
Abstract
The primary obstacle in detecting bloodstream infections is their often extremely low bacterial concentration. To accurately detect such infections, we developed a quantitative PCR assay, named LowLoad-qPCR, based on the combination of increased input DNA by random priming amplification method and the use of multiple probes sharing a single fluorophore. Here, we detail the key improvements over conventional qPCR and evaluate its performance using blood samples.
Similar content being viewed by others


Introduction
Sepsis is a major cause of critical illness and mortality worldwide, with over 30 million cases annually1. Rapid initiation of effective antibiotics is essential, as delays increase mortality by 7.6% per hour in septic shock2. However, bacteremia diagnosis remains challenging, as bacterial cells are vastly outnumbered by host components in blood, with bacterial concentrations often as low as 0.1–10 colony-forming units (CFU)/mL3. Blood culture (BC), the current gold standard, has limited sensitivity, a high false-negative rates, and requires more than 24 h for actionable results4.
Few clinically available alternatives exist for the direct detection of bloodstream pathogens in whole blood. Among them, T2Bacteria is the only Food and Drug Administration (FDA)-approved Nucleic Acid Amplification Tests (NAAT)-based assay, providing results in four hours via magnetic resonance technology5,6. Although highly sensitive, its implementation is limited by high cost, dedicated equipment, and narrow species coverage (six species)7. Metagenomic next-generation sequencing (mNGS) is an untargeted sequencing approach that identifies all nucleic acids in a sample, enabling simultaneous detection of diverse microbes. It offers broader detection, but is compromised by non-specific signals from circulating cell-free host DNA8. Moreover, several emerging platforms aim to enable rapid, culture-independent pathogen detection directly from whole blood. The Qvella FAST-ID BSI panel processes a single sample in under 1 h through selective blood-cell lysis, bacterial concentration, and PCR-based rRNA detection9. The LiDia Seq BSI/AMR test uses semiconductor sequencing to identify pathogens and resistance markers within 3–4 h (https://www.dnae.com/). HelixBind’s system applies electrostatic capture to detect pathogens from 6 mL of whole blood in under 3 h (https://www.helixbind.com), while the automated SepsiSTAT platform enables PCR-based detection and recovery of viable organisms from 12 mL of blood (https://momentumbio.co.uk). Nevertheless, despite their promise, these technologies remain limited by high costs, workflow complexity, and infrastructure requirements, which may hinder widespread implementation8. Given the lack of sensitive and cost-effective techniques for direct detection of bacteremia, we developed LowLoad-qPCR, an ultrasensitive assay designed to recover and detect bacteria at very low concentrations directly in whole blood.
Results and discussion
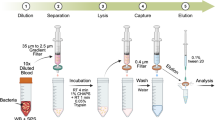
Escherichia coli, Pseudomonas aeruginosa, and Staphylococcus aureus were selected for LowLoad-qPCR development as representative and clinically relevant bloodstream pathogens. Maximizing bacterial recovery from blood is critical for detecting the causative agent of bacteremia. To achieve this, we compared two methods: The classical blood centrifugation and the Peps6-CaptoBAC kit (ApoH-Technologies, La Grande Motte) following manufacturer’s instructions10,11,12. A detailed overview of the full workflow, including sample processing, random-priming amplification and multi-probe detection, is provided in Fig. 1, which outlines all experimental steps and their rationale within the LowLoad-qPCR protocol. Capture efficiency was quantified for each species and method. Centrifugation yielded recovery rates of 19% for E. coli, 43.5% for P. aeruginosa, and 43% for S. aureus. In contrast, the Peps6-CaptoBAC kit achieved 8.74%, 7.86%, and 29.79%, respectively (Fig. 2a). Centrifugation outperformed the commercial kit (Peps6-CaptoBAC kit), offering higher efficiency and lower cost, key factors for adoption in clinical laboratories. Tae Hyun et al. used the same Peps6 magnetic bead–based bacterial recovery system reporting higher recovery percentages, which may be attributable to their use of starting concentrations of 5 CFU/mL, approximately 10,000-fold lower than our concentrations. Their recovery line plots likewise suggest that capture efficiency decreases as bacterial concentration increases12. Furthermore, these beads appear unable to bind several of the most prevalent species responsible for sepsis, representing a clear disadvantage relative to conventional centrifugation, which has been reported to yield recovery rates of 36–69%3,13,14. Importantly, those published results were obtained using specialized plasma-separation devices; despite conducting our experiments without any special devices, we observed similar outcomes3,14.

Overview of the LowLoad-qPCR workflow. a Schematic representation of the LowLoad-qPCR workflow using both bacterial recovery methods. After sample processing and DNA extraction, the workflow proceeds to downstream molecular analysis. b For each DNA extract obtained, two parallel molecular approaches are performed: (i) a conventional single-probe qPCR, and (ii) the LowLoad-qPCR workflow, which incorporates a random-priming amplification step followed by a multiplex TaqMan assay using multiple species-specific probes to enhance detection sensitivity.

Stepwise design of the LowLoad-qPCR protocol. a Comparison of bacterial recovery efficiencies from spiked blood for E. coli, P. aeruginosa, and S. aureus using both recovery methods. b Effectiveness of DNase treatment in removing extracellular S. aureus DNA without compromising detection of intracellular E. coli DNA. c Impact of random priming amplification on total DNA yield and Cq values for all three species. d, e Cq values and fluorescence intensities benefit from probe multiplexing on a single target. c Data shown as mean ± s.d. a, b, d, e Boxes and whiskers show interquartile range and maximum and minimum values of data; centre lines in boxes represent the median. b–g Each data point represents an independent experimental replicate (n = 3 per experiment).
Following the evaluation of the bacterial recovery methods, we next addressed the potential interference of circulating cell-free DNA (cfDNA), which can cause false-positive results15. cfDNA levels vary depending on physiological and pathological conditions15. In healthy individuals, cfDNA is efficiently cleared, but in malignancies, inflammation, or excessive cell death, accumulation occurs15. To eliminate possible circulating DNA, a DNase treatment was applied after blood centrifugation and prior to DNA extraction. To assess DNase efficacy and confirm it did not degrade intracellular bacterial DNA, Cq values were measured in matched samples with and without DNase containing a clinical E. coli strain (106 CFU/mL) and extracted S. aureus DNA (10 ng), both in PBS and blood plasma (Fig. 2b). For E. coli, Cq values remained unchanged, indicating bacterial DNA integrity was preserved. In contrast, significant Cq shifts (5.8 in PBS, 5.3 in plasma) were observed for S. aureus DNA, confirming effective removal. These results demonstrate that DNase treatment is able to remove a substantial portion of circulating DNA without compromising bacterial detection.
After optimizing the pre-qPCR workflow, we sought to enhance sensitivity by incorporating a random priming amplification step (EquiPhi29 DNA Amplification Kit, Thermo Fisher). The effect of this procedure was evident in both the increased average DNA yield [E. coli (from 1.18 ng/µL to 86.1 ng/ul), P. aeruginosa (from 3.17 ng/µL to 87.7 ng/ul), S. aureus (from 1.17 ng/µL to 89.4 ng/ul)] (Fig. 2c) and the reduction in mean quantification cycle (Cq) values [E. coli (from 23.8 to 9.9 Cq), P. aeruginosa (from 24.2 to 9.5 Cq), S. aureus (from 28.8 to 14.8 Cq)] (Fig. 2d). Although the reduction in Cq values may seem disproportionate to the < 100-fold increase in total DNA yield, these measurements are not directly comparable. The random-priming enrichment step does not amplify the genome uniformly; instead, specific regions are preferentially and more efficiently amplified. As a result, the copy number of the qPCR target could increase substantially more than the overall DNA yield, providing a plausible explanation for the marked improvement in Cq values.
In the second approach, multiple probes labelled with the same fluorophore were designed to target the same gene, aiming to enhance sensitivity and signal strength. This multiplexing reduced Cq values E. coli (from 18.8 to 16.5 Cq), P. aeruginosa (from 20.8 to 15.3 Cq), S. aureus (from 27.5 to 23.9 Cq) compared to single-probe assays (Fig. 2d). Maximum fluorescence intensity also improved across all species analysed E. coli (ΔRn = 602,923.7), P. aeruginosa (ΔRn = 656,660.2), and S. aureus (ΔRn = 417,406.7)] (Fig. 2e). Nevertheless, the apparent Cq reduction should be interpreted with caution. The three-probe configuration produced substantially steeper amplification curves than the single-probe reactions, and standard Cq determination methods, including fixed fluorescence thresholds or regression-based algorithms could therefore yield reductions in Cq values that arise from curve shape rather than genuine assay improvements. As a result, the observed Cq decreases may overestimate the true benefit of probe multiplexing. Although the three-probe strategy clearly enhances fluorescence intensity and improves signal robustness, the precise magnitude of the Cq reduction should not be considered quantitatively definitive.
Once the LowLoad-qPCR parameters were established, we validated the method by comparing its performance with the conventional qPCR. Assays were performed in blood from healthy volunteers and PBS, each spiked with serial dilutions (100, 101, 102, 104 and 106 CFU/ml) of E. coli, P. aeruginosa, and S. aureus. Although bacterial recovery experiments gave better results with blood centrifugation, validation was carried out using both blood recovery protocols (blood centrifugation and Peps6-CaptoBAC kit). Previous studies showed that processing large volumes of blood introduces a high human DNA background, which interferes with qPCR when amplifying low pathogen numbers. For example, Vutukuru et al. showed the importance of removing human background for detection of pathogens at < 10 CFU/ml16,17 As Peps6 magnetic beads specifically capture bacteria, their use could reduce human DNA and improve LowLoad-qPCR sensitivity, even if pathogen recovery is lower.
To assess how these improvements impact detection performance, we conducted a broader comparison using blood samples spiked with decreasing bacterial concentrations. LowLoad-qPCR consistently outperformed conventional qPCR across all species and matrices. In plasma (the matrix with highest bacterial recovery) the improvement was marked both in terms of earlier amplification and in reducing nondetections. While conventional qPCR frequently failed to amplify at ≤ 102 CFU/mL, LowLoad-qPCR produced reproducible positive results across the full dilution series. At higher bacterial loads where both methods yielded measurable Cq values, LowLoad-qPCR showed a substantial quantitative advantage, improving amplification by 3.7 to 12.5 Cq cycles, depending on the microorganism (Fig. 3a–c). Given the exponential nature of qPCR, these ΔCq values correspond to an approximately 12- to 6000-fold increase in effective detectable signal. To evaluate linearity, we performed regression analyses of Cq values against log10-transformed bacterial concentrations in plasma. LowLoad-qPCR displayed slopes of − 2.06 to − 3.08 cycles/log10 with high linearity (R2 = 0.96–0.97) across E. coli, P. aeruginosa and S. aureus, indicating a well-preserved proportional concentration–response relationship. Conventional qPCR showed slopes between − 2.21 and − 3.48, but for E. coli and P. aeruginosa only two data points were available due to dropout at lower concentrations.

Validation of LowLoad-qPCR for direct detection of bloodstream pathogens. a–c Cq values obtained with LowLoad-qPCR and conventional qPCR in blood from healthy volunteers and phosphate-buffered saline (PBS), each spiked with decreasing bacterial concentrations (106, 106, 106, 106 and 100 CFU/mL) of E. coli, P. aeruginosa, and S. aureus. Assays were performed using two bacterial recovery methods: standard blood centrifugation and the Peps6-CaptoBAC kit. The red dashed line represents the detection threshold. Data are shown as mean ± s.d. Each data point represents an independent experimental replicate (n = 3). d, e heatmaps display the percentage of positive replicates (n = 3 per condition) for E. coli, P. aeruginosa, and S. aureus spiked into PBS, plasma, or bead-treated blood and tested at five bacterial concentrations (106, 104, 102, 101 and 100 CFU/mL). LowLoad-qPCR consistently improved detection, particularly in low-burden and complex matrices. Black dividing lines separate bacterial species.
Detection rates were measured for each species, using both conventional qPCR and LowLoad-qPCR. As shown in the heatmap (Fig. 3d,e), LowLoad-qPCR consistently achieved higher detection rates across all concentrations. This improvement was especially notable at lower bacterial loads (≤ 102 CFU/mL), where conventional qPCR frequently failed. These patterns were consistent across all three species, demonstrating that LowLoad-qPCR reliably extends detection to lower bacterial concentrations compared to conventional qPCR, being the detection threshold (concentration at which at least 1 of 3 replicates yielded detection) of 1 CFU/mL for the three species with LowLoad-qPCR and ≥ 10,000 CFU/mL for the three species with conventional qPCR when bacteria were recovered from plasma (Supplementary Fig. 1 and Table 1). Regarding the data obtained from this study we could confirm that both probe multiplicity and random priming enhance sensitivity and robustness, particularly in low-input samples.
This study has several limitations that should be acknowledged. First, our experiments were conducted under controlled laboratory conditions, which may not fully reproduce the complexity of real clinical samples. For instance, S. aureus can be internalized by blood cells, potentially reducing recovery after centrifugation. In our experimental model, samples were processed within minutes of inoculation, thereby minimizing the time available for internalization; however, this may not fully reflect in vivo infection dynamics, and further validation with patient samples is warranted. A further limitation is that only three technical PCR replicates were performed for each experiment. Previous work has shown that even identical PCR reactions can exhibit substantial between-replicate variability due to intrinsic stochastic effects18. Nevertheless, our results were consistent across replicates, supporting the reliability of the data, but additional studies with a larger number of technical replicates will be needed to confirm and refine these findings and to support future clinical implementation in real patient workflows.
Importantly, this performance was achieved with an estimated cost of less than 20€ per sample, remaining the cost lower than many commercial molecular diagnostics, nevertheless further reductions would enhance suitability for low-resource environments.
In conclusion, LowLoad-qPCR could offer a sensitive approach for the direct detection of bacterial pathogens in blood, with a shorter turnaround time compared to conventional blood culture and lower costs compared to existing commercial assays. Through optimized recovery, host DNA depletion, and signal-enhancing strategies, the assay enables reliable detection down to 1 CFU/mL levels beyond the reach of conventional methods. While the workflow is not yet optimized for point-of-care, it demonstrates a promising framework for accelerating culture-independent detection. Further development and validation will enable high-throughput adaptation and integration with genomic tools, advancing precision diagnostics in infectious disease.
Methods
Bacterial isolates
Clinical strains of E. coli, P. aeruginosa, and S. aureus were isolated from bacteremic patient samples. These samples were collected by the Microbiology Service at the University Hospital Virgen del Rocío (Seville, Spain) and used for the development and evaluation of the LowLoad-qPCR. Identification of the isolates was performed using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) (Bruker, Germany)19.
Blood specimen collection
Blood samples were collected from 15 healthy volunteers. All specimens were collected in 5 mL EDTA vacuum tubes (Greiner Bio-One K2EDTA VACUETTE). The tubes were stored at 4 °C for no more than 48 h prior to use. The use of human tissue samples in all methods was carried out in accordance with relevant guidelines and regulations. The experimental protocols were reviewed and approved by the Ethics Committee of the University Hospital Virgen Macarena-Virgen del Rocío (approval code: SICEIA-2024-000003). Informed consent was also obtained from all subjects and/or their legal guardians prior to participation.
DNA extraction in PBS medium
Serial dilutions (100, 101, 102, 104 and 106 CFU/mL) of bacteria were prepared in PBS (Phosphate-Buffered Saline) from a bacterial suspension at 0.5 McFarland and centrifuged at 9400g for 1 min. The pellet obtained was resuspended in 200 µL of lysozyme (20 mg/mL) and incubated at 37 °C with shaking to degrade the cell walls. DNA extraction was performed with QIAamp DNA Mini Kit QIAGEN (Germany). Following the manufacturer’s instructions, 20 µL of proteinase K was added and incubated at 56 °C for 10 min to digest proteins. Next, 200 µL of lysis buffer (AL) was added to facilitate cell lysis and DNA release, and the samples were mixed thoroughly by vortexing for 15 s. After incubation, 200 µL of 96–100% ethanol was added to the tube and the mixture was transferred to a spin column (in a 2 mL collection tube). The column was centrifuged at 9400g for 1 min. This step ensures DNA binding to the membrane in the column. The flow-through was discarded, and the column was placed in a clean collection tube. 500 µL of washing buffer 1 (AW1) was added to the column. It was centrifuged at 9400g for 1 min. The flow-through was discarded. Then, 500 µL of washing buffer 2 (AW2) was added and centrifuged at 20,000g for 3 min. Finally, the spin column was transferred to a clean 1.5 mL Eppendorf tube. 100 µL of elution buffer (AE) was added directly to the membrane. After a 1-min incubation at room temperature, the column was centrifuged at 20,000g for 1 min to elute the DNA. The eluted DNA was stored at -20 °C until further analysis.
DNA extraction from blood using centrifugation
From bacterial suspensions at 0.5 McFarland (108 CFU/mL), serial dilutions of bacteria were prepared using PBS. 50 µL of those dilutions were used to inoculate 5 ml of blood in order to obtain different final bacterial concentrations (100, 101, 102, 104 and 106 CFU/mL). Each blood tube was used to be inoculated with a specific bacterial concentration, used as a unique replicate. Immediately, the blood samples containing bacteria were centrifuged at 500×g for 10 min (Eppendorf Centrifuge 5702/R). Following centrifugation, the blood separated into plasma, the buffy coat, and the erythrocyte layer. Plasma was carefully collected and transferred into a 1.5 mL Eppendorf tube. Next, plasma was centrifuged at 5000×g for 10 min, and the bacterial pellet was resuspended in 45 µL of supernatant. Following this, DNase treatment was performed in accordance with the TURBO DNA-free Kit protocol (Thermo Fisher Scientific, Waltham, Massachusetts). For that purpose, 5 µL of 10X Turbo Buffer and 2 µL of Turbo DNase enzyme (2 U/µL) was added. The samples were incubated for 30 min at 37 °C. After incubation, 5 µL of DNase inactivation reagent was added and samples were incubated at room temperature for 5 min. Next, 200 µL of lysozyme (20 mg/mL) was added and incubated for 30 min at 37 °C with shaking. DNA extraction was performed with QIAamp DNA Mini Kit (QIAGEN, Germany). With the aim of adjusting the proportion of reagent volumes, 25 µL of proteinase K was added and incubated at 56 °C for 10 min. Next, 280 µL of buffer AL was added and the samples were mixed thoroughly by vortexing for 15 s. After incubation, 280 µL of 96–100% ethanol was added to the tube and the mixture was transferred to a spin column (in a 2 mL collection tube). The column was centrifuged at 9400g for 1 min. The flow-through was discarded, and the column was placed in a clean collection tube. At this point, the QIAamp DNA Mini Kit protocol (QIAGEN, Germany), as described in the previous section, was followed.
DNA extraction from blood using Peps6 beads
Serial dilutions of the bacterial culture were prepared in PBS and dispensed into 5 mL of blood (100, 101, 102, 104 and 106 CFU/mL). Bacterial capture from blood was performed following the Peps6-CaptoBAC kit protocol (ApoH technologies, La Grande Motte). As indicated in the manufacturer’s protocol, 5 mL of buffer TTGB 2X was added to the blood to reach a final concentration of 1X. Next, 20 µL of Peps6 magnetic beads was added (1013 beads/mL) and thoroughly resuspended by pipetting. The samples were incubated for 30 min at 37 °C under proper agitation. After incubation, Peps6 magnetic beads are expected to be bound to the bacteria present in the blood. The sample tubes were placed on a magnetic rack for 10 min and the supernatant was discarded. Two washes were performed with PBS. Peps6 magnetic beads-bacteria association was resuspended in 200 µL of lysozyme (20 mg/mL) and incubated at 37 °C for 30 min with shaking. Following the QIAamp DNA Mini Kit (QIAGEN, Germany) protocol, 20 µL of proteinase K was added and incubated at 56 °C for 10 min. After incubation, 200 µL of buffer AL was added to the samples, incubated at 70 °C for 10 min and mixed thoroughly by vortexing for 15 s to ensure dissociation of the bacteria from Peps6 magnetic beads. To remove Peps6 magnetic beads, the samples were centrifuged for 1 min at 1000×g, and the supernatant was transferred to a new 1.5 mL Eppendorf tube. At this point, the QIAamp DNA Mini Kit (QIAGEN, Germany) protocol was then continued.
DNA quantification
The DNA obtained was quantified by using Qubit 1X dsDNA HS Assay kit (Thermo Fisher Scientific, Waltham, Massachusetts) in conjunction with the Qubit Flex Fluorometer (Fisher Scientific, Waltham, Massachusetts).
Capture efficiency
Capture efficiency was determined by CFU counts on blood agar plates (BD Columbia Agar with 5% Sheep Blood). To this end, blood samples were spiked with bacterial dilutions of 10³ CFU/mL, and two different extraction methods (blood centrifugation and Peps6 magnetic beads), as described in previous sections were applied. Initially, 100 µL of a blood sample containing a bacterial dilution of 10³ CFU/mL were plated onto a blood agar plate as spiked bacterial concentration control. Non spiked blood specimens were plated in blood agar plates as negative control for blood contamination.
For the evaluation of bacterial capture efficiency by blood centrifugation, 100 µL of the total 1.5 mL of recovered plasma were plated onto a blood agar plate. In the case of Peps6 magnetic beads, the final magnetic beads-bacteria association obtained after incubation and washing steps was resuspended in 1 mL of PBS, and 100 µL of this suspension were plated onto agar plates. Plates were incubated overnight at 37 °C. Following incubation, CFUs were counted, and capture efficiency was subsequently calculated:
Capture efficiency= ([Bacterial concentration in plasma or beads] / [Bacterial concentration in blood]) × 100.
Random priming amplification (RPA)
The random priming amplification was performed following the EquiPhi29 DNA Amplification Kit (Thermo Fisher Scientific, Waltham, Massachusetts) manufacturer´s protocol. The reaction was prepared in a final volume of 20.5 µL, containing: 12 µL of extracted bacterial DNA template, 2 µL of 10X EquiPhi29 buffer, 2 µL of Exo-Resistant Random primers, which are primers resistant to the exonuclease activity of the polymerase (500 µM), 2 µL of 10 mM deoxynucleotide triphosphates Mix (dNTPs), 0.5 µL of 0.1 M Dithiothreitol (DTT), 1 µL of Pyrophosphatase (PPase) (0.1 U/µL) and 1 µL of EquiPhi29 Polymerase (10 U/mL). First, DNA template, reaction buffer, random primers, dNTP mix, and DTT were added in PCR microtubes. This mix was incubated at 95 °C for 3 min and then immediately placed on ice. After incubation, PPase and EquiPhi29 polymerase were added to the denatured reaction mix. The reaction was incubated at 42 °C for 90 min, followed by an enzyme inactivation step at 65 °C for 10 min.
Quantitative PCR (qPCR)
The presence of species-specific genes was analysed using quantitative PCR (qPCR). The selected qPCR targets were uidA gene for E. coli, gyrB gene for P. aeruginosa, and nuc gene for S. aureus. The selection of gene targets for species-specific detection was based on their high specificity, stable genomic presence, and widespread use in molecular diagnostics. Each target—uidA for E. coli, gyrB for P. aeruginosa, and nuc for S. aureus—was amplified using species-specific primers combined with three hydrolysis probes per gene, all labelled with the same fluorophore (FAM) to allow multiplex detection within a single channel (Supplementary Tables 2 and Fig. 2). Probe sequences were either newly designed or adapted from validated qPCR protocols (see references in Supplementary Table 2), and were selected to bind distinct, non-overlapping regions of the amplicon. This design aimed to improve detection sensitivity under limiting DNA conditions, while maintaining signal specificity. All oligonucleotides were synthesized by Microsynth AG (Balgach, Switzerland) and prepared at a final working concentration of 10 µM.
For conventional qPCR (using DNA template and one probe), the reaction mixture contained 5 µL of bacterial DNA template, 1 µL of forward and 1 µL of reverse primers (10 µM), 1 µL of probe (10 µM), 4 µL of LightCycler Multiplex DNA Master (Roche, Switzerland), and nuclease-free water to a final volume of 20 µL.
For LowLoad-qPCR (using random priming amplification product and three probes), the reaction mixture contained 5 µL of RPA product, 1 µL each of forward and reverse primers (10 µM), 1 µL of each of the three probes (10 µM)- resulting in a total of 3 µL of probes-, 4 µL of LightCycler Multiplex DNA Master (Roche, Switzerland), and nuclease-free water to a final volume of 20 µL. Moreover, in order to evaluate the individual advantages of the multi-probe design and the isothermal PCR, qPCR was also performed using RPA product with a single probe (5 µL of RPA product, 1 µL each of forward and reverse primers (10 µM), 1 µL of probe (10 µM), 4 µL of LightCycler Multiplex DNA Master, and nuclease-free water to a final volume of 20 µL) and using bacterial DNA with all three probes (5 µL of bacterial DNA template, 1 µL each of forward and reverse primers (10 µM), 1 µL of each of the three probes (10 µM)- resulting in a total of 3 µL of probes-, 4 µL of LightCycler Multiplex DNA Master and nuclease-free water to a final volume of 20 µL).
For all conditions (conventional qPCR, LowLoad-qPCR, qPCR using RPA product with a single probe, and qPCR using bacterial DNA with all three probes), negative amplification controls were included. These controls contained exactly the same reagents as the assay reactions, except that nuclease-free water was used instead of DNA or RPA product. As expected, no amplification was observed in any of the negative controls. These data were not included in the figures to avoid presenting non-informative results.
All reactions were performed using a QuantStudio 5 Dx Real-Time PCR System (Applied Biosystems). The thermal protocol consisted of an initial incubation at 95 °C for 5 min, followed by 35 amplification cycles of 96 °C for 15 s, 60 °C for 20 s, and 72 °C for 20 s.
Software
All figures were created using Biorender. Data and plotting were carried out using Microsoft Excel (v.16.59) and R (v.4.1.0).
Data availability
All data from this study are available within the article. Additional data that support the findings of this study are available from the corresponding authors on reasonable request.
References
Huang, M., Cai, S. & Su, J. The pathogenesis of sepsis and potential therapeutic targets. Int. J. Mol. Sci. 20 (21), 5376 (2019).
Kumar, A. et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 34, 1589–1596 (2006).
Alizadeh, M. et al. Rapid separation of bacteria from blood—Chemical aspects. Colloids Surf. B Biointerfaces. 154, 365–372 (2017).
Miller, J. M. et al. A guide to utilization of the microbiology laboratory for diagnosis of infectious diseases: 2018 update by the infectious diseases society of America and the American society for microbiology. Clin. Infect. Dis. 67 (6), e1–e94 (2018).
Nguyen, M. H. et al. Performance of the T2Bacteria panel for diagnosing bloodstream infections: A diagnostic accuracy study. Ann. Intern. Med. 170 (12), 845–852 (2019).
Vrettou, C. S. et al. Accuracy of T2 magnetic resonance assays as point-of-care methods in the intensive care unit. J. Hosp. Infect. 139, 240–248 (2023).
Paggi, R. et al. Accuracy and impact on patient management of new tools for diagnosis of sepsis: Experience with the T2 magnetic resonance bacteria panel. Pathogens 10 (9), 1132 (2021).
Samuel, L. Direct-from-Blood detection of pathogens: A review of technology and challenges. J. Clin. Microbiol. 61 (7), e00231–e00221 (2023).
Khine, A. A. et al. Evaluating the analytical sensitivity of Qvella’s FAST(TM) ID system for early detection and identification of bloodstream infection in whole blood 27th ECCMID. Vienna, Austria, April 22–25 (2017).
Vutukuru, M. R. et al. A rapid, highly sensitive and culture-free detection of pathogens from blood by positive enrichment. J. Microbiol. Methods. 131, 105–109 (2016).
Kustanovich, A. et al. Life and death of Circulating cell-free DNA. Cancer Biol. Ther. 20 (8), 1057–1067 (2019).
Kim, T. H. et al. Blood culture-free ultra-rapid antimicrobial susceptibility testing. Nature 632 (8026), 893–902 (2024).
Vutukuru, M. R. & Mitra, N. Theoretical assessment data for the binding of sepsis causing pathogens to ApoH beads. Data Brief. 13, 18–21 (2017).
Buchanan, C. M. et al. Rapid separation of very low concentrations of bacteria from blood. J. Microbiol. Methods. 139, 48–53 (2017).
Stroun, M. et al. Isolation and characterization of DNA from the plasma of cancer patients. Eur. J. Cancer Clin. Oncol. 23 (6), 707–712 (1987).
Vutukuru, M. R. et al. A rapid, highly sensitive and culture-free detection of pathogens from blood by positive enrichment. J. Microbiol. Methods. 127, 59–61 (2016).
Silkie, S. S., Tolcher, M. P. & Nelson, K. L. J. Reagent decontamination to eliminate false-positives in Escherichia coli qPCR. Microbiol. Methods. 72 (3), 275e282 (2008).
Lievens, A., Van Aelst, S., Van den Bulcke, M. & Goetghebeur, E. Simulation of between repeat variability in real time PCR reactions. PLoS ONE. 7 (11), e47112 (2012).
Bizzini, A. & Greub, G. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry, a revolution in clinical microbial identification. Clin. Microbiol. Infect. 16 (11), 1614–1619 (2010).
Acknowledgements
The authors thank the 15 volunteers and the technicians for providing the blood samples and performing the extractions, respectively.
Funding
This study has been funded by Instituto de Salud Carlos III (ISCIII) through the projects PI23/01760 and co-funded by the European Union. PGE is supported by the Subprograme PFIS, Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Ciencia, Innovación y Universidades, Spain (FI24/00301). JMOR is supported by the Subprograme Miguel Servet, Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Ciencia, Innovación y Universidades, Spain (CP24/00137). GMG has received funding from the Andalusia Government in the grants for human resources reinforcement in research activity (B-0006-2019).
Author information
Authors and Affiliations
Contributions
PGE: Formal analysis, Investigation, Methodology, Writing – original draft; GMG: Conceptualization, Formal analysis, Generation of figures and schemes, Funding acquisition, Writing – review and original draft; JMC: Resources, Supervision, Writing – review and editing; JAL: Resources, Writing – review and editing; JMOR: Conceptualization, Formal analysis, Investigation, Methodology, Supervision, Validation, Funding acquisition, Writing – review and editing. All authors discussed the results and edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gómez Estévez, P., Cisneros, J.M., Lepe, J.A. et al. LowLoad-qPCR as a novel clinical strategy for detecting low-load bacteremia. Sci Rep 16, 4163 (2026). https://doi.org/10.1038/s41598-025-34230-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-34230-w
