
Abstract
Sequence-dependent mechanical properties of DNA could play essential roles in nuclear processes by affecting histone-DNA interactions. Previously, we found that the DNA entry site of the first nucleosomes from the transcription start site (+ 1 nucleosome) in budding yeast enriches AA/TT steps, but not the exit site, and the biased presence of AA/TT in the entry site was associated with the transcription levels of yeast genes. Because AA/TT is a rigid dinucleotide step, we considered that AA/TT causes DNA unwrapping. However, our previous MNase-seq experiments with reconstituted nucleosomes left some doubt regarding this interpretation, owing to its high exonuclease activity. Furthermore, MNase cleavage did not provide direct evidence of its structural state. In this study, Förster resonance energy transfer (FRET) measurements were used to investigate salt-induced conformational changes in nucleosomal DNA containing AA/TT repeats at the entry site. We observed that the AA/TT region wrapped around the histone core was as likely as other DNA sequences at physiological salt concentrations. However, it unwrapped at a lower salt concentration, indicating weaker electrostatic interactions with the histone core. Ethidium-induced nucleosome disruption assay showed that the intercalator had greater access to DNA with AA/TT at the entry site. Taken together, these results suggest that AA/TT at the entry sites induces DNA unwrapping from the histone core on the promoter side, which may promote transcriptional activation in response to the approach of transcription-related proteins.
Similar content being viewed by others
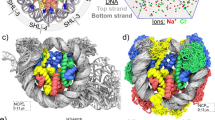


Introduction
Nucleosomes are the basic, structural units of eukaryotic chromatin used as packages of genetic materials. It is a protein-DNA complex in which approximately 147 bp of DNA wraps approximately 1.65 times around the core histones H2A, H2B, H3, and H41. Nucleosome positioning is strictly regulated in the genome in various ways, such as post-translational modifications and different histone variants, chromatin remodeling, and histone chaperoning2,3,4. DNA sequences are also fundamentally important for positioning, as they have intrinsically different structures and mechanical properties depending on their base composition5,6. For example, 10 bp periodic oscillation of AT-rich elements exists in nucleosome positioning sequences7,8,9. The shallow minor groove of the sequences leads to preferential interactions between histones and the DNA minor groove10,11. The DNA sequence affects the DNA wrapping/unwrapping equilibrium and consequently changes the accessibility of transcription factors in the nucleosome12. Nucleosomal DNA sequences are thus widely believed to be related to biological processes such as transcription and replication by changing the stability of histone-DNA interactions. However, how the particular DNA sequence element regulates a specific biological process remains unknown.
Regarding the transcription, nucleosomes act as physical barriers to the passage of RNA polymerase13,14,15. The + 1 nucleosome, the first nucleosome downstream of the transcription start site, is particularly crucial for transcriptional regulation because it is the first obstacle encountered by RNA polymerase16,17, resulting in the pause of RNA polymerase II at the promoter-proximal regions18. RNA polymerase II stalls at the entry site of the + 1 nucleosome in most Drosophila genes19,20. Epigenetic marks, such as histone variants (e.g. H2A.Z) and histone modifications, dynamically regulate the barrier’s strength in cells20,21,22,23. However, the role of the DNA sequence at the + 1 nucleosome is still poorly understood.
Previously, we analyzed the DNA sequences of + 1 nucleosomes of budding yeast to explore the role of DNA sequences in transcription24. We noticed that AA/TT dinucleotides were enriched in the entry sites but not in the exit sites. AA/TT content in the + 1 nucleosome of yeast was associated with gene transcription levels. We then conducted an MNase-seq experiment on 20 reconstituted + 1 nucleosomes containing AA/TT or TA repeat sequences. We found that AA/TT was more susceptible to MNase cleavage than other DNA sequences. Since AA/TT is known as a rigid DNA step that is resistant to DNA bending5,6, we hypothesized that the presence of AA/TT may cause DNA unwrapping at the entry site. However, there was an ambiguity about whether AA/TT affects the conformation of the DNA entry site because of the complexity of the enzymatic reaction, such as the simultaneous occurrence of end- and exo-type cleavage, strong sequence dependence of the enzyme, and multiple rounds of cleavage during the reaction time24,25. In particular, DNA entry sites with any sequence were quickly cleaved out by its exo-type cleavage activity in our experiment. Therefore, we were limited to discussing the sequence dependence of regions more than 37 bp inside the DNA end. In addition, the MNase assay can only assess the reactivity of MNase to nucleosomal DNA, which challenges the direct acquisition of structural information.
In this study, we used the Förster resonance energy transfer (FRET) method to directly observe the conformational state of DNA. We used nucleosomal DNA containing AA/TT or TA repeats at the entry/exit site of the Widom 601 sequence as a model system to evaluate the effects of the DNA sequence. We monitored the structural changes via salt-induced nucleosomal DNA unwrapping to examine how electrostatic interactions were affected by the sequence at the entry site. We also conducted an intercalator-induced nucleosome disruption assay to evaluate the sequence-dependent DNA accessibility. These analyses provide insights into the molecular mechanism underlying the role of the DNA sequence element in gene activation via the + 1 nucleosome.
Results
The entry site with AA/TT adopts a regular wrapped conformation, but the histone-DNA interactions are weak
Previously, we found an abundance of AA/TT in the first 15–20 bp of the entry site24. To analyze the effect on the DNA structure of the sequence in this region, we designed nucleosomal DNA containing 15 bp AA/TT or TA repeats as a control on one side of the Widom 601 sequence9. To measure DNA unwrapping with FRET, we added a donor fluorescent dye at the position near the dyad and an acceptor fluorescent dye at one end of the DNA. The positions of the dyes and the substituted sites are shown in Fig. 1a. The exact sequences are listed in Tables S1 and S2. We refer to the nucleosome containing Widom 601 as 601 NCP, and those containing AA/TT and TA repeat sequences as AA and TA NCPs, respectively. As will be explained later, we prepared another variant with a long but discontinuous AA/TT repeats (referred to as ImperfectAA).

FRET measurement of the DNA unwrapping of NCPs with different DNA sequences at the entry site. (a) The DNA sequences and the positions of fluorescent dyes for the FRET analysis are shown in the left panel. Green and yellow stars indicate the positions of the donor and acceptor dyes, respectively. The distribution of fluorescent dyes, calculated by FPS software25, are denoted as green and yellow dots with the structure of the nucleosome (PDB ID: 3lz0) in the middle and the right panel. The positions mutated to AA/TT or TA repeats are denoted in red in the middle panel. In the right panel, AA/TT repeats and C/G of Imperfect AA NCP are shown in red and cyan, respectively. (b) The mean distance and standard deviation of the distances obtained from the FRET measurement at 0 mM KCl. (c,d) The mean distance (c) and standard deviation (d) of the distances titrated by KCl for NCPs. The inset graph shows the zoom-in of the 0–300 mM range.
To examine FRET, we used a time-correlated single photon counting (TCSPC) method to measure the fluorescence lifetime. A representative fluorescence decay curve is shown in Fig. S1. This method is suitable for analyzing molecules with different conformations exhibiting multiple fluorescence lifetimes. We assumed the distance between the two dyes followed a Gaussian distribution and calculated their mean distance (\(\:<{R}_{DA}>\)) and the standard deviation (\(\:{\sigma\:}_{DA}\)). This assumption was used to analyze the sub-ensemble of data in single-molecule FRET using the TCSPC module25,26. The \(\:<{R}_{DA}>\) indicates the degree of DNA unwrapping. The larger \(\:{\sigma\:}_{DA}\) indicates the local conformational flexibility of the DNA. The breaks in histone-DNA interactions accompanied by the DNA unwrapping cause the \(\:{\sigma\:}_{DA}\) increase.
Figure 1b (left) shows \(\:<{R}_{DA}>\) for 0 mM KCl. NCPs of 601, TA, and AA were in the same order. However, AA NCP had slightly larger \(\:<{R}_{DA}>\:\)than 601 and TA NCPs. This might reflect a small conformational deviation in the AA NCP. The value of \(\:{\sigma\:}_{DA}\) of AA NCP (Fig. 1b, right) was between 601 and TA NCPs (around 1.0 nm). To evaluate the degree of \(\:{\sigma\:}_{DA}\) only resulting from the fluctuations of the linker to the dyes, we obtained simulated \(\:{\sigma\:}_{DA}\) using the AAV method25. It was 1.3 nm assuming that the Alexa Flour 647 and Alexa Flour 488 dyes with 10-atoms-flexible-linkers are separated by 5 nm. This value is comparable to the experimental values, indicating that DNA-histone interactions in all variants of the Widom 601 sequence were strong enough to maintain the wrapped conformation. The slight variations among the sequences probably resulted from differences in the intrinsic flexibility of DNA.
Next, KCl titration was conducted to examine the effects of salt concentration (Fig. 1c,d). A moderate salt concentration can weaken the repulsion between the negatively charged phosphate backbones, allowing the end of the DNA to be closer to the second turn of the nucleosomal DNA. Indeed, we observed a decrease in \(\:<{R}_{DA}>\) by approximately 0.2 nm for all sequences from 0 to 150 mM KCl (see the inlet graph of Fig. 1c). However, \(\:{\sigma\:}_{DA}\) increased by approximately 0.2 nm at 150 mM KCl (Fig. 1d), indicating that reduced interactions of histone-DNA caused a gradual increase in DNA flexibility. This salt-dependent behavior was observed in the salt concentration range for all NCPs. However, sequence dependence of DNA unwrapping emerged at concentrations higher than 150 mM. \(\:<{R}_{DA}>\) of AA NCP showed a sharp rise from 250 mM KCl. \(\:{\sigma\:}_{DA}\) of AA NCP also increased rapidly. In contrast, \(\:<{R}_{DA}>\) of 601 and TA NCPs were stable until around 400 mM KCl and \(\:{\sigma\:}_{DA}\) of them increased slowly. In conclusion, the unwrapping of the AA NCP at a lower salt concentration than that of the other NCPs indicates that AA/TT repeats reduce the electrostatic interactions between proteins and DNA more effectively than other sequences.
To evaluate whether AA/TT directly weakened the interactions at the interface between histones and DNA, we designed a variant called ImperfectAA (Fig. 1a). ImperfectAA NCP had more AA/TT steps, but C/G was inserted at the interaction sites between histones and DNA. Our FRET analysis (Fig. 1b–d) showed that the deviations of \(\:<{R}_{DA}>\) and \(\:{\sigma\:}_{DA}\) of ImperfectAA NCP from those of 601 and TA NCPs were small in the range of 0-150 mM, as seen in AA NCP. However, the increases in \(\:<{R}_{DA}>\) and \(\:{\sigma\:}_{DA}\) of ImperfectAA NCP at a higher salt concentration were less than those of AA NCP, especially at a salt concentration lower than 500 mM KCl. This indicates that the sequence at the protein-DNA interface is crucial for maintaining proper interactions.
In summary, at a low salt concentration of around 150 mM KCl, which corresponds to the physiological salt concentration, the AA/TT repeat at the entry site can be incorporated into the regular nucleosomal DNA conformation with a slightly deviated conformation. However, the entry sites with AA/TT repeats underwent salt-induced DNA unwrapping more readily than those without the repeats, indicating that the AA/TT element weakened the electrostatic histone-DNA interactions.
AA/TT at the entry site makes nucleosomes highly intercalator-susceptible
We could not obtain information on the accessibility of the DNA entry site from our previous MNase cleavage experiments24, which suffered from the intense exonuclease activity. To complement this, we employed an ethidium intercalation method to assess DNA accessibility. Ethidium is a classic double-stranded DNA-intercalating reagent with low sequence specificity. The binding of ethidium bromide disrupts nucleosome structure, which initially releases one copy of the H2A-H2B dimer27. This release is induced by the intercalation to the core particle DNA28. We added different concentrations of ethidium bromide to 601, TA, and AA NCPs (Fig. 2). In this experiment, these three NCPs were selected because AA and TA NCPs have the same number of substitutions against Widom 601. We analyzed the fraction of intact NCPs using native polyacrylamide gel electrophoresis after five hours of incubation at room temperature.

Ethidium-induced nucleosome disruption. (a) Structure of the 601 NCP (PDB ID: 3lz0). The entry/exit site of 601 NCP was mutated to either AA or TA repeat sequence and is shown in red. (b) Fractions of the intact NCPs (θ) after five hours of incubation of 601, AA, and TA NCP with different concentrations of ethidium. The mean value and the standard deviation obtained from the three repetitive experiments are shown. (c) Representative native PAGE gel images. The band of intact AA NCPs disappears more quickly than those of 601 and TA NCPs. The images were cropped. Whole gel images for three repetitive experiments are shown in Fig. S5.
The intact AA NCP decreased at intercalator concentrations lower than 601 and TA, whereas 601 and TA NCPs did not show significant differences, as shown in Fig. 2b,c. The band shift of NCPs in native polyacrylamide gel electrophoresis can be owing to the different positioning of the histone octamer, unwrapping of DNA ends, and/or dissociation of the nucleosome components27,29,30. In addition, the binding of ethidium to NCP could cause a band shift due to the neutralization of the negative charge of the DNA by ethidium intercalation. Considering the above, our band shift is probably due to a combination of these phenomena. To rule out the possibility of ethidium having a higher affinity for AA/TT, we measured the affinity of ethidium for the bare AA/ TT repeat DNA sequence by visible-light absorbance. This was slightly lower than that of 601 and TA repeats (Fig. S2). Thus, our data indicate that AA/TT at the entry site makes ethidium more accessible to the DNA by inducing local nucleosomal DNA unwrapping.
Discussion
The DNA sequence is essential not only because it is the source of the protein sequences but also because it provides the mechanical code through its sequence-dependent conformation and flexibility of DNA5,6,31,32. The AA/TT step is an inherently stiff sequence element, whearas the TA step is flexible5,6,33. Previously we revealed that AA/TT steps are abundant at the entry site of the + 1 nucleosome, and further bioinformatics analysis showed that the presence of AA/TT at the entry site is associated with the transcriptional level24. However, its effect on nucleosome structure remains unclear. In the present study, we found that the entry site of DNA adopts a typical well-wrapped conformation, with a slight conformational deviation at physiological salt concentrations. However, DNA unwrapping of the entry site containing AA/TT repeats occurs more easily than in other sequences in a salt-sensitive manner. This suggests that in the AA/TT region, the electrostatic interactions between histones and DNA are weak; therefore, they can be easily perturbed by binding to other molecules. Indeed, the presence of AA/TT increased the binding of an intercalator.
The entry site of nucleosomes may function as a transcriptional regulator because it is the first barrier to the transition of polymerase II and is located near the H3, H4, and H2A histone tails that are subjected to epigenetic modifications. A recent cryo-EM analysis illustrated how RNA polymerase II passes through a nucleosome, in which two of the four significant barriers exist at the entry site: SHL(-6) and SHL(-5)15. The barrier to RNA polymerase passage in cells can be relaxed in several ways, such as by post-translational modifications of histones and the incorporation of histone variants into nucleosomes, etc35,36. Some histone tail modification enzymes use the entry-exit site as their binding site37,38,39,40. For example, Set2, which catalyzes histone H3 K36 methylation, binds to the unwrapping state of the nucleosome by associating with the entry-exit sites, including the residues on H3 αN-helix37,39. The DNA unwrapped is essential for the binding of these enzymes to nucleosomes.
Finally, we proposed a model for how the presence of AA/TT at the entry site of the + 1 nucleosome could influence gene activation (Fig. 3). We showed that nucleosomes with AA/TT at the entry site formed regular, wrapped conformation at physiological salt concentrations. This indicates that during DNA storage, the DNA in the + 1 nucleosomes is protected from various stresses, such as UV radiation, as in other sequences. However, the weak interaction of such + 1 nucleosomes at the promoter side, which is close to transcription-related proteins, such as RNA polymerase II and histone modification enzymes, may promote the transcription reactions via DNA unwrapping. The nucleosomal DNA unwrapping in the + 1 nucleosome has also been pointed out to trigger FACT recruitment to the transcribed chromatin, and it spreads toward downstream nucleosomes with the help of Chd134. We believe DNA sequences and other cellular factors, such as histone chaperones and chromatin remodelers, would coordinately regulate the initial stage of transcription in this way. Future studies, including the measurement of transcription rate in nucleosomes with varying AA/TT content at the entry sites, will elucidate the biological consequences.

Proposed model of the role of the AA/TT element via the + 1 nucleosome during transcription. AA/TT at the entry site of the + 1 nucleosome could cause the asymmetric unwrapping of the DNA from the promoter side when the transcription-related proteins approach it and might eventually stimulate transcription from the proper direction.
Methods
Preparation of DNAs
Unlabeled DNA fragments containing Widom 601 or its derivatives (Table S1) were prepared by PCR amplification using the high-fidelity DNA polymerase KOD -Plus- (TOYOBO). To prepare fluorescent dsDNA (Table S2), Alexa Fluor 488- and Alexa Fluor 647-labeled oligonucleotides corresponding to 91 and 55 bp, respectively, with four nucleotide overhangs at each end, were individually synthesized (Thermo Fisher Scientific). Then, the complementary pairs of the oligonucleotides were annealed and ligated with T4 DNA Ligase (Nippon Gene) at 16 ºC. Both unlabeled and fluorescently labeled dsDNA were purified by native polyacrylamide gel electrophoresis using a Prep Cell system (Bio-Rad).
Preparation of NCPs
NCPs with different sequences were prepared as previously described41. His-tagged human histones H2A, H2B, H3.1, and H4 were individually expressed in E. coli cells and purified by affinity chromatography using Ni-NTA agarose (QIAGEN). After removal of the His-tags with thrombin treatment, the histones were purified using Mono S column chromatography (Cytiva) and freeze-dried. To prepare the histone octamer, equal amounts of H2A, H2B, H3.1, and H4 were mixed in a buffer containing 50 mM Tris-HCl (pH 7.5), 7 M guanidine-HCl, and 20 mM 2-mercaptoethanol and dialyzed with a refolding buffer containing 10 mM Tris-HCl (pH 7.5), 2 M NaCl, and 2 mM 2-mercaptoethanol. The octamer was purified on a Hiload 16/60 Superdex200 prep grade column (Cytiva). The octamer was then mixed with DNA fragments in a high-salt buffer containing 10 mM Tris-HCl pH 7.5, 2 M KCl, 1 mM EDTA, and 1 mM DTT. In this step, a 1:9 mixture of fluorescently labeled dsDNA and unlabeled dsDNA was used to prepare fluorescently labeled NCP. The NCPs were reconstituted using the salt dialysis method, in which the salt concentration was gradually reduced to 250 mM KCl. Finally, the reconstituted NCPs were purified using a Prep Cell system. The purity of the obtained NCP was confirmed using 6 % native polyacrylamide gel electrophoresis (Fig. S3).
FRET analysis
Forty microliters of 2.5 µM of the dual fluorescently labeled NCPs were titrated with KCl by mixing them with 2.5 µL of 1.5, 2.5, or 3.5 M KCl in 20 mM Tris-HCl (pH 7.5) and 1 mM DTT. A 1.00 MHz repetitive pulsed laser at a wavelength of 467 nm (Horiba NanoLED) was applied to them. The timing of the fluorescence emission was recorded up to 10,000 photon counts at a wavelength of 525 nm with a 16 nm bandpass in a DeltaFlex TCSPC system (Horiba Jobin Yvon Inc).
Fluorescence intensity decay with FRET (\(\:{I}_{with\:FRET}\left(t\right)\)) and without FRET (\(\:{I}_{without\:FRET}\left(t\right)\)) is generally expressed as follows42:
where \(\:{I}_{0}\) is the intensity at t = 0, \(\:{\tau}_{Di}\) is the \(\:i\)th lifetime of the donor without FRET, and \(\:P\left({R}_{DA}\right)\) is the probability distribution of the distance between the fluorophores, \(\:{R}_{DA}\). As we explain later, we used two lifetimes of the donor without FRET, \(\:{\tau}_{Dl}\) and \(\:{\tau}_{Ds}\). The fractional amplitudes of the lifetimes corresponding to \(\:{\tau}_{Dl}\) and \(\:{\tau}_{Ds}\) are \(\:{\alpha}_{l}\) and \(\:{\alpha}_{s}\), respectively. Equations (1) and (2) can be written as
\(\:{R}_{0}\) for the Alexa Fluor 488 and Alexa Fluor 647 pair was set to 5.5 nm. It was assumed that the distances followed a Gaussian distribution. \(\:P\left({R}_{DA}\right)\) can be expressed as follows:
where \(\:{<R}_{DA}>\) and \(\:{\sigma}_{DA}\) are the mean and the standard deviation of \(\:{R}_{DA}\). To account for the variation in the incorporation rate of the acceptor dye or the reconstitution rate of the NCPs during preparation, we used the following fitting function for the decay, \(\:I\left(t\right)\):
where, \(\:{f}_{with\:FRET}\) and \(\:{f}_{without\:FRET}\) are the fractional amplitudes of the components with and without FRET, respectively42. We applied the values at 0 mM KCl to all the titration data. The values of \(\:{f}_{without\:FRET}\) were approximately 0.05, and did not seem to vary significantly with the sequence.
In addition, we measured the fluorescence decay of 2.5 µM of 601 NCP labeled only with the donor using the same procedure as for the dual-labeled NCPs to obtain the lifetimes without FRET. Because a part of Alexa Fluor 488 molecules on the protein surface is reported to have a shorter lifetime due to fluorescence quenching, we fitted each curve with the following double exponential model43:
where \(\:{\tau}_{Dl}\) and \(\:{\tau}_{Ds}\) are the longer and shorter lifetimes, respectively, and \(\:{\alpha}_{l}\) and \(\:{\alpha}_{s}\) correspond to the fractional amplitudes of \(\:{\tau}_{Dl}\) and \(\:{\tau}_{Ds}\), respectively. \(\:{\alpha}_{l}\) was about 0.91. The obtained \(\:{\tau}_{Dl}\), \(\:{\tau}_{Ds}\) and \(\:{\alpha}_{l}\) are shown in Fig. S4. The lifetime of Alexa Fluor 488 in the aqueous solution was reported to be 4.1 ns by the manufacturer (Thermo Fisher Scientific), and the value is almost the same as our \(\:{\tau}_{Dl}\).
Our own Python scripts were used to calculate these values and create plots. Inter-dye distances were computed using the AAV method with FPS software25. We used the dye-linker parameters for Alexa Fluor 488 and Alexa Fluor 647, reported in reference44.
Ethidium-induced nucleosome disruption assay
Two microliters of 50 ng/µL unlabeled NCP were mixed with 2 µL of 0–30 pg/µL ethidium bromide (Nakalai tesque) in 20 mM Tris-HCl (pH 7.5) and 1 mM DTT and was incubated at room temperature for 5 h. Electrophoresis on a 6 % native polyacrylamide gel was performed in 0.5 ✕ TBE (45 mM Tris-Borate, 1 mM EDTA) buffer for 60 min at 150 V. After staining the gel with ethidium bromide, images were captured using a FluorChem FC2 Imager (ProteinSimple). Band intensities were quantified using the Band/Peak Quantification macro of ImageJ45.
Data availability
Data are available at a reasonable request to T. S. and H. Kono.
Change history
20 March 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-93097-z
References
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997).
Jiang, C. & Pugh, B. F. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 10, 161–172 (2009).
Teif, V. B. & Clarkson, C. T. Nucleosome positioning. Encyclopedia Bioinf. Comput. Biology: ABC Bioinf. 2, 308–317 (2019).
Struhl, K. & Segal, E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 20, 267–273 (2013).
Fujii, S., Kono, H., Takenaka, S., Go, N. & Sarai, A. Sequence-dependent DNA deformability studied using molecular dynamics simulations. Nucleic Acids Res. 35, 6063–6074 (2007).
Packer, M. J., Dauncey, M. P. & Hunter, C. A. Sequence-dependent DNA structure: dinucleotide conformational maps. J. Mol. Biol. 295, 71–83 (2000).
Brogaard, K., Xi, L., Wang, J. P. & Widom, J. A map of nucleosome positions in yeast at base-pair resolution. Nature 486, 496–501 (2012).
Satchwell, S. C., Drew, H. R. & Travers, A. A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 191, 659–675 (1986).
Lowary, P. T. & Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 (1998).
Chua, E. Y. D., Vasudevan, D., Davey, G. E., Wu, B. & Davey, C. A. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 40, 6338–6352 (2012).
Rohs, R. et al. The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253 (2009).
North, J. A. et al. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res. 40, 10215–10227 (2012).
Gaykalova, D. A. et al. Structural analysis of nucleosomal barrier to transcription. Proc. Natl. Acad. Sci. U S A. 112, E5787–E5795 (2015).
Chen, Z. et al. High-resolution and high-accuracy topographic and transcriptional maps of the nucleosome barrier. Elife 8, e48281 (2019).
Kujirai, T. et al. Structural basis of the nucleosome transition during RNA polymerase II passage. Sci. (1979). 362, 595–598 (2018).
Rhee, H. S. & Pugh, B. F. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483, 295–301 (2012).
Farnung, L. Nucleosomes unwrapped: structural perspectives on transcription through chromatin. Curr. Opin. Struct. Biol. 82, 102690 (2023).
Jimeno-González, S., Ceballos-Chávez, M. & Reyes, J. C. A positioned + 1 nucleosome enhances promoter-proximal pausing. Nucleic Acids Res. 43, 3068–3078 (2015).
Weber, C. M., Ramachandran, S. & Henikoff, S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell. 53, 819–830 (2014).
Mavrich, T. N. et al. Nucleosome organization in the Drosophila genome. Nature 453, 358 (2008).
Bagchi, D. N., Battenhouse, A. M., Park, D. & Iyer, V. R. The histone variant H2A.Z in yeast is almost exclusively incorporated into the + 1 nucleosome in the direction of transcription. Nucleic Acids Res. 48, 157–170 (2020).
Bowman, G. D. & Poirier, M. G. Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 115, 2274–2295 (2015).
Lorch, Y., Maier-Davis, B. & Kornberg, R. D. Histone acetylation inhibits RSC and stabilizes the + 1 nucleosome. Mol. Cell. 72, 594–600 (2018).
Luo, D. et al. MNase, as a probe to study the sequence-dependent site exposures in the + 1 nucleosomes of yeast. Nucleic Acids Res. 46, 7124–7137 (2018).
Kalinin, S. et al. A toolkit and benchmark study for FRET-restrained high-precision structural modeling. Nat. Methods 9, 1218–1225 (2012).
Gansen, A. et al. High precision FRET studies reveal reversible transitions in nucleosomes between microseconds and minutes. Nat. Commun. 9, 4628 (2018).
McMurray, C. T. & Van Holde, K. E. Binding of ethidium bromide causes dissociation of the nucleosome core particle. Proc. Natl. Acad. Sci. 83, 8472–8476 (1986).
McMurray, C. T., Small, E. W. & van Holde, K. E. Binding of ethidium to the nucleosome core particle. 2. Internal and external binding modes. Biochemistry 30, 5644–5652 (1991).
Arimura, Y., Tachiwana, H., Oda, T., Sato, M. & Kurumizaka, H. Structural analysis of the hexasome, lacking one histone H2A/H2B dimer from the conventional nucleosome. Biochemistry 51, 3302–3309 (2012).
Takizawa, Y. et al. Cryo-EM structure of the nucleosome containing the ALB1 enhancer DNA sequence. Open. Biol. 8, 170255 (2018).
Yamasaki, S., Terada, T., Kono, H., Shimizu, K. & Sarai, A. A new method for evaluating the specificity of indirect readout in protein–DNA recognition. Nucleic Acids Res. 40, e129–e129 (2012).
Harteis, S. & Schneider, S. Making the Bend: DNA tertiary structure and Protein-DNA interactions. Int. J. Mol. Sci. 15, 12335–12363 (2014).
Nelson, H. C. M., Finch, J. T., Luisi, B. F. & Klug, A. The structure of an oligo(dA).Oligo(dT) tract and its biological implications. Nature 330, 221–226 (1987).
Jeronimo, C. et al. FACT is recruited to the + 1 nucleosome of transcribed genes and spreads in a Chd1-dependent manner. Mol. Cell. 81, (2021).
Li, B., Carey, M. & Workman, J. L. The role of chromatin during transcription. Cell 128, 707–719 (2007).
Petesch, S. J. & Lis, J. T. Overcoming the nucleosome barrier during transcript elongation. Trends Genet. 28, 285–294 (2012).
Endo, H. et al. Nucleosome surface containing nucleosomal DNA entry/exit site regulates H3-K36me3 via association with RNA polymerase II and Set2. Genes Cells. 17, 65–81 (2012).
Chio, U. S. et al. Cryo-EM structure of the human sirtuin 6–nucleosome complex. Sci. Adv. 9, eadf7586 (2023).
Bilokapic, S. & Halic, M. Nucleosome and ubiquitin position Set2 to methylate H3K36. Nat. Commun. 10, 1–9 (2019).
Spangler, C. J. et al. Structural basis of paralog-specific KDM2A/B nucleosome recognition. Nat. Chem. Biol. 19, 624–632 (2023).
Kujirai, T. et al. Methods for preparing nucleosomes containing histone variants. Methods Mol. Biol. 1832, 3–20 (2018).
Lakowicz, J. R. Time-resolved energy transfer and conformational distributions of biopolymers. in Principles of Fluorescence Spectroscopy 477–506 (Springer, Boston, MA, 2006) https://doi.org/10.1007/978-0-387-46312-4_14.
Peulen, T. O., Opanasyuk, O. & Seidel, C. A. M. Combining graphical and analytical methods with molecular simulations to analyze time-resolved FRET measurements of labeled macromolecules accurately. J. Phys. Chem. B. 121, 8211–8241 (2017).
Höfig, H., Gabba, M., Poblete, S., Kempe, D. & Fitter, J. Inter-dye distance distributions studied by a combination of single-molecule FRET-filtered lifetime measurements and a weighted accessible volume (wAV) algorithm. Molecules 19, 19269–19291 (2014).
Ohgane, K. & Yoshioka, H. Quantification of gel bands by an image J macro, band/peak quantification tool. Preprint at. https://doi.org/10.17504/protocols.io.7vghn3w (2019).
Acknowledgements
We thank M. Koyama and W. Nagakura for the initial sample preparations. We also thank W. S. Chan, H. Ishida, A. Kumar, A. Matsumoto, and S. Sakuraba for their valuable discussions. This work was supported by JSPS KAKENHI (grant numbers JP18H05534, JP22K06176, and JP24H00884), and BINDS from AMED (grant numbers JP23ama121024 and JP23ama121009).
Author information
Authors and Affiliations
Contributions
T.S., D.L., H. Kurumizaka, and H. Kono planned and designed the research project. T.S., S.S., and J.K. prepared the samples. T.S. performed the native PAGE experiment, and T.S., M.Y., and K.A. performed the FRET experiment. T.S. and H. Kono wrote and reviewed the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in Figure 1 where the range of X in the inlet of panel (c) was not 0-300 mM as explained in the legend. The original Figure 1 and accompanying legend appear below. The Supplementary Information 1 file published with this Article contained errors. The original Supplementary Information 1 file is provided below. In addition, in the Results’ section, under the subheading “AA/TT at the entry site makes nucleosomes highly intercalator-susceptible”, “To rule out the possibility of ethidium having higher affinity for AA/TT, we measured the affinity of ethidium for the bare AA/TT repeat DNA sequence by UV absorbance.” now reads: “ To rule out the possibility of ethidium having a higher affinity for AA/TT, we measured the affinity of ethidium for the bare AA/ TT repeat DNA sequence by visible-light absorbance.”
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sunami, T., Luo, D., Sato, S. et al. FRET analysis of the unwrapping of nucleosomal DNA containing a sequence characteristic of the + 1 nucleosome. Sci Rep 15, 2169 (2025). https://doi.org/10.1038/s41598-025-86075-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86075-y
