
Abstract
Memory consolidation is thought to rely on hippocampo-cortical dialogue orchestrated by three cardinal sleep oscillations: cortical slow oscillations, thalamic spindles, and hippocampal sharp-wave ripples. However, how hippocampal outputs are routed to specific cortical targets and dynamically regulated across brain states remains unclear. Here, we performed simultaneous multisite recordings from the dorsal and ventral hippocampus, and frontal and retrosplenial cortex in rats alternating between wakefulness and sleep. Frontal slow oscillations operated as a global temporal scaffold, resetting thalamic circuits and initiating spindle volleys in both anterior and posterior cortex, while retrosplenial slow oscillations more effectively recruited hippocampal ripples. Ripple-spindle reflected direct anatomical connectivity, as dorsal ripples preferentially enhanced retrosplenial spindles, while ventral ripples engaged mainly frontal spindling. When ripples synchronized across dorsal and ventral hippocampus, associated slow oscillations and spindles were attenuated, hippocampal spiking decreased locally and redistributed to the opposite pole, and cortical neuronal responses were suppressed in their corresponding anatomical pathways. These effects indicate that dorso-ventral ripple synchrony gates, rather than amplifies, hippocampo-cortical communication. This gating effect was brain-state dependent, as dorsal-driven reactivation persisted across vigilance states, whereas ventral pathways were more prominent during sleep. Together, these results outline a hierarchical architecture in which slow oscillations provide a global clock, spindles implement anatomically specific reactivation channels, and ripple synchrony dynamically gates hippocampal output, likely shaping the precision and specificity of memory consolidation within a highly variable neural substrate.
Similar content being viewed by others


Introduction
Memory consolidation critically depends on precise interactions between cortical and hippocampal networks during sleep1,2, orchestrated by canonical sleep oscillations: cortical slow oscillations, thalamocortical spindles, and hippocampal sharp-wave ripples3. These oscillations are hierarchically organized during sleep4, and, according to the active systems consolidation model5, this nested structure supports memory consolidation by allowing slow oscillations to temporally organize spindles, which in turn facilitate the emergence of ripples, thereby enabling effective hippocampo-cortical communication1. Although the temporal coordination among these rhythms is well-established3,6, their anatomical specificity and the dynamics of their propagation across distinct brain regions remain largely unexplored.
Additionally, the hippocampus exhibits significant functional heterogeneity along its septo-temporal axis7. Extensive research has demonstrated a clear functional division, with the dorsal CA1 (CA1d) primarily involved in spatial navigation and contextual memory encoding, whereas the ventral CA1 (CA1v) predominantly participates in emotional processing and motivational behavior8,9,10. These hippocampal domains differ substantially in their intrinsic circuits and extrinsic connectivity; CA1d robustly innervates regions such as the retrosplenial cortex (RSC), posterior parietal cortex, and dorsal subiculum11,12,13, while CA1v projects extensively to the medial prefrontal cortex (PFC), nucleus accumbens, and amygdala14,15,16. This anatomical specialization suggests that ripple events originating from different hippocampal domains could selectively engage distinct cortical networks. Indeed, ripple oscillations can arise at various positions along the hippocampal septo-temporal axis, spreading septally or temporally, thus potentially activating different cortical circuits17. However, it remains unknown whether this directional propagation affects cortical processing, particularly regarding neural activity patterns that are essential for memory processing in cortical target areas.
Anatomical tracing studies indicate a striking topographical divergence in hippocampal outputs: the ventral hippocampus sends robust direct projections to the PFC18,19, whereas the dorsal hippocampus preferentially innervates the RSC20. The CA1v–PFC pathway is primarily excitatory, originating from glutamatergic pyramidal neurons in CA1v and subiculum that monosynaptically excite PFC neurons21. By contrast, CA1d-RSC, though also arising from glutamatergic cells (e.g. in dorsal subiculum)22 . Moreover, there are direct long-range inhibitory projections that have been established23. In vivo studies show that ripple-associated bursts from the dorsal hippocampus strongly activate RSC interneurons while suppressing RSC excitatory cells, indicating a feed-forward inhibitory effect of CA1d on retrosplenial networks22. Functionally, this anatomical dichotomy suggests that ventral hippocampal output can directly drive or synchronize prefrontal networks, possibly supporting contextual memory retrieval and emotional regulation24, whereas dorsal hippocampal output modulates retrosplenial activity via inhibition, potentially gating neocortical activity during offline memory consolidation. Consistent with this, silencing the CA1d-RSC pathway impairs the formation of context memories22, highlighting how excitatory CA1v–PFC versus inhibitory CA1d-RSC connections differentially integrate hippocampal information into cortical processes.
Ripple events have traditionally been considered central to memory reactivation and consolidation processes25,26,27. More recent findings demonstrate that hippocampal ripples can couple with neocortical ripples, thereby facilitating memory trace transfer from hippocampal to association cortical networks28,29,30. In the hippocampus, ripples can occasionally occur simultaneously across the dorsal and ventral poles8,17,31, although their incidence is seemingly low and the implications of their coordination remain unclear. According to the active systems consolidation framework, coordinated ripple events should enhance cortical reactivation by promoting broader hippocampal-cortical interaction32. Contrary to these predictions, our findings indicate a larger coincidence than expected merely by chance, and a paradoxical effect of hippocampal ripple synchrony: instead of amplifying cortical reactivation, dorsal-ventral ripple coordination appears to redistribute hippocampal activity and consequently suppress cortical engagement. These unexpected observations suggest that coordinated ripple episodes may dynamically modulate hippocampo-cortical interactions during sleep, indicating that hippocampal ripples might act not as broad facilitators, but as selective gating mechanisms that influence cortical information processing, particularly relevant for memory consolidation.
Results
Hierarchical organization of sleep oscillations
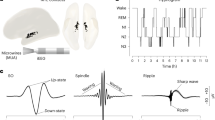
To characterize the interaction structure of canonical sleep oscillations, we performed simultaneous recordings of neuronal activity from anatomically interconnected hippocampal (CA1d and CA1v) and cortical regions (RSC and PFC, Table S1, Fig. S1) in rats resting after performing a spatial task that engages hippocampal-prefrontal interactions. Neural activity was monitored across interleaved task and sleep sessions (Table S2). Hence, we extracted cortical slow oscillations (SOs), cortical spindles, and hippocampal sharp-wave ripples to assess their precise temporal coordination (Fig. 1a). SOs appeared to be the master clock for hippocampo-cortical dynamics. Aligning spindle probability to the PFC SO onset (i.e.; down-state) revealed a biphasic sequence in which both PFC and RSC spindling dipped below baseline just before the onset (–1.35 ± 0.1 z) and then rebounded in a high-gain burst with similar dynamics, but with larger amplitude frontally than parietally (PFC, 5.45 ± 0.3; RSC, 4.23 ± 0.2 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 4, Fig. 1b). When the reference was switched to the RSC SO, the trough was practically abolished, and the subsequent spindle burst was largely reduced, but still slightly larger frontally (PFC, 2.64 ± 0.2; RSC, 2.21 ± 0.2 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3, Fig. 1b). Overall, SO evoked stronger spindling in the frontal cortex (two-way ANOVA, F(1,521) = 172.7, p = 2.8 × 10− 24; post-hoc Tukey comparison, p = 1.1 × 10− 10), indicating that frontal waves broadcast globally whereas retrosplenial waves tune activity locally. Spindle onset latencies relative to SOs were comparable between regions (two-way ANOVA, F(1,521) = 2.0, p = 0.15; post-hoc Tukey comparison, p = 0.15), and consistently positive (Wilcoxon signed-rank test against zero, z = 19.8, 343.75 ± 7.2 ms, p = 2.5 × 10− 87).

Hierarchical temporal organization of sleep oscillations across hippocampus and neocortex. (A) Example traces of simultaneously recorded local field potentials (LFPs) from ventral CA1 (CA1v), dorsal CA1 (CA1d), retrosplenial cortex (RSC), and prefrontal cortex (PFC) during nREM sleep (rat PF24, session 9), illustrating canonical sleep oscillatory events. Highlighted segments denote representative ripples in CA1v and CA1d (gray shading), a cortical spindle in RSC (blue shading), and a slow oscillation (SO) in PFC (red shading). Below each LFP trace, the corresponding band-pass filtered signals are shown (ripples: 100–250 Hz; spindles: 7–15 Hz; SOs: 0.1–4 Hz). Horizontal scale bar: 1 s; vertical scale bar: 0.5 mV. Insets; horizontal scale bar: 25 ms; vertical scale bar: LFP 250 µV, ripple 50 µV. (B) Temporal coupling of spindle occurrence to SOs in cortex. Z-scored spindle occurrence in PFC (blue) and RSC (orange) aligned to PFC SO onset and RSC SO onset. Spindle latencies were similar between pairs (343.75 ± 7.2 ms, Wilcoxon signed rank test against zero, z = 19.8, p = 2.5 × 10− 87). (C) Temporal coupling of hippocampal ripple occurrence to cortical SOs. Z-scored ripple occurrence in CA1d (magenta) and CA1v (teal) aligned to PFC SO onset and RSC SO onset. Ripple latencies were similar between pairs (138.6 ± 17.7 ms, Wilcoxon signed rank test against zero, z = 6.8, p = 1.2 × 10− 11). (D) Temporal coupling of cortical spindle occurrence to hippocampal ripple onset. Z-scored spindle occurrence in PFC (blue) and RSC (orange) aligned to CA1d and CA1v ripple onset. Spindle latencies were different between pairs (PFC, 332.9 ± 57.1 ms; RSC, -20.7 ± 53.2 ms, Wilcoxon signed rank test, z = 4.9, p = 8.5 × 10− 7). In all panels, shaded areas indicate standard error of the mean, and dashed vertical lines mark the onset (t = 0) of the reference event. Asterisks indicate the time bins in which the two traces differ significantly (Wilcoxon signed-rank test across sessions and animals at each bin). P values were corrected for multiple comparisons across all bins using the Benjamini–Hochberg FDR procedure. Animals recorded in CA1d (n = 18), CA1v (n = 13), RSC (n = 19), and PFC (n = 20).
In addition, SOs biased ripple generation with distinct septo-temporal preferences. PFC SOs were preceded by a gradual ripple buildup culminating in a brief dorsal-larger peak at the SO onset (CA1d, 2.29 ± 0.2 z; CA1v, 0.94 ± 0.2 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 3; Fig. 1c). RSC SOs recruited both hippocampal poles with similar dynamics, yet again dorsal hippocampus responses dominated (CA1d, 3.22 ± 0.2 z; CA1v, 2.47 ± 0.3 z; Wilcoxon signed-rank test, FDR-corrected, p < 0.001; Fig. 1c). Overall, SO-evoked ripple bursts were stronger in the dorsal when compared to ventral hippocampus (two-way ANOVA, F(1,412) = 22.4, p = 3.0 × 10− 6; post-hoc Tukey comparison, p = 2.2 × 10− 6), suggesting that SO preferentially propagated into the dorsal hippocampus. Ripple onset latencies relative to SOs were shorter in the dorsal compared to the ventral hippocampus (two-way ANOVA, F(1,412) = 5.3 p = 0.02; post-hoc Tukey comparison, p = 0.02), suggesting that SO reaches earlier the dorsal hippocampal pole.
To further confirm this hierarchical sequence, we then used hippocampal ripples as a reference point and analyzed subsequent cortical spindle activity (Fig. 1d). Dorsal ripples preferentially launched retrosplenial spindle bursts while only modestly engaging frontal spindles (RSC, 2.48 ± 0.1 z; PFC, 1.08 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3; Fig. 1d), whereas ventral ripples produced cortical responses with similar time course, yet dominated by the frontal cortex (PFC, 2.35 ± 0.2 z; RSC, 1.70 ± 0.2 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 4; Fig. 1d), consistent with known anatomical hippocampo-cortical connectivity patterns33. Spindle latencies relative to ripple onsets were consistently longer in frontal than retrosplenial cortex (PFC, 332.9 ± 57.1 ms; RSC, -20.7 ± 53.2 ms, Wilcoxon signed rank test, z = 4.9, p = 8.5 × 10− 7), matching anatomical distance. In addition, sleep oscillations exhibited robust cross-frequency phase modulation. Indeed, SOs strongly modulated spindle occurrence, with spindles preferentially emerging during the up-state, and frontal spindles consistently leading retrosplenial spindles (Fig. S2). Cortical spindles modestly but significantly modulated hippocampal ripple incidence, with ripples most frequently occurring around the trough of the spindle cycle (Fig. S2). Moreover, the coincident occurrence of the distinct sleep oscillations (such as slow oscillations, spindles, and ripples) was generally infrequent (< 0.1 Hz, Table S3), necessitating extended recording periods to reliably capture their interactions. Collectively, these findings establish that frontal SOs operate as a global temporal scaffold, retrosplenial SOs selectively tune hippocampal engagement, and ripple–spindle coupling reflects direct anatomical connectivity, together revealing a hierarchical and topographically organized oscillatory framework that structures hippocampo-cortical communication during sleep.
Regional neuronal recruitment by sleep oscillations
Sleep oscillations effectively regulated neuronal firing throughout the hippocampo-cortical circuit (Table S4). For example, dorsal ripples simultaneously modulated neuronal discharge locally in the hippocampus (Fig. 2a) and distally in the cortex (Fig. 3a). Alignment to PFC SO down-state onset revealed distinct regional firing patterns. CA1d exhibited robust triphasic activity, with a pronounced activation preceding the SO (1.99 ± 0.1 z), swift strong suppression during the cortical down-state (− 1.91 ± 0.1 z), and a significant posterior rebound (0.63 ± 0.01 z, Fig. 2b). Conversely, CA1v showed a weaker response with biphasic modulation, characterized by moderate pre-SO excitation (0.85 ± 0.1 z) and subsequent modest suppression (− 0.83 ± 0.1 z), without clear rebound (Fig. 2b). All response phases were significantly stronger in dorsal hippocampus (Wilcoxon signed-rank test, FDR-corrected, p < 10− 4; Fig. 2b). Changing the reference to RSC SO maintained a similar, but attenuated triphasic structure in CA1d (peak, 1.72 ± 0.1 z; trough, − 0.85 ± 0.1 z; rebound, 0.71 ± 0.1; Fig. 2b) and a reduced, biphasic pattern in CA1v (peak, 1.19 ± 0.1 z; trough, − 0.68 ± 0.1 z; Fig. 2b). Regardless of the SO trigger, dorsal spiking was consistently stronger than ventral neuronal activation (Wilcoxon rank-sum test, z = 4.1, p = 4.82 × 10− 5), confirming the larger influence of SOs into the dorsal hippocampus.

Neuronal recruitment in the hippocampus by sleep oscillations. (A) Heatmaps show normalized firing rates of individual neurons in dorsal (CA1d, left) and ventral (CA1v, right) hippocampus, aligned to the onset (t = 0 ms) of CA1d ripples. Each row represents the average activity of a single neuron, sorted by response latency, with color indicating firing rate (z-score). (B) Z-scored mean firing rates of CA1d (orange) and CA1v (teal) neurons aligned to the onset of PFC (top) and RSC (bottom) slow oscillations (SOs). (C) Normalized firing rates aligned to the onset of PFC (top) and RSC (bottom) spindles. (D) Normalized firing rates aligned to the onset of CA1d ripples (top) and CA1v ripples ( bottom). In all panels, shaded areas represent the standard error of the mean. Dashed vertical lines indicate event onset (t = 0). Asterisks indicate the time bins in which the two traces differ significantly (Wilcoxon signed-rank test across sessions and animals at each bin). P values were corrected for multiple comparisons across all bins using the Benjamini–Hochberg FDR procedure. Animals recorded in CA1d (n = 18), CA1v (n = 13), RSC (n = 19), and PFC (n = 20).

Neuronal recruitment in the cortex by sleep oscillations. (A) Heatmaps showing normalized firing rates of individual neurons in PFC (left) and RSC (right), aligned to the onset of CA1d ripples (t = 0 ms). Each row represents a single neuron, sorted by response latency, with color indicating firing rate (z-score). (B) Z-scored mean firing rates of PFC (blue) and RSC (orange) neurons aligned to the onset of PFC SOs (top) and RSC SOs (bottom). (C) Normalized firing rates aligned to PFC (top) and RSC (bottom) spindle onsets. (D) Normalized firing rates aligned to the onset of CA1d ripples (top) and CA1v ripples (bottom). Shaded areas represent the standard error of the mean. Dashed vertical lines indicate event onset. Asterisks indicate the time bins in which the two traces differ significantly (Wilcoxon signed-rank test across sessions and animals at each bin). P values were corrected for multiple comparisons across all bins using the Benjamini–Hochberg FDR procedure. Animals recorded in CA1d (n = 18), CA1v (n = 13), RSC (n = 19), and PFC (n = 20).
Cortical regions demonstrated consistent triphasic firing patterns in response to SOs (Fig. 3b). PFC SO evoked large spiking triphasic responses in both retrosplenial and prefrontal neurons, with initial excitation preceding the down-state (PFC, 2.21 ± 0.1 z; RSC, 1.57 ± 0.1 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 3), pronounced inhibition at the SO onset (PFC, − 4.44 ± 0.3 z; RSC, − 2.53 ± 0.2 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 4), and clear rebound excitation (PFC, 0.37 ± 0.1 z; RSC, 0.43 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p > 0.05, Fig. 3b). Similarly, RSC SO discharged retrosplenial and prefrontal neurons, with the initial large peak (PFC, 0.70 ± 0.1 z; RSC, 1.36 ± 0.1 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 4), followed by marked inhibition at the down-state onset (PFC, − 2.35 ± 0.2 z; RSC, − 2.82 ± 0.2 z; Wilcoxon signed-rank test, FDR-corrected, p > 0.05), and later rebound (PFC, 0.35 ± 0.1 z; RSC, 0.79 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 4; Fig. 3b). Spiking activity triggered by PFC SOs was consistently stronger than that observed following RSC SOs (Wilcoxon rank-sum test, z = 4.5, p = 6.7 × 10⁻¹⁴), confirming that frontal SOs are more effective drivers of cortical neuronal discharge than retrosplenial waves. Neuronal spiking across hippocampo-cortical circuits was strongly modulated by the phase of SOs, with maximal phase-locking occurring around the decaying phase of the up-state (Supplementary Fig. S3). Together, these observations suggest that frontal SOs broadcast globally and preferentially recruit the dorsal hippocampus, whereas retrosplenial SOs remain local but are nonetheless sufficient to entrain both hippocampal poles, again with a dorsal bias.
Cortical spindles exerted modest control of neuronal discharge. Frontal spindles elicited small monophasic excitation in both dorsal and ventral hippocampus (CA1d, 0.33 ± 0.1 z; CA1v, 0.28 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p > 0.05, Fig. 2c). CA1d displayed contrasting patterns, with pronounced biphasic modulation by retrosplenial spindles (Fig. 2c), initially exhibiting significant pre-spindle suppression (− 1.08 ± 0.1 z), followed by rebound excitation (1.56 ± 0.1 z). Conversely, CA1v showed exclusively weak monophasic excitation in response to RSC spindles (0.52 ± 0.1 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 4, Fig. 2c). Thus, cortical spindles weakly influenced the ventral hippocampus. Cortically, spindles triggered biphasic responses in cortical neurons. Indeed, frontal spindles elicited biphasic responses in both PFC and RSC with pre-spindle inhibition (PFC, − 0.92 ± 0.1 z; RSC, − 0.52 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3), resulting from the SO down-state preceding spindling, followed by modest activation (PFC, 0.55 ± 0.1 z; RSC, 0.39 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3; Fig. 3c). Similarly, RSC spindles evoked biphasic modulation of both local and distal neuronal discharge with prominent early suppression (PFC, − 1.1 ± 0.1 z; RSC, − 0.8 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p > 0.05) and modest subsequent excitation (PFC, 0.39 ± 0.1 z; RSC, 0.58 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p > 0.05, Fig. 3c). The magnitude and temporal pattern of this response were remarkably similar across regions. Furthermore, cortical spindles organized neuronal firing into distinct oscillation-phases, albeit with weaker modulation than SOs and with effects that varied across regions. (Fig. S3). Collectively, spindle oscillations weakly modulate local cortical spiking dynamics34 and selectively influence hippocampal activity through a preferential dorsal-parietal axis.
Hippocampal ripples differentially controlled neuronal reactivation across the hippocampo-cortical network. Neuronal discharge in CA1d most prominently increased during local dorsal ripples, demonstrating a robust and sharply timed peak (18.14 ± 0.8 z), eliciting a largely minor response in CA1v (1.29 ± 0.1 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 4; Fig. 2d), probably reflecting residual ripple propagation between hippocampal poles17. Ventral ripples evoked a similarly selective, yet less prominent, neuronal activation locally in CA1v (11.27 ± 1.0 z), accompanied by a strongly reduced response in CA1d (2.2 ± 0.2 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 4; Fig. 2d). In cortical regions, ripples generated biphasic responses, albeit with different connection-specific profiles. Dorsal ripples caused significant spiking responses of comparable magnitude in both cortical regions (RSC, 1.04 ± 0.1 z; PFC, 0.84 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p > 0.05; Fig. 3d). A different pattern was generated by ventral ripples, which evoked substantial modulation of PFC spiking (1.57 ± 0.2 z), while RSC showed weaker firing modulation (0.46 ± 0.1 z, Wilcoxon signed-rank test, FDR-corrected, p < 10− 4; Fig. 3d). Interestingly, the ventral hippocampus was most effective in engaging cortical neurons, particularly in the frontal cortex (one-way ANOVA F(1,1570) = 22.4, p = 2.3 × 10− 10, post-hoc Tukey comparison, p = 2.8 × 10− 10). These results reveal a topographic organization during nREM sleep, in which the dorsal hippocampus acts as a preferential hub over the ventral hippocampus, being robustly modulated by the three cardinal sleep oscillations. Conversely, ventral hippocampal ripples dominated cortical activation, specifically in the prefrontal cortex.
Ripple synchrony gates hippocampo-cortical reactivation
Despite their distinct connectivity profiles and differential dynamics, the dorsal and ventral hippocampal poles exhibited substantial coordination during ripple events (Fig. 4a). Hippocampal ripples showed low intrinsic rates (0.53 ± 0.1 Hz) and brief mean durations (57.27 ± 0.1 ms). Under such conditions, if ripples were independently generated in the dorsal and ventral poles, purely random overlap would be expected in a minor fraction of events (about 3%). In contrast, we observed synchronization in a larger proportion of ripples (> 12%) between the dorsal and ventral hippocampal poles, suggesting active septo-temporal coordination rather than chance temporal alignment (Wilcoxon signed-rank test against 3%, z = 9.1, p = 7.4 × 10− 20).

Effect of coordinated versus independent ripples on hippocampo-cortical connectivity. (A) Example simultaneous LFP recordings from CA1v and CA1d (rat PF40, session 5), showing example independent (gray shading) and coordinated (pink shading) ripple events, with corresponding filtered ripple band signals below. Horizontal scale bar: 1 s; vertical scale bar: 0.5 mV. Insets; horizontal scale bar: 25 ms; vertical scale bar: LFP 250 µV, ripple 50 µV. (B) Left: Distribution of CA1v ripple onset times relative to CA1d ripple onset during coordinated events, indicating the mean delay (6 ms, red arrow). Right: Violin plot of the mean ripple delay for CA1d→CA1v propagation across events (Wilcoxon signed-rank test against 0, z = 6.5, p = 7.3 × 10− 11). (C) Z-scored occurrence of slow oscillations (SOs) in PFC (top) and RSC (bottom), time-locked to the onset of independent (black) and coordinated (red) CA1d (left) and CA1v (right) ripples. (D) Z-scored occurrence of spindle events in PFC (top) and RSC (bottom), time-locked to ripple onset as in (C). In (C) and (D), solid lines show the mean, shaded areas represent standard error of the mean, and asterisks indicate time points with significant differences. Dashed vertical lines mark ripple onset (t = 0). Asterisks indicate the time bins in which the two traces differ significantly (Wilcoxon signed-rank test across sessions and animals at each bin). P values were corrected for multiple comparisons across all bins using the Benjamini–Hochberg FDR procedure. Animals recorded in CA1d (n = 18), CA1v (n = 13), RSC (n = 19), and PFC (n = 20).
To further investigate intra-hippocampal synchrony and its functional consequences on cortical circuits, we classified sharp wave ripples according to their temporal overlap as independent ripples (CA1d, 84 ± 0.3% of events; CA1v, 87.7 ± 0.3% of events) and coordinated CA1d–CA1v ripples (CA1d, 15 ± 0.7% of events; CA1v, 12.2 ± 0.7% of events). As expected by their relative abundance, independent ripples were more frequent than coordinated ripples (Wilcoxon signed-rank test, z = -13.5, p = 1.5 × 10− 41, Fig. S4). The duration of coordinated ripples was slightly longer than independent events in both hippocampal poles (Wilcoxon signed-rank test, z = 6.1, p = 10− 9, Fig. S4), pointing to intra-hippocampal synchrony as a candidate mechanism regulating the intrinsic structure of ripple episodes. To assess whether ripples were synchronized by propagation across the septo-temporal axis17, we computed the dorso-ventral ripple crosscorrelogram (Fig. 4b). Indeed, when using CA1d as temporal reference, we found that the distribution of ventral onsets was sharply centered at short positive delays (5.90 ± 0.4 ms). Also, the distribution of mean delays indicated that in most cases (77.6%, 83 out of 107 sessions), coordinated ripples spread from dorsal to ventral pole (Fig. 4b), also consistent with the larger amplitude of coordinated ripples specifically in the ventral hippocampus (Fig. S4).
Because cortical SOs can effectively trigger hippocampal ripple events35, we next analyzed SOs immediately preceding ripples, distinguishing between independent and coordinated ripple episodes. Peak amplitude, onset latency, and total duration of the cortical SOs preceding ripples were comparable between independent and coordinated events (Fig. S5). Interestingly, the rising slope of SOs preceding coordinated ripples was modestly yet significantly shallower compared to independent ripples (Wilcoxon rank-sum test, z = -1.6, p < 0.03; Fig. S5). Remarkably, frontal firing rates during both SO down and up states preceding coordinated ripples were higher compared to those preceding independent ripple events (Wilcoxon rank-sum test, z = 41.4, p < 10− 6, Fig. S5). These findings suggest that a gradual transition into the SO down-state, combined with increased cortical discharge during SOs, likely enhances the probability of dorsal hippocampal burst propagation ventrally, thus generating a coordinated ripple.
To determine the impact of ripple synchrony on hippocampo-cortical dynamics, we first studied ripple coordination with cortical SOs, yet explicitly segregating ripples into independent and coordinated events. Unexpectedly, this separation uncovered selective interferent cortical dynamics. In fact, coordinated ripple events consistently led to significantly depressed cortical SO associations compared to independent ripples (Fig. 4c). Specifically, the cortical SO response typically triggered by CA1d was the most affected when ripples coordinated (Wilcoxon signed-rank test, FDR-corrected, p < 10− 4, Fig. 4c), indicating an attenuated hippocampo-cortical communication. Conversely, CA1v ripples, which generally triggered weaker cortical responses, showed no further reduction in SO activity under dorsal-ventral synchrony (Wilcoxon signed-rank test; FDR-corrected, p > 0.05, Fig. 4c). Moreover, synchronised ripple events consistently led to significantly weaker spindle responses compared to independent ripples (Wilcoxon signed-rank test; FDR-corrected, p < 10− 4, Fig. 4d). Spindling suppression was more pronounced for the synaptically connected hippocampo-cortical regions. That is, decreased coupling was deeper in CA1d-RSC and CA1v-PFC pathways than in the CA1v-RSC and CA1d-PFC pairs (Wilcoxon signed-rank test; z = 4.2, p = 2.9 × 10− 5, Fig. 4d). In addition, no significant differences were apparent in the latencies of evoked responses between independent (191.5 ± 45.6 ms) and coordinated ripples (152.4 ± 55.7 ms, Wilcoxon signed-rank test; z = 0.5, p = 0.6, Fig. 4d), suggesting that coordinated and independent ripples engage chiefly the same hippocampo-cortical pathways, yet with different synaptic strength.
Because ripple propagation along the hippocampal septo-temporal axis was directionally biased, we reasoned that the temporal correlations between sleep oscillations would possibly differ according to such spreading direction within the hippocampus. Nevertheless, regardless of the hippocampal leading pole, spindle-evoked responses in the cortex were strikingly similar (Wilcoxon signed-rank test, FDR-corrected, p > 0.05, Fig. S6). Likewise, SO responses in both cortical regions remained nearly identical regardless of ripple propagation direction (Wilcoxon signed-rank test, FDR-corrected, p > 0.05, Fig. S6). Thus, cortical spiking activity during the slow oscillation (SO) biases the hippocampal network toward either focal reactivation (independent ripples) or septotemporal synchronization (coordinated ripples), thereby linking cortical dynamics to the emergence of ripple synchrony. Furthermore, ripple synchrony seems to operate as a network-level gating mechanism that regulates hippocampal–cortical connectivity.
Hippocampal synchrony modulates ripple-driven reactivation
Remarkably, ripples coordination prominently altered hippocampal spiking patterns. In CA1d, independent ripples elicited the highest local firing peak (15.8 ± 0.9), whereas coordinated ripples produced a significantly smaller local spiking response (5.8 ± 0.4 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3), but simultaneously induced a robust activation in CA1v (1.88 ± 0.2 z), which was absent during independent CA1d events (0.39 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3, Fig. 5a). A mirror pattern emerged for CA1v ripples, where independent events generated the maximal local response in CA1v (10.1 ± 1.1 z), while coordinated ripples resulted in a substantially reduced local response (3.6 ± 0.4 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3), yet recruited CA1d (3.94 ± 0.3 z), which was otherwise modestly activated (0.93 ± 0.1 z; Wilcoxon signed-rank test, FDR-corrected, p < 10− 3, Fig. 5a). Robust activation of the distal pole was observed primarily during coordinated ripples, underscoring the role of active septo-temporal drive in ripple propagation.

Ripple type modulation of ripple-triggered spiking across hippocampal–cortical networks. Mean z-scored firing rates of CA1d, CA1v, RSC, and PFC neurons time-locked to CA1d ripple onset (A) or to CA1v ripple onset (B). In all panels, black traces show independent ripples (event confined to the triggering pole, with no near-simultaneous ripple in the opposite pole), and red traces show coordinated ripples (near-simultaneous dorsal–ventral co-occurrence). Shaded bands indicate the SEM; the dashed vertical line marks ripple onset (t = 0 ms). Asterisks denote time bins with significant differences between conditions (paired Wilcoxon signed-rank test across sessions; Benjamini–Hochberg FDR correction across bins for each trace pair). Animals recorded: CA1d (n = 18), CA1v (n = 13), RSC (n = 19), PFC (n = 20).
Neural activation at the distal pole induced by coordinated ripples was consistently smaller than that evoked by independent local events (Wilcoxon signed-rank test, z = 7.8, p = 6.5 × 10− 15), suggesting partial recruitment or a reduction in overall excitatory drive during coordinated ripples. This was further supported by the temporal profile of local spiking responses, since the peak response latency remained unchanged between ripple type (independent, 36.19 ± 0.9 ms; coordinated, 38.31 ± 1.0 ms; Wilcoxon signed-rank test, z= -1.6, p = 0.12), while decay time constants were significantly slower for coordinated ripples (independent, − 13.7 ± 0.7 s⁻¹; coordinated, − 6.76 ± 0.5 s⁻¹; Wilcoxon signed-rank test; z = -6.5, p = 5.8 × 10⁻¹¹), consistent with their longer duration (Fig. S4). These results suggest a diminished excitatory barrage during coordinated events, which likely accounts for the observed depression of local spiking responses. Further analysis of excitation–inhibition dynamics revealed that, despite the overall reduction in activation, the balance between excitatory and inhibitory activity remained stable. Specifically, we classified putative interneurons and pyramidal cells based on action potential waveform features (Fig. S7) and calculated the excitatory-to-inhibitory spiking ratio. Excitation increased relative to inhibition during ripple episodes in both dorsal and ventral hippocampus, and this ratio was consistently maintained for both independent and coordinated ripples (Fig. S8), indicating that excitation–inhibition balance is preserved during ripple synchrony even when overall excitatory drive is reduced.
Next, we evaluated the effect of ripple synchrony on the neuronal spiking of the cortical network. We aligned RSC spiking activity to independent ripples detected exclusively in CA1d and to coordinated dorsal-leading ripple pairs (Fig. 5b). Independent CA1d ripples drove a sizable RSC spike burst (1.14 ± 0.1 z), whereas the same cortical population response was significantly suppressed (0.32 ± 0.1 z) when the dorsal ripple propagated to the ventral pole (Wilcoxon signed-rank test; FDR-corrected, p < 10− 4, Fig. 5b). Ripple coordination was ineffective in modifying the PFC spiking profile associated to CA1d. We computed the complementary comparison for PFC, with independent ripples detected only in CA1v. Similarly, coordinated ripples were unable to modulate the modest RSC discharge correlated with CA1v activation. Conversely, CA1v ripples elicited a prominent response in PFC (1.35 ± 0.2 z), which significantly depressed at the peak (0.86 ± 0.1 z) when the ventral ripple propagated dorsally (Wilcoxon signed-rank test; FDR-corrected, p < 10− 4, Fig. 5b). Together, these patterns show that dorsal–ventral synchrony redistributes, rather than augments, hippocampal excitation, sharply dampening local spiking while spreading a weaker volley to the distant pole, suggesting that coordinated ripples act as an intrinsic gate that tempers hippocampo-cortical output.
Brain state and other factors jointly modulate neuronal Spike timing
Contrasting with thalamic spindles and cortical SOs, which are expressed exclusively during sleep, sharp-wave ripples emerge during both nREM sleep and quiet wakefulness27. To test whether ripple-driven hippocampo-cortical transfer is brain state-dependent, we compared population firing aligned to ripple events recorded in nREM and quiet wake during the same recording sessions (Fig. S9). When referenced to CA1d ripples, local neuronal activation remained strong and sharply timed in both states, but was significantly larger during sleep (sleep, 16.56 ± 0.8 z; wake, 13.30 ± 0.7 z; Wilcoxon signed-rank test; FDR-corrected, p < 10− 4, Fig. S9), suggesting potentiation of the dorsal ripple generator36. Downstream responses were nonetheless brain state-invariant, with CA1v (sleep, 0.95 ± 0.1 z, wake, 0.85 ± 0.1 z; Wilcoxon signed-rank test; FDR-corrected, p > 0.05), RSC (sleep, 0.96 ± 0.1 z, wake, 0.75 ± 0.1 z; Wilcoxon signed-rank test; FDR-corrected, p > 0.05), and PFC (sleep, 0.68 ± 0.1 z, wake, 0.71 ± 0.1 z; Wilcoxon signed-rank test; FDR-corrected, p > 0.05,) showing comparable activation profiles, consistent with the lack of a significant main effect of state (Fig. S9).
An unanticipated pattern emerged when correlations were triggered by CA1v ripples. Indeed, the local ventral reactivation burst was markedly reduced during wakefulness (sleep, 10.6 ± 1.1 z, wake, 5.68 ± 0.5 z; Wilcoxon signed-rank test; FDR-corrected, p < 10− 4), and both dorsal hippocampal (sleep, 2.03 ± 0.2 z, wake, 0.89 ± 0.1 z; Wilcoxon signed-rank test; FDR-corrected, p < 10− 4) and PFC excitation (sleep, 1.51 ± 0.2 z, wake, 0.57 ± 0.1 z; Wilcoxon signed-rank test; FDR-corrected, p < 10− 4) were also strongly attenuated (Fig. S9). RSC maintained its biphasic response pattern, yet both phases were reduced during wakefulness, the initial activation (sleep, 0.26 ± 0.1 z, wake, 0.47 ± 0.1 z; Wilcoxon signed-rank test; FDR-corrected, p < 10− 4), followed by the significant suppression (sleep, -0.22 ± 0.1 z, wake, 0.07 ± 0.07 z; Wilcoxon signed-rank test; FDR-corrected, p < 10− 4, Fig. S9). Thus, ripple-mediated hippocampo-cortical communication is asymmetric across brain states, with dorsal ripples transmitting effectively during sleep and wake, whereas ventral ripples reach cortical targets chiefly during nREM.
Finally, we conducted a comprehensive statistical analysis to assess the joint influence of multiple factors on neuronal firing during ripple events. Hence, we analyzed neuronal firing rates during hippocampal ripple events using linear mixed-effects models (Table S5), which revealed that, despite a large number of observations yielding high statistical significance for most main effects and interactions, the experimental factors studied together accounted for only a modest proportion of the variance in neuronal firing rates (~ 6%, Table S6). An ANOVA marginal test determined the fixed effects significance (Table S7), confirming what we showed in the previous sections when considering parameters individually. Collectively, these findings suggest that ripple-driven neuronal recruitment across hippocampo–cortical networks is influenced by multiple interacting factors, consistent with complex mechanisms governing hippocampo–cortical communication. Among these, ripple synchrony appears to be an important contributor to spike-timing coordination across hippocampal and cortical circuits.
Discussion
Cortical dynamics fundamentally depend on the orchestrated interaction of canonical sleep oscillations: cortical slow oscillations, thalamo-cortical spindles, and hippocampal sharp-wave ripples2. Yet, the organizational logic and the functional rules guiding this cross-structure dialogue remain only partially understood. Our results provide a hierarchical and mechanistically nuanced view of how these oscillatory events coordinate the flow of information within the hippocampo-cortical network, which may have important implications for cortical memory consolidation. Specifically, we reveal that SOs originating in frontal cortex serve as a global temporal scaffold, resetting thalamic circuits and launching spindle volleys in both anterior to posterior cortex, consistent with prior findings in both rodents and humans3,37,38. Spindles, in turn, act as region-specific channels, with retrosplenial spindles triggering robust, biphasic suppression–rebound modulation of dorsal hippocampus ripples, while frontal spindles provide weaker, monophasic excitation that influences both hippocampal poles. This regional selectivity establishes anatomically matched feedback loops, which set the stage for precisely targeted neuronal reactivation.
Our work extends the active systems consolidation model5,6 by showing that hippocampo-cortical interactions are not simply global synchronizations but are routed through distinct anatomical pathways, forming a multilayered network in which specificity and selectivity are paramount. In this architecture, hippocampal ripples act as content carriers whose cortical targets mirror their septo-temporal origin, as dorsal ripples preferentially recruit the retrosplenial cortex, whereas ventral ripples more broadly engage both frontal and parietal regions. This pole-specific recruitment aligns with gene expression and anatomical data suggesting a blend of discrete functional domains and continuous gradients along the hippocampal long axis7,39. Such anatomical specificity supports the idea that different hippocampal subregions may convey spatial versus emotional and motivational content, consistent with the well established role of dorsal hippocampus in spatial navigation and the ventral pole involvement in affective processing10,40.
A central finding of our study is that synchrony between dorsal and ventral hippocampal poles during ripple episodes does not amplify but rather suppresses cortical reactivation. Indeed, when ripples occur simultaneously in both hippocampal poles, a phenomenon we observed to be more frequent than expected by chance, local excitatory drive at the initiating pole is decreased and redistributed rather than globally enhanced. This is evident as a marked reduction in local neuronal discharge and a collapse of spindle and SO responses in the cortex, despite preserved ripple waveform and unchanged excitatory-inhibitory balance at the source. These results challenge the widespread assumption that increased hippocampal synchrony universally strengthens memory replay and instead reveal that dorso-ventral ripple coordination acts as a dynamic gate32,41. Rather than broadly promoting replay, this gating mechanism ensures that only the most relevant memory traces, likely those with the strongest initial activation, are integrated into cortical networks at high gain, while broader, lower-gain coordinated events suppress nonspecific or potentially interfering reactivation.
Our data further highlight that the microstructure of preceding cortical SOs can bias the hippocampal network toward either local, high-gain replay or septo-temporally coordinated, lower-gain events. This finding supports a model in which slow waves and their transitions not only group spindles and ripples within up-states42,43 but also dynamically gate the specificity of hippocampal output to cortex. This is also consistent with recent observation that CA1d and RSC operate as weakly coupled excitable systems capable of bidirectional perturbation, with the RSC potentially serving as a gateway that enables hippocampal ripples to propagate SO down-states to downstream cortical regions via cortico-cortical pathways44. This alternation between up- and down-states may reflect alternating windows for active consolidation and homeostatic recalibration45. The PFC, in particular, exhibits pronounced firing suppression during SO down-states, aligning with its proposed role as a site of long-term associative memory storage and plasticity46.
We also show that ripple-driven communication is strongly modulated by brain state, but primarily through its interactions with other factors. For example, CA1d ripples are effective in driving cortical targets during both sleep and quiet wakefulness, whereas CA1v ripples preferentially amplify hippocampo-cortical coupling during nREM sleep. This is consistent with work demonstrating privileged reactivation of hippocampal ensembles during sleep47,48 and suggests that nREM may provide optimal network conditions for the integration of emotionally or motivationally significant information, often processed via the ventral hippocampus40. We thus propose a dual-level control, in which SO phase and brain state act globally, while ripple synchrony provides a local, dynamic gating mechanism for hippocampal-cortical transfer.
Overall, these insights refine the active systems consolidation framework, moving beyond simple models where higher synchrony entails stronger replay. Instead, our results argue that hippocampal synchrony operates as a selective brake, protecting the cortex from indiscriminate activation. This dynamic gating allows the hippocampo-cortical network to flexibly prioritize high-value traces, likely spatial in dorsal pathways, and emotional-motivational in ventral connections, and to suppress potentially spurious activation. By mapping how SOs establish a global temporal scaffold, spindles specify which hippocampal pole engages with cortical targets, and ripple synchrony ultimately determines the effectiveness of hippocampo-cortical connectivity, we provide a functional anatomical blueprint for how memory consolidation could be organized. This is conceptually consistent with findings from large-scale neural population studies, which show that experimentally induced or natural variability often explains only a modest proportion of neural firing rate variance, with the majority attributable to intrinsic and trial-level fluctuations49,50,51. In our data, linear mixed-effects modeling confirmed that fixed experimental factors and their interactions explain approximately 6% of the total variance in firing rates, with the remainder reflecting trial-by-trial variability and minor (< 1%) contributions from animal and session identity. This highlights the importance of effect size and context, not just statistical significance, when interpreting large-scale neural datasets52,53,54.
By integrating the hierarchy of oscillatory interactions, our findings help to shed light on previous discrepancies regarding the functional significance of hippocampal synchrony and extend the systems consolidation model to account for dynamic, context-dependent gating. Indeed, the selective amplification of hippocampo-cortical axes during sleep may explain the preferential consolidation of emotional or motivational memories observed in some behavioral studies6,55. Our approach underscores the necessity of hierarchical statistical modeling and transparent reporting of explained variance to ensure mechanistic interpretations are both meaningful and generalizable. Looking forward, future studies could employ simultaneous recordings from upstream inputs (e.g., CA3 or entorhinal cortex) and targeted manipulations, such as optogenetic silencing or chemogenetic modulation, to delineate the cellular and circuit mechanisms underlying ripple-driven gating56. Single-cell imaging approaches might further reveal how specific neuronal ensembles participate in or are excluded from cortical reactivation during coordinated versus independent ripple events, providing insight into the microcircuit logic of this dynamic gate28.
In humans, brain oscillations and inter-regional synchrony are routinely probed non-invasively using multiple modalities (MEG, EEG, fMRI, etc.), thus offering a bridge between our findings and the human literature. Magnetoencephalograpy studies have demonstrated that canonical sleep rhythms like spindles and slow waves, traditionally observed in scalp EEG, can be resolved into their cortical generators and network interactions in the human brain57. Furthermore, simultaneous EEG-fMRI experiments have revealed that transient electrophysiological events are mirrored by coordinated hemodynamic fluctuations across the brain. For instance, the appearance of sleep spindles is associated with the emergence of a global low-frequency (< 0.1 Hz) BOLD oscillation in light sleep, whereas deep slow-wave sleep is marked by higher-frequency (> 0.1 Hz) BOLD fluctuations linked to slow wave58. This coupling between EEG-defined events and fMRI signals indicates that large-scale neural synchrony can be inferred through hemodynamic imaging. Even optical methods like functional near-infrared spectroscopy (fNIRS) corroborate these findings, as studies have found that the pronounced neuronal synchronization of slow-wave sleep corresponds to a reduction in spontaneous cortical blood-flow oscillations compared to lighter sleep or wake states59. Together, these human neuroimaging studies underscore that the coordinated oscillatory dynamics and cross-regional synchrony we observe are also reflected in the living human brain during various sleep stages, despite the challenges inherent to non-invasive brain recordings.
In sum, our findings are consistent with a working model of cortical functional communication during sleep in which cortical slow oscillations orchestrate precise ripple–spindle coupling dynamics, hippocampal synchrony acts as a context-sensitive gate for cortical engagement, and the interplay of anatomical cortical topography, brain state, and local network configuration ensures selective, efficient, and topographically precise coordination of cortical circuits. This multilayered control mechanism provides a flexible substrate for shaping the precision and content of hippocampo-cortical exchanges and may bias which interactions are preferentially stabilized within large-scale networks.
Materials and methods
Animals
Twenty adult male Sprague-Dawley rats (postnatal days 40–60, weighing 300–350 g) were obtained from the Center for Innovation on Biomedical Experimental Models (CIBEM, https://cibem.bio.puc.cl/) at the Pontificia Universidad Católica de Chile. Rats were housed in a temperature-controlled room (22 ± 1 °C) under a 12-hour light/dark cycle (lights on at 7:00 a.m.) with food and water provided ad libitum. All experimental procedures were approved by the Scientific Ethical Committee for the Care of Animals and the Environment (CEC-CAA, protocol 220512003). We confirm that all methods were carried out in accordance with relevant guidelines and regulations. We confirm that all methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Habituation
Animals were habituated to the housing environment for three days, then handled daily by the experimenter for three to five additional days. Subsequently, rats were habituated to the experimental environment by placing them on a towel-covered drum in the recording room for 30–60 min daily over approximately one week.
Recording implant assembly
The recording drive was designed using Autodesk Fusion software and 3D-printed on a Fusion360 F410 printer. Sixteen or 32 tungsten standard tapered-tip electrodes (MicroProbes, USA; impedances of 1, 3, and 5 MΩ) were assembled onto a custom-fabricated drive targeted to brain regions of interest based on a rat brain stereotaxic atlas60. Each electrode was connected to an EIB-18 or EIB-36 PCB card (capacity of 18 or 36 channels, including ground and reference). Ground and reference channels were connected to stainless steel screws placed into the skull during surgery. The entire assembly was covered with copper mesh to minimize electrical noise.
Stereotaxic surgery
Rats were anesthetized with isoflurane (4% induction, 1.5–2% maintenance) and secured in a stereotaxic frame (Stoelting Inc.). Body temperature was maintained at 35–37 °C using a homeothermic blanket, and animals received hourly hydration with glucosaline solution (0.9% NaCl, 2.5% dextrose). Following a scalp incision, four craniotomies (~ 1 mm diameter each) were performed in the right hemisphere at predetermined stereotaxic coordinates (CA1d: -4.0 AP, 2.0 ML, 2.7 DV; CA1v: -5.7 AP, -5.5 ML, 7.2 DV; RSC: -4.0 AP, 0.7 ML, 2.2 DV; PFC: 2.5 AP, -0.7 ML, 4.2 DV, all in mm). Two additional craniotomies anterior to bregma accommodated the ground and reference electrodes, while four more craniotomies (two parietal contralateral, one parietal ipsilateral, and one posterior to lambda) secured the implant with additional screws. The dura was carefully removed in the craniotomies, cortical surfaces were moistened with mineral oil, and electrodes were carefully inserted. Craniotomies were sealed with silicone elastomer or wax, and the recording drive was fixed using dental acrylic. Postoperative care involved daily subcutaneous injections of enrofloxacin (10 mg/kg) and meloxicam (1 mg/kg) for three consecutive days. Animals recovered for at least seven days before recordings began.
Electrophysiological recordings
Following recovery, electrophysiological recordings were conducted over 10 consecutive days during the light phase in a Faraday-shielded enclosure. Rats were placed on a towel-covered platform for 60–90-minute sessions. The EIB-18 or EIB-36 boaard was connected via a 16- or 32 channel headstage (Intan Technologies) to an amplifier (Intan RHD Recording System, Intan Technologies, USA), with one electrode serving as reference. Video recordings synchronized with the amplifier clock aided in brain state identification. Signals were sampled at 20 kHz using RHX Data Acquisition software (Intan Technologies) and later converted to MATLAB format using the LAN toolbox for further analysis.
Histology
After the recording protocol finished, rats were anesthetized with isoflurane (4% induction, 1.5–2% maintenance), and electrolytic lesions (5 µA for 10 s) marked electrode locations. After 48 h of recovery, animals were anesthetized to a surgical plane with ketamine (300 mg/kg) and xylazine (30 mg/kg, intraperitoneally [i.p.]) and euthanized by transcardial perfusion with 0.9% saline followed by 4% paraformaldehyde. Brains were postfixed overnight, transferred to PBS-azide, and sectioned coronally using a vibratome (World Precision Instruments, USA). Sections were Nissl-stained and examined with a Nikon Eclipse CI-L microscope to verify electrode placements.
Brain state identification
We used a previously described method61,62 Briefly, brain states (quiet wakefulness, nREM sleep, and REM sleep) were identified via visual inspection of raw LFP from the CA1d region, spectral analysis (Fourier spectrogram), and behavioral observation from video recordings. States were classified in 10-second epochs: quiet wakefulness exhibited mixed-frequency activity (> 20 Hz) with minimal movement; nREM sleep displayed continuous low-frequency activity (0.1–4 Hz) without movement; REM sleep showed pronounced theta activity (5–10 Hz) and muscle atonia. State scoring employed custom MATLAB scripts.
Sleep oscillations detection
Ripples were identified by band-pass filtering LFP (100–250 Hz, zero-phase shift non-causal filter, 0.5 Hz roll-off), rectifying, and low-pass filtering at 20 Hz (4th order Butterworth). Signals were z-score normalized, and events exceeding a 3.5 SD threshold (CA1d) or 2.5 SD (CA1v) lasting ≥ 50 ms were identified and visually verified. Onset and offset time-points were determined by the crossing of a 1 SD threshold, with a 50 ms refractory period to prevent duplicates. Spectral analysis using 7-cycle Morlet wavelets (adapted from a previously used method32 were used to determine ripple frequency, amplitude, and duration. Spindles were identified using LFP band-pass filtering (7–15 Hz, 4th order Butterworth), root mean square envelope smoothing (100 ms Gaussian), and segments exceeding 1 SD for ≥ 500 ms (adapted from a previously used method63. Slow oscillations were isolated by low-pass filtering LFP (0.1–4 Hz, 2nd and 5th order filters, respectively). Oscillations were identified by zero crossings, with thresholds set for positive peaks (> 85th percentile) and negative troughs (< 40th percentile). Putative slow oscillations were further filtered to periods of nREM sleep (adapted from previously used methods38,64.
Spike sorting
Extracellular signals were band-pass (600–5000 Hz) filtered and the spike waveforms with either negative or positive peaks exceeding were extracted. Single units were sorted using a custom manual clustering program (Kilosort2, https://github.com/MouseLand/Kilosort) and were distinguished based on principal components. Putative pyramidal cells and interneurons were further separated by principal component analysis of the unit waveforms and mean firing rat. A unit cluster was classified as a single-unit activity if the refractory period (time between two consecutive spikes) was at least 1.5 ms. When the recording quality and the spike sorting did not allow unambiguous single-unit isolation, the spike cluster was conservatively classified as multiunit activity (MUA).
Linear mixed-effects model
We conducted a comprehensive statistical analysis to evaluate how the multiple factors examined in the study jointly influence neuronal firing during ripple events, thereby illustrating the complex interactions that we previously described individually across the article. Hence, we analyzed neuronal firing rates during hippocampal ripple events using linear mixed-effects models, incorporating ripple type, brain state, ripple initiation pole, and recording region as fixed factors. Animal identity and recording session were included as random effects to account for repeated measures and nested data structures (Table S5). Linear mixed-effects modeling revealed that, despite a large number of observations yielding high statistical significance for most main effects and interactions, the experimental factors studied together accounted for only a modest proportion of the variance in neuronal firing rates (fixed-effects marginal R² = 0.052, conditional R² = 0.058, Table S6). Variance decomposition indicated that animal and session random effects accounted for less than 1% of the total variance, with the majority arising from within-session (trial-to-trial) variability (Table S6). After obtaining the parameter estimates, we performed an ANOVA marginal test to determine the fixed effects significance (Table S7). Interestingly, the most significant effect was yielded by the ripple type*ripple initiation pole interaction (p = 3.82 × 10− 54). Moreover, robust main effects of recording region (p = 7.3 × 10− 26), ripple hippocampal pole (p = 1.4 × 10− 5), and especially ripple type (p = 7.0 × 10− 43) were observed, along with significant higher-order interactions (Table S7), such as recording region*ripple type*ripple initiation pole (p = 9.89 × 10⁻50, Fig. 5a). Importantly, the effect of brain state alone was not significant (p = 0.16), yet brain state contributed to several significant interactions, such as recording region*brain state*ripple initiation pole (p = 2.64 × 10⁻15, Fig. 5c).
Statistical analysis
Group differences with a single, independent factor were examined with a one-way ANOVA when the data met parametric assumptions (normality and homogeneity of variances); if normality was violated the non-parametric Kruskal–Wallis test was substituted. For repeated observations, a repeated-measures ANOVA was applied when residuals were normally distributed. If only two repeated levels were compared, a paired-samples t-test was used; when the normality assumption was violated a Wilcoxon signed-rank test (two levels) or a Friedman test (≥ 3 levels) replaced the parametric test. Sphericity was assessed with Mauchly’s test, and the Greenhouse–Geisser correction was applied when sphericity was violated. Post-hoc pairwise comparisons after any omnibus ANOVA were adjusted with the Bonferroni procedure. Linear relations between two continuous, normally distributed variables were assessed with Pearson’s r; otherwise Spearman’s ρ was used. The threshold for statistical significance was set at two-tailed α = 0.05. Effect sizes were reported for all main tests (Cohen’s d for t-tests, partial η² for ANOVAs, and r or ρ for correlations). The decay time constant (b) was calculated by fitting the data to a single-term exponential decay model of the form y = a*exp(b*t) using least-squares regression. The fitting and statistical analyses were performed using the MATLAB software package (MathWorks).
Data availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
References
Diekelmann, S. & Born, J. The memory function of sleep. Nat. Rev. Neurosci. 11, 114–126 (2010).
Sirota, A., Csicsvari, J., Buhl, D. & Buzsáki, G. Communication between neocortex and hippocampus during sleep in rodents. Proc. Natl. Acad. Sci. U S A. 100, 2065–2069 (2003).
Staresina, B. P. et al. Hierarchical nesting of slow oscillations, spindles and ripples in the human hippocampus during sleep. Nat. Neurosci. 18, 1679–1686 (2015).
Steriade, M. Grouping of brain rhythms in corticothalamic systems. Neuroscience 137, 1087–1106 (2006).
Born, J. & Wilhelm, I. System consolidation of memory during sleep. Psychol. Res. 76, 192–203 (2012).
Klinzing, J. G., Niethard, N. & Born, J. Mechanisms of systems memory consolidation during sleep. Nat. Neurosci. 22(10), 1598–1610 (2019).
Strange, B. A., Witter, M. P., Lein, E. S. & Moser, E. I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 15, 655–669 (2014).
Sosa, M., Joo, H. R. & Frank, L. M. Dorsal and ventral hippocampal sharp-wave ripples activate distinct nucleus accumbens networks. Neuron 105, 725–741.e8 (2020).
Moser, E., Moser, M. B. & Andersen, P. Spatial learning impairment parallels the magnitude of dorsal hippocampal lesions, but is hardly present following ventral lesions. J. Neurosci. 13, 3916–3925 (1993).
Fanselow, M. S. (ed, H. W.) Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65 7–19 (2010).
Koike, B. D. V. et al. Electrophysiological evidence that the retrosplenial cortex displays a strong and specific activation phased with hippocampal theta during paradoxical (REM) sleep. J. Neurosci. 37, 8003–8013 (2017).
Nitzan, N. et al. Propagation of hippocampal ripples to the neocortex by way of a subiculum-retrosplenial pathway. Nat. Commun. 11 (2020).
Van Groen, T. & Wyss, J. M. Extrinsic projections from area CA1 of the rat hippocampus: Olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J. Comp. Neurol. 302, 515–528 (1990).
Swanson, L. W. A direct projection from Ammon’s horn to prefrontal cortex in the rat. Brain Res. 217, 150–154 (1981).
Rosene, D. L. & Van Hoesen, G. W. Hippocampal efferents reach widespread areas of cerebral cortex and amygdala in the rhesus monkey. Sci. (1979). 198, 315–317 (1977).
Swanson, L. W. & Cowan, W. M. An autoradiographic study of the organization of the efferet connections of the hippocampal formation in the rat. J. Comp. Neurol. 172, 49–84 (1977).
Patel, J., Schomburg, E. W., Berényi, A., Fujisawa, S. & Buzsáki, G. Local generation and propagation of ripples along the septotemporal axis of the hippocampus. J. Neurosci. 33, 17029–17041 (2013).
Jay, T. M., Glowinski, J. & Thierry, A. M. Selectivity of the hippocampal projection to the prelimbic area of the prefrontal cortex in the rat. Brain Res. 505, 337–340 (1989).
Hoover, W. B. & Vertes, R. P. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 212, 149–179 (2007).
Sugar, J., Witter, M. P., van Strien, N. M. & Cappaert, N. L. M. The retrosplenial cortex: intrinsic connectivity and connections with the (para)hippocampal region in the rat. An interactive connectome. Front. Neuroinform. 5, 10798 (2011).
Sesack, S. R. & Pickel, V. M. In the rat medial nucleus accumbens, hippocampal and catecholaminergic terminals converge on spiny neurons and are in apposition to each other. Brain Res. 527, 266–279 (1990).
Opalka, A. N., qiang Huang, W., Liu, J., Liang, H. & Wang, D. V. Hippocampal ripple coordinates retrosplenial inhibitory neurons during slow-wave sleep. Cell. Rep. 30, 432–441e3 (2020).
Jinno, S. et al. Neuronal diversity in GABAergic long-range projections from the hippocampus. J. Neurosci. 27 (2007).
Liu, X. & Carter, A. G. Ventral hippocampal inputs preferentially drive corticocortical neurons in the infralimbic prefrontal cortex. J. Neurosci. 38, 7351 (2018).
Wilson, M. A. & McNaughton, B. L. Dynamics of the hippocampal ensemble code for space. Science 261, 1055–1058 (1993).
Foster, D. J. & Wilson, M. A. Reverse replay of behavioural sequences in hippocampal place cells during the awake state. Nature 440, 680–683 (2006).
Buzsáki, G. Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 25, 1073–1188 (2015).
Khodagholy, D., Gelinas, J. N. & Buzsáki, G. Learning-enhanced coupling between ripple oscillations in association cortices and hippocampus. Sci. (1979). 358, 369–372 (2017).
Norman, Y. et al. Hippocampal sharp-wave ripples linked to visual episodic recollection in humans. Science. 365 (2019).
Vaz, A. P., Inati, S. K., Brunel, N. & Zaghloul, K. A. Coupled ripple oscillations between the medial temporal lobe and neocortex retrieve human memory. Sci. (1979). 363, 975–978 (2019).
Morici, J., Paiva-Lima, I., Silva, A., Pronier, É. & Girardeau, G. Dorso-ventral hippocampus neural assemblies reactivate during sleep following an aversive experience. BioRxiv 2024.06.13.598918 https://doi.org/10.1101/2024.06.13.598918 (2025).
Logothetis, N. K. et al. Hippocampal-cortical interaction during periods of subcortical silence. Nature 491, 547–553 (2012).
Qiu, S. et al. Whole-brain spatial organization of hippocampal single-neuron projectomes. Science (1979). 383 (2024).
Sejnowski, T. J. & Destexhe, A. Why do we sleep? Brain Res. 886, 208–223 (2000).
Staresina, B. P., Niediek, J., Borger, V., Surges, R. & Mormann, F. How coupled slow oscillations, spindles and ripples coordinate neuronal processing and communication during human sleep. Nat. Neurosci. 26, 1429–1437 (2023).
Ylinen, A. et al. Sharp wave-associated high-frequency oscillation (200 hz) in the intact hippocampus: network and intracellular mechanisms. J. Neurosci. 15, 30–46 (1995).
Siapas, A. G. & Wilson, M. A. Coordinated interactions between hippocampal ripples and cortical spindles during slow-wave sleep. Neuron 21, 1123–1128 (1998).
Mölle, M., Yeshenko, O., Marshall, L., Sara, S. J. & Born, J. Hippocampal sharp wave-ripples linked to slow oscillations in rat slow-wave sleep. J. Neurophysiol. 96, 62–70 (2006).
Cembrowski, M. S. et al. Spatial gene-expression gradients underlie prominent heterogeneity of CA1 pyramidal neurons. Neuron 89, 351–368 (2016).
Jimenez, J. C. et al. Anxiety cells in a hippocampal-hypothalamic circuit. Neuron 97, 670–683e6 (2018).
Ramirez-Villegas, J. F., Logothetis, N. K. & Besserve, M. Diversity of sharp-wave-ripple LFP signatures reveals differentiated brain-wide dynamical events. Proc. Natl. Acad. Sci. U S A. 112, E6379–E6387 (2015).
Mölle, M. & Born, J. Slow oscillations orchestrating fast oscillations and memory consolidation. Prog Brain Res. 193, 93–110 (2011).
Neske, G. T. The slow oscillation in cortical and thalamic networks: mechanisms and functions. Front. Neural Circuits. 9, 172130 (2016).
Swanson, R. A. et al. Topography of putative bi-directional interaction between hippocampal sharp-wave ripples and neocortical slow oscillations. Neuron 113, 754–768e9 (2025).
Vyazovskiy, V. V. & Harris, K. D. Sleep and the single neuron: the role of global slow oscillations in individual cell rest. Nat. Rev. Neurosci. 14(6), 443–451 (2013).
Euston, D. R., Gruber, A. J. & McNaughton, B. L. The role of medial prefrontal cortex in memory and decision making. Neuron 76, 1057–1070 (2012).
Roux, L., Hu, B., Eichler, R., Stark, E. & Buzsáki, G. Sharp wave ripples during learning stabilize the hippocampal spatial map. Nat. Neurosci. 20(6), 845–853 (2017).
Peyrache, A., Khamassi, M., Benchenane, K., Wiener, S. I. & Battaglia, F. P. Replay of rule-learning related neural patterns in the prefrontal cortex during sleep. Nat. Neurosci. 12, 919–926 (2009).
Averbeck, B. B., Latham, P. E. & Pouget, A. Neural correlations, population coding and computation. Nat. Rev. Neurosci. 7, 358–366 (2006).
Renart, A. et al. The asynchronous state in cortical circuits. Sci. (1979). 327, 587–590 (2010).
Stringer, C. et al. Spontaneous behaviors drive multidimensional, brainwide activity. Science (1979) 364 (2019).
Schielzeth, H. & Nakagawa, S. Nested by design: model fitting and interpretation in a mixed model era. Methods Ecol. Evol. 4, 14–24 (2013).
Wasserstein, R. L., Schirm, A. L. & Lazar, N. A. Moving to a world beyond p < 0.05. Am. Stat. 73, 1–19 (2019).
Cumming, G. The new statistics: why and how. Psychol. Sci. 25, 7–29 (2014).
Payne, J. D., Stickgold, R., Swanberg, K. & Kensinger, E. A. Sleep preferentially enhances memory for emotional components of scenes. Psychol. Sci. 19, 781–788 (2008).
Stark, E. et al. Pyramidal cell-interneuron interactions underlie hippocampal ripple oscillations. Neuron 83, 467–480 (2014).
Brancaccio, A., Tabarelli, D., Bigica, M. & Baldauf, D. Cortical source localization of sleep-stage specific oscillatory activity. Sci. Rep. 10, 1–15 (2020).
Song, C., Boly, M., Tagliazucchi, E., Laufs, H. & Tononi, G. fMRI spectral signatures of sleep. Proc. Natl. Acad. Sci. U S A. 119, e2016732119 (2022).
Näsi, T. et al. Spontaneous hemodynamic oscillations during human sleep and sleep stage transitions characterized with near-infrared spectroscopy. PLoS One. 6, e25415 (2011).
Paxinos, G. & Watson, C. The Rat Brain in Stereotaxic Coordinates. (2013).
Valdivia, G. et al. Sleep-dependent decorrelation of hippocampal spatial representations. iScience. 27 (2024).
Peyrache, A., Lacroix, M. M., Petersen, P. C. & Buzsáki, G. Internally organized mechanisms of the head direction sense. Nat. Neurosci. 18, 569–575 (2015).
Peyrache, A., Battaglia, F. P. & Destexhe, A. Inhibition recruitment in prefrontal cortex during sleep spindles and gating of hippocampal inputs. Proc. Natl. Acad. Sci. U S A. 108, 17207–17212 (2011).
Shin, J. D. & Jadhav, S. P. Prefrontal cortical ripples mediate top-down suppression of hippocampal reactivation during sleep memory consolidation. Curr. Biol. 34, 2801–2811e9 (2024).
Acknowledgements
We are grateful to Jan Born for reading the manuscript and providing insightful comments. This manuscript was prepared with the assistance of AI-based tools (ChatGPT). ChatGPT was used exclusively for improving the clarity, grammar, and overall flow of the text, as well as for providing guidance on statistical analyses, including verification and refinement of the statistical treatment of results, particularly the linear mixed-effects models. In all cases, the authors thoroughly reviewed, edited, and validated any AI-generated content. ChatGPT was not used for core research tasks such as experimental design, data acquisition, primary data analysis, interpretation of results, or drawing scientific conclusions. The authors take full responsibility for all content of the manuscript.
Funding
This work was supported by grants fondecyt 1230589 and Anillos ACT 210053.
Author information
Authors and Affiliations
Contributions
PF designed research and wrote the manuscript; NE performed experiments and analysis. GL, MC, GF, AA and AL-V performed experiments. The authors declare no competing interest.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lazcano, G., Caneo, M., Aguilera, A. et al. Hippocampal synchrony dynamically gates cortical connectivity across brain states. Sci Rep 16, 3497 (2026). https://doi.org/10.1038/s41598-025-31725-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-31725-4
