., Lorenzini, E. & Ciarrocchi, A. BRD4 and cancer: going beyond transcriptional regulation. Mol. Cancer. 17, 164 (2018).
Kamiya, H. et al. Low blood level of tumour suppressor miR-5193 as a target of immunotherapy to PD-L1 in gastric cancer. Br J Cancer. 202310.1038/s41416-023-02532-3.
Chen, H. et al. JMJD6 K375 acetylation restrains lung cancer progression by enhancing METTL14/m6A/SLC3A2 axis mediated cell ferroptosis. J. Transl Med. 23, 233 (2025).
Zhu, Y. et al. Interferon-α2a induces CD4(+) T cell apoptosis and suppresses Th1/Th17 responses via upregulating IRF1-mediated PDL1 expression in dendritic cells from behcet’s uveitis. Clin. Immunol. 250, 109303 (2023).
Wang, J. et al. BRD4-IRF1 axis regulates chemoradiotherapy-induced PD-L1 expression and immune evasion in non-small cell lung cancer. Clin. Transl Med. 12, e718 (2022).
Tian, B. et al. BRD4 couples NF-κB/RelA with airway inflammation and the IRF-RIG-I amplification loop in respiratory syncytial virus infection. J Virol ; 91. (2017).
Ebine, K. et al. Interplay between interferon regulatory factor 1 and BRD4 in the regulation of PD-L1 in pancreatic stellate cells. Sci. Rep. 8, 13225 (2018).
Kim, S. L., Choi, H. S. & Lee, D. S. BRD4/nuclear PD-L1/RelB circuit is involved in the stemness of breast cancer cells. Cell. Commun. Signal. 21, 315 (2023).
Delmore, J. E. et al. BET bromodomain Inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011).
Chen, S. et al. JMJD6 in tumor-associated macrophage regulates macrophage polarization and cancer progression via STAT3/IL-10 axis. Oncogene 42, 2737–2750 (2023).
Casey, S. C. et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016).
Bu, L. L. et al. STAT3 induces immunosuppression by upregulating PD-1/PD-L1 in HNSCC. J. Dent. Res. 96, 1027–1034 (2017).
Liang, M. Q., Yu, F. Q. & Chen, C. C-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am. J. Transl Res. 12, 379–388 (2020).
Jahangiri, A., Dadmanesh, M. & Ghorban, K. STAT3 Inhibition reduced PD-L1 expression and enhanced antitumor immune responses. J. Cell. Physiol. 235, 9457–9463 (2020).
Lastwika, K. J. et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in Non-Small cell lung cancer. Cancer Res. 76, 227–238 (2016).
Moon, J. W. et al. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci. Rep. 7, 17810 (2017).
Mellman, I., Coukos, G. & Dranoff, G. Cancer immunotherapy comes of age. Nature 480, 480–489 (2011).
Sharma, P. & Allison, J. P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015).
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science 348, 56–61 (2015).
Tang, Y. et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget 6, 14209–14219 (2015).
Jeanson, A. et al. Efficacy of immune checkpoint inhibitors in KRAS-Mutant Non-Small cell lung cancer (NSCLC). J. Thorac. Oncol. 14, 1095–1101 (2019).
Topper, M. J. et al. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 17, 75–90 (2020).
J, B., MK, G. & CH, W. TNM Classification of Malignant Tumors, 8th Edition. (2017).
Imamura, T. et al. Overexpression of ZRF1 is related to tumor malignant potential and a poor outcome of gastric carcinoma. Carcinogenesis 39, 263–271 (2018).
Takashima, Y. et al. Overexpression of Tetraspanin31 contributes to malignant potential and poor outcomes in gastric cancer. Cancer Sci. 113, 1984–1998 (2022).
Kiuchi, J. et al. Overexpression of YEATS4 contributes to malignant outcomes in gastric carcinoma. Am. J. Cancer Res. 8, 2436–2452 (2018).
Arakawa, H. et al. Overexpression of SHANK2 contributes malignant outcomes as a Hippo pathway regulator in gastric cancer. Am. J. Cancer Res. 15, 363–374 (2025).
Komatsu, S. et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 30, 1139–1146 (2009).
Kashimoto, K. et al. Overexpression of TRIM44 contributes to malignant outcome in gastric carcinoma. Cancer Sci. 103, 2021–2026 (2012).
Nishimura, Y. et al. Overexpression of YWHAZ relates to tumor cell proliferation and malignant outcome of gastric carcinoma. Br. J. Cancer. 108, 1324–1331 (2013).
Ohashi, T. et al. Overexpression of PBK/TOPK relates to tumour malignant potential and poor outcome of gastric carcinoma. Br. J. Cancer. 116, 218–226 (2017).
Aratani, K. et al. Overexpression of CTEN relates to tumor malignant potential and poor outcomes of adenocarcinoma of the esophagogastric junction. Oncotarget 8, 84112–84122 (2017).
Tsuda, H. Individualization of breast cancer based on histopathological features and molecular alterations. Breast Cancer. 15, 121–132 (2008).
Author information
Authors and Affiliations
Contributions
RI and SK designed, and SK and AS reviewed the research; RI, HK, HA, YT, KN, MY, and SH performed cell cultures, molecular biology experiments; SK, TO, TI, JK, KN, HS, TA, TK, HK, HF, TI, HT and AS provided clinical specimens and performed clinical data analyses. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All experimental methods were carried out in accordance with relevant guidelines and regulations, such as the Declaration of Helsinki. Written informed consent was obtained from all patients to use their tissue specimens and blood samples. This study was approved by the institutional review boards of Kyoto Prefectural University of Medicine (ERB-C-319-1).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.








Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ishida, R., Komatsu, S., Kamiya, H. et al. Overexpression of JMJD6 drives immune evasion via the BRD4–IRF1–PD-L1 axis and promotes malignancy in gastric cancer. Sci Rep 16, 1069 (2026). https://doi.org/10.1038/s41598-025-30705-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-30705-y

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}