
Abstract
Endometriosis (EM) is a chronic disease that can cause pain and infertility in patients. As is well known, immune cell infiltrations (ICIs) play important roles in the pathogenesis of EM. However, the pathogenesis and biomarkers of EM that can be used in clinical practice and their relationship with ICIs still need to be elucidated. The gene expression datasets of EM and the healthy control were obtained from the Gene Expression Omnibus (GEO). To identify the central modules and explore the correlation between the gene network and EM, weighted gene co-expression network analysis (WGCNA) was executed. The hub genes were screened using machine learning. The qRT-PCR results showed that only CHMP4C and KAT2B differentially expressed in ectopic tissues compared to the normal. Subsequently, the samples were clustered based on the expression of CHMP4C and KAT2B. Depending on the differential expression genes of the two 2rG Clusters, the samples were divided into two gene Clusters. Significant differences in immune cell infiltrations were observed among the two 2rG Clusters and the two gene Clusters. Furthermore, varied immune checkpoint genes were shown to be correlated with EM. The qRT-PCR results showed that the two genes were significantly related to the ICI genes in EM. Hub genes CHMP4C and KAT2B are involved in the pathogenesis of EM by regulating ICI.
Similar content being viewed by others

Introduction
Endometrial glands and stroma are present outside of the uterine cavity in endometriosis (EM), which is thought to be a persistent, painful, and oestrogen-dependent condition1. The ectopic tissue is predominantly located in the pelvic peritoneum and ovaries, whose development promotes inflammation and adhesions which lead to pain or infertility2,3. Globally, 5–10% of women within reproductive years are affected by EM; 50–80% of affected women have pelvic pain and 50% of them are diagnosed with infertility4. Usually, EM is clinically divided into three types, peritoneal (superficial), ovarian and adenomyosis externa (deep)5,6,7. Laparoscopy combined with histological examination is currently regarded as the gold standard for EM diagnosis. However, laparoscopic is associated with risks such as trauma, adhesion, and decreased fertility, and it fails to achieve the goal of early detection8. According to statistics, the average interval from the onset of endometriosis symptoms to diagnosis is 6.7 years9, and in some cases, it may even take as long as 12 years to make a diagnosis and implement corresponding treatment measures10, resulting in the missed opportunity of the optimal treatment time. Therefore, it is essential to seek non-invasive biomarkers for the early diagnosis of EM.
Retrograde menstruation has been regarded as a reliable and intuitive hypothesis of the etiology. It was proposed that endometrial cells may be implanted in the peritoneal cavity via lymphatic or hematogenous dissemination11,12. However, the exact pathogeny and molecular mechanism remain to be mysterious. The genetic/epigenetic theory postulates that genetic and/or epigenetic changes occur in ectopic endometrial cells that are exposed to abnormal environments, such as inflammation, oxidative stress and immunology cytokines13. It was revealed that inflammation upregulated the expression of progesterone receptor (PR)-C, which antagonized the anti-inflammatory PR-B. The researchers then inferred that inflammation may promote the progression of EM by upregulating PR-C levels14. There are also studies indicating that apolipoprotein E and peroxisome proliferator-activated receptor γ, as well as phospholipase A2 group II/V and the hydroxysteroid 11b-dehydrogenase were up-regulated markedly in ectopic tissue compared with eutopic tissue15. Researchers also propose that long non coding RNA (lncRNA) can be associated with the occurrence and development of EM16,17. However, the above researches mainly focused on the role of single genetic or epigenetic changes in EM, which inevitably limited the understanding of the intrinsic relationships between different genes in EM. Bioinformatics analysis was utilized in this study to compare the gene expression profiles of EM patients and healthy individuals to identify more candidate genes and to understand the intrinsic relationships between genes in EM more comprehensively. Immune cell infiltration (ICI) has been considered an essential process in the pathogenesis of EM. Numerous studies showed that immune cells and cytokines secreted by them promoted inflammation and led to the lesions in EM18,19. It was shown that neutrophil infiltration significantly increased in early ectopic tissues to secrete IL-8 to induce inflammation20,21. It was found that the immune system, including various immune cells and their secreted cytokines, was also closely related to the development of EM22.Thus, to regard the immune mechanisms of EM, ICI was also taken into consideration and analyzed.
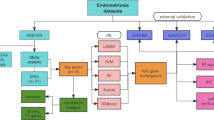
Bioinformatics analysis has been recently considered an effective method for analyzing candidate genes. The expression data of GSE6364, GSE7305, and GSE51981 were first downloaded from Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs) are identified through the “limma” package in R. The weighted gene co-expression network analysis (WGCNA) was then used to identify gene modules related to EM. The 2rG gene signature consisting of CHMP4C and KAT2B was thereafter constructed by machine learning (Lasso, RF and SVM) and qRT-PCR, and this signature was considered as the EM biomarker and used to further divide EM samples into two 2rG clusters by unsupervised clustering. To verify the above clustering, consensus clustering was conducted based on the DEGs between the 2rG clusters. In order to further verify the relationship between these biomarkers and EM, in addition to further examining the ICI characteristics of each cluster, the correlation between 2 hub genes and ICI was also verified through qRT-PCR.
Materials and methods
Data collection and preparation
The Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) provided the gene expression datasets in this study. All of the datasets are available for free download. We searched “endometriosis” in the GEO and acquired three eligible Endometriosis (EM) profiles (GSE6364, GSE7305, and GSE51981) on homo sapiens for deep analysis based on GPL570 (Affymetrix Human Genome U133 Plus 2.0 Array). The dataset GSE6364 consists of 16 Normal samples and 21 EM samples. The dataset GSE7305 is composed of 10 Normal samples and 10 EM samples. And the dataset GSE51981 comprises 71 Normal samples and 77 EM samples. In total, 97 Normal samples and 108 EM samples were collected. Additionally, we obtained the raw microarray data file and ran the analysis using R (version 3.6.1).
Identification of differentially expressed genes (DEGs)
The “limma” (linear models for microarray data) package of the Bioconductor was used to screen the DEGs. To find DEGs, the selection criterion |log2FC|≥0.7 and P<0.05 were used. DEGs with log2FC ≥ 0.7 were defined as up-regulated genes, whereas those with log2FC ≤-0.7 were defined as down-regulated genes. The ggplot2 package was used to create the volcanic maps for further study. The “ClusterProfiler” R package (version 3.2.11)23 which deals with domains including biological processes, cellular components, and molecular functions was used to conduct function enrichment for DEGs. For the purpose of assessing the differences between ectopic and normal groups, Gene set enrichment analysis (GSEA) was utilized. In order to reveal the results of the enrichment, we chose the words with a count ≥ 5 and P value <0.05.
Weighted gene co-expression network analysis (WGCNA)
WGCNA is a method that studies the associations between genes and phenotypes by constructing gene co-expression networks24. By the approach of WGCNA25, we analyzed 21,654 genes in 205 samples with R (version 3.6.1), identified the core modules and explored the correlation between gene network and EM, as well as the central genes in the network. According to the METree, a different sample with too high expression was given up. Afterwards, 12 was selected as the power value in the output file relying on diagrams of scale independence and mean connectivity to make it closer to a scale-free network. Using a dynamic tree cut package, similar expression profiles were divided into the same gene modules preliminarily, and these gene modules were combined subsequently. After the value of height was taken as 0.2, 5414 genes were divided into 10 modules. The heat map depicts the association between different modules and EM, and the green-yellow module containing 291 genes was identified, which was associated highly with EM. After that, we assessed the genes associated with EM and the green-yellow module’s module membership, respectively, and found that there was a strong association between them.
Two EM-related hub gene
To represent the common component of DEGs, we created Venn diagrams. There were 198 overlapped genes in 463 DEGs selected in the volcano map and 291 genes obtained after WGCNA analysis. Three statistical feature selection methods were applied in the analysis of 198 DEGs: the least absolute shrinkage and selection operator (LASSO), random forest (RF), and support vector machine (SVM). LASSO is a machine learning technique that combines variable selection and regularization and can improve prediction accuracy26. RF is a prediction algorithm that does not impose restrictions on variable conditions, enabling it to provide predictions without significant changes27. The SVM algorithm classifies data by constructing a hyperplane. It uses regularization terms to eliminate the overfitting problem in the model28. After that, utilizing Venn analysis, 5 hub genes (CHMP4C, SPZ1, KAT2B, GPC6, and EPCAM) were found in the overlap of results of LASSO, SVM, and RF regression analysis. Subsequently, Only CHMP4C and KAT2B were shown to be downregulated in EM by qRT-PCR.
Unsupervised clustering based on two hub genes
Based on the expression of two hub genes in each sample, EM was identified using unsupervised clustering analysis, and R (version 3.6.1) was used to develop subgroups for future investigation. A 2-cluster solution was retained after consensus clustering algorithms were used to assess the number of clusters and their stability. The aforementioned procedures were repeated up to 1000 times using the “ConsensusClusterPlus” program in order to ensure the classification’s stability29.
Difference analysis between two 2rG clusters
Using a single sample gene set enrichment analysis (ssGSEA) algorithm, the abundance of 36 immune signatures within the different clusters was quantified. These immune signatures included activated B cells, activated CD4 T cells, macrophages, etc. Subsequently, in order to determine DEGs between 2rG Clusters, the “limma” R package was used. The significance criteria for determining DEGs were P value < 0.05 and |log2FC|≥1.5. Finally, the 95 DEGs were obtained.
Consensus clustering based on 95 DEGs
Using the consensus clustering algorithm above, we examined the number of gene clusters as well as their stability based on the expression of 95 DEGs. K = 2 was taken and samples were classified into 2 gene Clusters. The same estimation of ICI was conducted between the two gene clusters, using a single sample gene set enrichment analysis (ssGSEA) algorithm.
Collection and analysis of immune-checkpoint blockade genes
We got a set of immune checkpoint genes from some reviews30,31. Then relevant data, including functional information and detailed clinical annotations, were searched in the GeneCards (https://www.genecards.org/). The R (version 3.6.1) was applied to deal with the gene expression profiles in samples. With the analysis of 2rG Clusters and gene Clusters performed, we obtained numerous immune checkpoints correlated with EM patterns.
Single-gene gene set enrichment analysis
In order to explore the prospective functions of core gene, we conducted GSEA on single gene using org.Hs.eg.db and clusterProfiler packages. Subsequently, according to the expression value of core gene, the correlation coefficients of core gene with all genes were ranked. The threshold for significant enrichment was set at a P value < 0.05.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed using three pairs of matched eutopic and ectopic endometrial tissue. Informed consent was obtained from all participants prior to biopsy and the use of human tissues was approved by the Institutional Research Ethics Committees of Shandong Provincial Hospital Affiliated to Shandong University. Written informed consent was obtained from all participants before being included in the study.
The marker of each ICI signature was selected from Supplementary Tables 1 and also shown in Supplementary Table 2. Total RNA was extracted from 50 mg of normal and ectopic tissues using the Trizol reagent (AG21101, Accuate Biology, Wuhan, China) and then reverse-transcribed into cDNA using a reverse transcription reagent kit (AG11728, Accuate Biology, Wuhan, China) to prepare the PCR system. PCR enzymes were bought from Jiangsu CWBio Company and PCR reaction was performed using the CFX Connect™ PCR system (Bio-Rad, Hercules, USA). The primers were synthesized by Shanghai BioSune Company and shown in Supplementary Table 2. Furthermore, SPSS 18.0 was used to calculate the correlation coefficient between the two hub genes and their most related ICI signatures.
Results
Selection and analysis of DEGs
The gene expression datasets were obtained from the GEO dataset. mRNA sequencing (GSE6364, GSE7305, and GSE51981) of EM were acquired on homo sapiens for deep analysis. To identify the applicability of these three EM queues, PCA was conducted. The PCA results (Fig. 1A) showed great differences among the three groups, and the samples within the group had good repeatability. Batch calibration was then performed, and the results were shown in Fig. 1B. The limma package was used to analyze the DEGs (including upregulated genes and downregulated genes) between the normal and ectopic groups. The ggplot2 package was utilized to draw the volcanic map (Fig. 1C). The enrichment analysis was conducted to investigate the biological behaviors of these DEGs using GSEA. The results were shown in Fig. 1D and E.

Annotation of endometriosis-associated genes and pathway analysis of normal groups and ectopic groups. (A) GSE6364, GSE7305, and GSE51981 were presented. (B) We conducted batch correction. (C) The volcano map of differential expression genes. |log2FC| ≥0.7 and P < 0.05 were identified as significantly altered. (D, E) GSEA analysis of normal groups and ectopic groups, respectively.
Combining WGCNA analysis to identify the EMs-related genes
To determine the co-expression modules, WGCNA was conducted using co-expression networks. Based on log10-transformed RNA-seq fractional counts, samples were clustered using the Euclidean Distance-Based Hierarchical Clustering (Fig. 2A) with the basic patient information added below the resulting tree. To construct a WGCNA network, the soft thresholding power β was calculated, and the coexpression similarity was raised to compute adjacency. In WGCNA, the function pickSoftThreshold was used to analyze the network topology. The soft thresholding power β = 12 was used in the subsequent analysis because the scale independence reached 0.8 (Fig. 2B) and the average connectivity was high (Fig. 2C). The one-step network construction function of the WGCNA R package was used to build the gene network and to identify modules. According to the results shown in Fig. 2D-F, the first set of modules was identified using the Dynamic Tree Cut algorithm, then the correlated modules (r > 0.2) were merged (Merged Dynamic); ten modules were identified in EMs (Fig. 2F). The gene co-expression was summarized by Eigengenes (i.e., the first component expression of genes in each module) and the correlations between each Eigengene were calculated. The green-yellow module containing 291 genes showed the highest negative correlation with EMs (Fig. 2F). These results allowed the selection of modules that were interesting for further analysis. After that, we evaluated the correlation significance between gene significance for EM and module membership in the green-yellow module, respectively, and it was concluded that there existed a great correlation between the gene with the module and the gene with EMs (Fig. 2G).

Weighted gene co-expression network analysis (WGCNA) and module trait relationship. (A) Sample cluster dendrogram of 205 samples. (B,C,D) Diagrams of scale independence and mean connectivity, in which 12 was selected as power value in the output file on clustering of module eigengenes. (E, F) Similar expression profiles were divided into the same gene modules preliminarily, which were combined subsequently. (G) The heat map depicts the association between different modules and endometriosis, in which the green-yellow module containing 291 genes was selected. (H) Correlation significance between gene significance for endometriosis and module membership in the greenyellow module was presented in.
Screening of the hub genes
We found that 198 genes were overlapping between module genes with DEGs (Fig. 3A). The important genes related to EMs were classified by SVM, Lasso, and RF to select the genes of interest, and 22, 27, and 50 genes were obtained respectively. Among these genes related to EMs, 5 hub genes (CHMP4C, SPZ1, KAT2B, GPC6, and EPCAM) were overlapping between SVM, Lasso, and RF (Fig. 3B). The qRT-PCR results shown that only CHMP4C and KAT2B were downregulated in EM samples compared to Normal samples (Fig. 3C). Eventually, CHMP4C and KAT2B were identified as hub genes.

Identification of EM-associated hub genes. (A) The venn diagram of 463 DEGs and 291 genes from WGCNA. (B) The venn diagram of SVM, Lasso and RF. (C) Relative mRNA expression of 5 hub genes and genes of ICI signatures between normal and ectopic tissues. * p < 0.05, ** p < 0.01, *** p < 0.001.
Two 2rG clusters divided by two hub genes
Based on the expression of the 2 hub genes (CHMP4C and KAT2B), the Consensus Clustering Method was used to cluster the samples in order to determine the immune status of the different EM samples (Fig. 4A). Additionally, we found that when K = 2 was chosen for the consensus clustering analysis (Fig. 4B), the cumulative distribution function (CDF) curve fluctuates minimum. As a result, two clusters were found and given the names 2rG Cluster 1 and 2rG Cluster 2.

Clustering of samples based on expression characteristics of two hub genes. (A) K = 2 was selected for consensus clustering analysis to reduce the interference between subgroups, so the samples were divided into two 2rG Clusters. (B) The cumulative distribution function (CDF) curve. (C) ICI characteristics of the two 2rG Clusters. (D) The correlation between ICI characteristics and CHMP4C. (E) The correlation between ICI characteristics and KAT2B. (F-H) The expression of checkpoint genes of 2rG Clusters. * p < 0.05, ** p < 0.01, *** p < 0.001.
Immune status and difference analysis for the two 2rG clusters
The variations in ICI across the two clusters were then analyzed using a variety of techniques (Fig. 4C). Between the two clusters, we discovered substantial changes in 25 immunological markers in EMs. The result showed that 2rG Cluster 1 showed the lower infiltration and 2rG Cluster 2 showed the higher infiltration, including Activated B cell, Activated dendritic cell, CD56bright natural killer cell, Immature B cell, Immature dendritic cell, MDSC, Macrophage, Mast cell, Monocyte, Natural killer T cell, Plasmacytoid dendritic cell, Regulatory T cell, T follicular helper cell, Type1 T helper cell, APC co-inhibition, APC co-stimulation, CCR, Check point, HLA, Parainflammation, T cell co-inhibition, T cell co-stimulation, Type I IFN Reponse, Type II IFN Reponse, Natural killer cell (Fig. 4C). We also analyzed the association of CHMP4C and KAT2B with these 36 immune signatures (Fig. 4D and E). We found that these hub genes had a significant correlation with numerous immune signatures. In summary, the immunity of EM patients is significantly influenced by CHMP4C and KAT2B. We assessed the association with the immunological checkpoints since the two clusters varied considerably in terms of ICI. 2rG Cluster 2 had the higher expression of PDCD1, PDCD1LG2, ICOS, ICOSLG, TNFSF14, TNFSF4, CD70, CD48, SIRPA, BTN2A1 and CD209. 2rG Cluster 1 had the lower expression of them (Fig. 4F-H). Above all, we found that 2rG Cluster 2 showed the higher infiltration and higher expression of immune checkpoints and 2rG Cluster 1 showed the lower. It reaffirmed that the expression of CHMP4C and KAT2B contributed significantly to the immunological control of the EMs microenvironment.
Two gene clusters obtained by the different expression of DEGs
We compared different genes in the two different 2rG Clusters and obtained 95 DEGs for further analysis. Figure 5A and B depicted the volcano plot and heat map of DEGs, respectively. Then, using the acquired 95 DEGs to categorize patients into various genomic subgroups, we conducted consensus clustering analysis to further confirm this regulatory mechanism (Fig. 6A). The unsupervised clustering approach showed two unique genomic symptoms that were consistent with the clustering grouping of cluster patterns. We termed these two gene clusters Gene Cluster 1 and Gene Cluster 2, respectively. The differences in ICI between the two gene clusters were then analyzed using a variety of techniques (Fig. 6B). Between the two gene clusters, there were significant variations in 23 immunological markers in EMs. The result showed that Gene Cluster 2 showed higher infiltration by Activated CD8 T cell, Activated dendritic cell, CD56dim natural killer cell, Gamma delta T cell, Immature B cell, Immature dendritic cell, Macrophage, Monocyte, Natural killer T cell, Natural killer cell, Neutrophil, Plasmacytoid dendritic cell, T follicular helper cell, Type1 T helper cell, Type17 T helper cell, APC co-stimulation, CCR, Check point, Cytolytic activity, Inflammation promoting, T cell co-inhibition, T cell co-stimulation, Type I IFN Reponse (Fig. 6B). We also looked at the relationship with the immunological checkpoints as the two gene clusters differed greatly in terms of ICI. Gene Cluster 2 had higher expression of TNFRSF18, HAVCR2, PVR, CD96, TMIGD2, CD47, SIRPA, IDO1 and CD200 (Fig. 6C-E). Above all, we found that Gene Cluster 2 showed higher infiltration and higher expression of immune checkpoints and Gene Cluster 1 showed lower. The aforementioned findings confirmed once more that the expression of two hub genes contributed significantly to molding various ICI scenarios.

Analysis of the differences between the two 2rG Clusters. (A) The Volcano plot of DEGs (P Value < 0.05 and |log2FC|≥1.5). (B) The heat map of DEGs.

Clustering of samples based on expression characteristics of the 95 DEGs. (A) K = 2 was selected for consensus clustering analysis to reduce the interference between subgroups, so the samples were divided into two Gene Clusters. (B) ICI characteristics of the two Gene Clusters. (C-E) The expression of checkpoint genes in the two Gene Clusters. * p < 0.05, ** p < 0.01, *** p < 0.001.
qRT-PCR analysis
The qRT-PCR was performed to validate the difference of 2 hub genes between the normal and ectopic tissues using 3 pairs of matched eutopic and ectopic endometrial tissue and the correlation between the hub genes and ICI. The results showed that expression levels of only CHMP4C and KAT2B of ectopic tissues were significantly lower than those of normal tissues (Fig. 3C). Moreover, the expression level of the two genes was consistent with the ICI signatures in Fig. 7A-B. For example, ASB2, IL17A, and HLA-E, markers of Type 2 T helper cell, Type 17 T helper cell and HLA, showed a significant correlation with CHMP4C (r = 0.578, 0.753 and 0.800), which was consistent in Fig. 4D. The correlation value between Type 2 T helper cell, Type 17 T helper cell, and HLA was 0.13, 0.50, and − 0.34. ssGSEA shown that CHMP4C mainly involved in Cytokine-cytokine receptor interaction (pvalue = 0.0017) and KAT2B mainly involved in Oxidative phosphorylation (pvalue = 0.00067) (Fig. 7C-D). Eventually, we concluded CHMP4C and KAT2B were hub biomarkers participating in the pathogenesis of EM by regulating of immune cell infiltration.

Results of qRT-PCR analysis and single gene GSEA. (A-B) The correlation between CHMP4C and KAT2B and the ICI signatures. (C) GSEA analysis of CHMP4C. The results shown that CHMP4C mainly involved in Cytokine-cytokine receptor interaction. (D) GSEA analysis of KAT2B. The results shown that KAT2B mainly involved in Oxidative phosphorylation. * p < 0.05, ** p < 0.01, *** p < 0.001.
Discussion
As an inflammatory disease involving the reproductive system, the presentation of Endometriosis (EM) is highly heterogenic and can be insidious, which may result in chronic pain symptoms and affect patients’ fertility32. Other than the decline in life quality, some observational studies indicated that EM may be connected with some types of ovarian cancer33,34. Thus, though it is thought to be a benign gynaecological disease, EM deserves more attention, especially in its prognosis. In terms of the presence of the endometrium-like tissue outside the uterus, retrograde menstruation35, coelomic metaplasia36, and lymphatic and vascular metastasis37 are thought to be the origin. The first theory—that ectopic endometrial cells and tissue fragments undergoing neuroangiogensis cause localized inflammation, fibrosis/scarring, and pain in order to survive, attach to, and penetrate pelvic structures—is the one that is most widely accepted. Therefore, EM arises from a mixture of genes, environment and abnormal immune responses38.
Macrophages (MΦ)39,40,41, dendritic cells (DC)42 and mast cells (MC)43 may lose their normal functions during menstruation, along with the increased natural killing cells (NK), eosinophils (EN), neutrophils (NT) and their products such as CD16, NKp46, pro-inflammatory factors and VEGF44,45,46,47,48,49,50,51, bringing about the impairment of eutopic endometrium and the raised risk of infertility. The change of B cell amount in EM has been proven to be elevated reflected by the raised immunoglobulin and sCD23, which is more distinct in the early stage52. Meanwhile, CD8 + T cells are responsible for supervising the whole body’s immune status and destroying possible harmful cells through various pathways53. The number and location of CD8 + T lymphocytes are normal in eutopic endometrium in EM patients, but they rise in ectopic lesions and are no longer under the humoral control of estrogen and progesterone54,55. The expression mode of pro-inflammatory cells, Th2 and Th17, and their byproducts tend to be positively correlated with the severity of the disease in CD4 + T cells, another adaptive immune cell cluster stimulated by APC. The majority of these cells are increasing in ectopic endometrium, peripheral blood, or peritoneal fluid. CD4 + The expression mode of pro-inflammatory cells, Th2 and Th17, and their byproducts tend to be positively correlated with the severity of the disease. T cells are another adaptive immune cell cluster stimulated by APC and can be further divided into T helper cells (Th1, Th2, Th9, Th17, and Th22) and Treg. The majority of these cells are present as increasing in ectopic endometrium, peripheral blood, or peritoneal fluid56,57. The GSEA analysis showed that KEGG pathways linked to immune signals, such as the chemokine, complement, and cytokine-receptor interaction, were primarily enriched in EM samples, highlighting once more the critical role that immune cells play in the etiology of EM.
Two DEGs (CHMP4C and KAT2B) were screened by numerous bioinformatics methods and qRT-PCR. Based on the two biomarkers, the EM samples could effectively be divided into subgroups with significant differences in immune infiltration, which provides guidance for the classification of EM. Moreover, the correlation with ICI were further validated using qRT-PCR. The results suggest that CHMP4C and KAT2B were correlated with multiple immune cells, suggesting that they may participate in the pathogenesis of EM by regulating the infiltration of immune cells. CHMP4C is a protein acting as an imperative regulator in kinetochore-microtubule interactions of abscission58,59, while the abscission checkpoint could be relayed if CHMP4C was phosphorylated by Aurora B60. Importantly, CHMP4C can be up-regulated by p53 to increase exosome secretion and enhances autophagy, which retards the cell division and promotes the communication with dendritic cells of the immune system61. Activated B cell were associated with CHMP4C the most in our study, indicating that CHMP4C participates in the pathogenesis of EM via promoting communication between ectopic tissue cells and activated B cells. Previous studies have found that the number of B cells in the peritoneal fluid (PF) of patients with EM increases62,63. Moreover, a study conducted on a mouse model demonstrated that the elimination of B cells using Ibrutinib could suppress the activity and growth of endometriotic lesions64. CHMP4C is downregulated in EM. According to our research findings, CHMP4C is negatively correlated with activated B cells. Therefore, we conclude that CHMP4C participates in the pathogenesis of EM by upregulating B cells. Wild et al. first observed the presence of antibodies in the endometrium through indirect immunofluorescence, suggesting that B cells are involved in EM65. In addition, cytokines secreted by B cells (such as interleukin-6, interleukin-17, and interferon-γ) are also involved in the pathogenesis of endometriosis66. In addition, through single-gene GSEA, we found that CHMP4C was mainly involved in cytokine-cytokine receptor interaction, which made the cytokines secreted by B cells further contribute to the occurrence and development of EM. KAT2B, lysine acetyltransferase 2B, is a histone acetyltransferase that is involved in multiple post-translational modifications67. KAT2B engaged in the regulation of liver fibrosis and breast cancer metastasis68,69. Furthermore, KAT2B participates in the pro-inflammatory polarization in response to oxidized low-density lipoprotein70 and in many inflammatory processes of some diseases via regulation of Interleukin-10 and inflammasome71,72. KAT2B could act as a diagnosis and prognosis biomarker in varied diseases, such as non-small cell lung cancer and colorectal cancer73,74. In our study, KAT2B mainly involved in oxidative phosphorylation. It has been found in previous studies that oxidative phosphorylation is an important pathogenesis mechanism of EM75.
ICI is regarded as the cause of pathophysiology changes in EM76. It has been shown that varied immune cells trigger and maintain lesions in EM by driving inflammation, such as macrophages, FOXP3 + regulatory T cells, and mast cells77,78,79. Consistent with prior research, clusters based on the five biomarkers exhibited high correlations with ICI, indicating that immune cells, as well as inflammatory reactions, plays an essential role in EM, which further verified the reliability and accuracy of our classification. Blockade of immune checkpoint has been proven to exert therapeutic values in varied diseases, such as cervical cancer80,81. In our study, a significantly higher expression of immune checkpoint genes was shown in both 2rG Cluster 2 and gene Cluster 2. ICI means poor prognosis in patients76. The inhibition of immune checkpoint may contribute to the treatment of this Cluster in EM. Thus, we obtained the high immune infiltration subtype through hub genes, providing a more accurate treatment strategy for patients with EM.
A key limitation of our study is the relatively small sample size used for qRT-PCR validation. While the results provide preliminary support for the differential expression of the identified genes, the limited sample size may reduce the statistical power and generalizability of the findings. This could potentially lead to an underestimation or overestimation of the true biological effects. To address this limitation, future studies should incorporate larger and more diverse sample cohorts to confirm these results. Moreover, additional experimental approaches, such as functional assays or in vivo models, would provide deeper insights into the roles of these genes in EM.
Conclusion
In summary, this study demonstrated the significant correlation between EM and immune cell infiltration, and finally identified two effective biomarkers to provide promising new biomarkers for newly subtyping and therapy of EM.
Data availability
The original contributions presented in this work are included in the article/Supplementary Materials. Further inquiries can be direct to the corresponding author.
References
Olive, D. L. & Schwartz, L. B. Endometriosis. N. Engl. J. Med. 328(24), 1759–1769 (1993).
Giudice, L. C. & Kao, L. C. Endometriosis. Lancet (London England). 364(9447), 1789–1799 (2004).
Peiris, A. N., Chaljub, E. & Medlock, D. Endometr. JAMA 320(24), 2608. https://doi.org/10.1001/jama.2018.17953. (2018).
Taylor, H. S., Kotlyar, A. M. & Flores, V. A. Endometriosis is a chronic systemic disease: Clinical challenges and novel innovations. Lancet (London England) 397(10276), 839–852. https://doi.org/10.1016/S0140-6736(21)00389-5 (2021).
Nisolle, M. & Donnez, J. Peritoneal endometriosis, ovarian endometriosis, and adenomyotic nodules of the rectovaginal septum are three different entities. Fertil. Steril. 68(4), 585–596 (1997).
Koninckx, P. R., Ussia, A., Adamyan, L., Wattiez, A. & Donnez, J. Deep endometriosis: Definition, diagnosis, and treatment. Fertil. Steril. 98(3), 564–571. https://doi.org/10.1016/j.fertnstert.2012.07.1061 (2012).
Kho, R. M. et al. Surgical treatment of different types of endometriosis: Comparison of major society guidelines and preferred clinical algorithms. Best Pract. Res. Clin. Obstet. Gynecol. 51, 102–110. https://doi.org/10.1016/j.bpobgyn.2018.01.020 (2018).
Goncalves, M. O. et al. Systematic evaluation of endometriosis by transvaginal ultrasound can accurately replace diagnostic laparoscopy, mainly for deep and ovarian endometriosis. Hum. Reprod. 36(6), 1492–1500 (2021).
Wang, L., Zheng, W., Mu, L. & Zhang, S. Z. Identifying biomarkers of endometriosis using serum protein fingerprinting and artificial neural networks. Int. J. Gynaecol. Obstet. 101(3), 253–258. https://doi.org/10.1016/j.ijgo.2008.01.018 (2008).
Anastasiu, C. V. et al. Biomarkers for the noninvasive diagnosis of endometriosis: State of the art and future perspectives. Int. J. Mol. Sci. 21(5), 1750. https://doi.org/10.3390/ijms21051750 (2020).
Hey-Cunningham, A. J. et al. Endometrial stromal cells and immune cell populations within lymph nodes in a nonhuman primate model of endometriosis. Reproductive Sci. (Thousand Oaks Calif) 18(8), 747–754. https://doi.org/10.1177/1933719110397210 (2011).
Burney, R. O. & Giudice, L. C. Pathogenesis and pathophysiology of endometriosis. Fertil. Steril. 98(3), 511–519. https://doi.org/10.1016/j.fertnstert.2012.06.029 (2012).
Koninckx, P. R. et al. Pathogenesis of endometriosis: The genetic/epigenetic theory. Fertil. Steril. 111(2), 327–340. https://doi.org/10.1016/j.fertnstert.2018.10.013 (2019).
Bukulmez, O., Hardy, D. B., Carr, B. R., Word, R. A. & Mendelson, C. R. Inflammatory status influences aromatase and steroid receptor expression in endometriosis. Endocrinology 149(3), 1190–1204 (2008).
Sanchez, A. M. et al. The endometriotic tissue lining the internal surface of endometrioma: Hormonal, genetic, epigenetic status, and gene expression profile. Reproductive Sci. (Thousand Oaks Calif). 22(4), 391–401. https://doi.org/10.1177/1933719114529374 (2015).
Yap, K. L. et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38(5), 662–674. https://doi.org/10.1016/j.molcel.2010.03.021 (2010).
Hudson, Q. J. et al. The role of long non-coding RNAs in endometriosis. Int. J. Mol. Sci. 22(21). https://doi.org/10.3390/ijms222111425 (2021).
Wu, J., Xie, H., Yao, S. & Liang, Y. Macrophage and nerve interaction in endometriosis. J. Neuroinflamm. 14(1), 53. https://doi.org/10.1186/s12974-017-0828-3 (2017).
Wang, L. et al. A history of endometriosis is Associated with decreased peripheral NK cytotoxicity and increased infiltration of uterine CD68 macrophages. Front. Immunol. 12, 711231. https://doi.org/10.3389/fimmu.2021.711231 (2021).
Lin, Y. J., Lai, M. D., Lei, H. Y. & Wing, L. Y. C. Neutrophils and macrophages promote angiogenesis in the early stage of endometriosis in a mouse model. Endocrinology 147(3), 1278–1286 (2006).
Monsanto, S. P. et al. Surgical removal of endometriotic lesions alters local and systemic proinflammatory cytokines in endometriosis patients. Fertil. Steril. 105(4). https://doi.org/10.1016/j.fertnstert.2015.11.047 (2016).
Burns, K. A. et al. Early Endometriosis in females is directed by Immune-mediated estrogen receptor α and IL-6 cross-talk. Endocrinology 159(1), 103–118. https://doi.org/10.1210/en.2017-00562 (2018).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 16(5), 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Langfelder, P. & Horvath, S. WGCNA: An r package for weighted correlation network analysis. BMC Bioinf. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Yang, C., Delcher, C., Shenkman, E. & Ranka, S. Machine learning approaches for predicting high cost high need patient expenditures in health care. BioMed. Eng. Online. 17, 131. https://doi.org/10.1186/s12938-018-0568-3 (2018).
Ellis, K. et al. A random forest classifier for the prediction of energy expenditure and type of physical activity from wrist and hip accelerometers. Physiol. Meas. 35, 2191–2203. https://doi.org/10.1088/0967-3334/35/11/2191 (2014).
Liu, Q., Gu, Q. & Wu, Z. Feature selection method based on support vector machine and shape analysis for high-throughput medical data. Comput. Biol. Med. 91, 103–111. https://doi.org/10.1089/omi.2011.0118 (2017).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 26(12), 1572–1573. https://doi.org/10.1093/bioinformatics/btq170 (2010).
Burugu, S., Dancsok, A. R. & Nielsen, T. O. Emerging targets in cancer immunotherapy. Semin Cancer Biol. 52(Pt 2), 39–52. https://doi.org/10.1016/j.semcancer.2017.10.001 (2018).
Morad, G., Helmink, B. A., Sharma, P. & Wargo, J. A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 184(21), 5309–5337. https://doi.org/10.1016/j.cell.2021.09.020 (2021).
Zondervan, K. T., Becker, C. M. & Missmer, S. A. Endometriosis. N. Engl. J. Med. 382(13), 1244–1256. https://doi.org/10.1056/NEJMra1810764 (2020).
Kajiyama, H. et al. Endometriosis and cancer. Free Radic. Biol. Med. 133, 186–192. https://doi.org/10.1016/j.freeradbiomed.2018.12.015 (2019).
Guo, S. W. Cancer-associated mutations in endometriosis: Shedding light on the pathogenesis and pathophysiology. Hum. Reprod. Update. 26(3), 423–449. https://doi.org/10.1093/humupd/dmz047 (2020).
Sampson, J. A. The development of the implantation theory for the origin of peritoneal endometriosis. Am. J. Obstet. Gynecol. 40(4), 549–557 (1940).
Gruenwald, P. Origin of endometriosis from the mesenchyme of the celomic walls. Am. J. Obstet. Gynecol. 44(3), 470–474 (1942).
Jerman, L. F. & Hey-Cunningham, A. J. The role of the lymphatic system in endometriosis: A comprehensive review of the literature. Biol. Reprod. 92(3), 64. https://doi.org/10.1095/biolreprod.114.124313 (2015).
Ramírez-Pavez, T. N. et al. The role of peritoneal macrophages in Endometriosis. Int. J. Mol. Sci. 22(19). https://doi.org/10.3390/ijms221910792 (2021).
Berbic, M. et al. Macrophage expression in endometrium of women with and without endometriosis. Hum. Reprod. 24(2), 325–332. https://doi.org/10.1093/humrep/den393 (2009).
Khan, K. N. et al. Escherichia coli contamination of menstrual blood and effect of bacterial endotoxin on endometriosis. Fertil. Steril. 94(7), 2860–2863e2863. https://doi.org/10.1016/j.fertnstert.2010.04.053 (2010).
Takebayashi, A. et al. Subpopulations of macrophages within Eutopic Endometrium of endometriosis patients. Am. J. Reprod. Immunol. 73(3), 221–231. https://doi.org/10.1111/aji.12331 (2015).
King, A., Gardner, L. & Loke, Y. W. Evaluation of oestrogen and progesterone receptor expression in uterine mucosal lymphocytes. Hum. Reprod. 11(5), 1079–1082. https://doi.org/10.1093/oxfordjournals.humrep.a019300 (1996).
Sugamata, M., Ihara, T. & Uchiide, I. Increase of activated mast cells in human endometriosis. Am. J. Reproductive Immunol. (New York N Y : 1989). 53(3), 120–125 (2005).
Donnez, J., Smoes, P., Gillerot, S., Casanas-Roux, F. & Nisolle, M. Vascular endothelial growth factor (VEGF) in endometriosis. Hum. Reprod. 13(6), 1686–1690. https://doi.org/10.1093/humrep/13.6.1686 (1998).
Nagorsen, D., Marincola, F. M. & Panelli, M. C. Cytokine and chemokine expression profiles of maturing dendritic cells using multiprotein platform arrays. Cytokine 25(1), 31–35. https://doi.org/10.1016/j.cyto.2003.08.012 (2004).
Takehara, M. et al. Vascular endothelial growth factor A and C gene expression in endometriosis. Hum. Pathol. 35(11), 1369–1375. https://doi.org/10.1016/j.humpath.2004.07.020 (2004).
Lousse, J. C. et al. Increased activation of nuclear factor-kappa B (NF-κB) in isolated peritoneal macrophages of patients with endometriosis. Fertil. Steril. 90(1), 217–220. https://doi.org/10.1016/j.fertnstert.2007.06.015 (2008).
Schulke, L., Manconi, F., Markham, R. & Fraser, I. S. Endometrial dendritic cell populations during the normal menstrual cycle. Hum. Reprod. 23(7), 1574–1580. https://doi.org/10.1093/humrep/den030 (2008).
Mor, G., Cardenas, I., Abrahams, V. & Guller, S. Inflammation and pregnancy: The role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 1221(1), 80–87. https://doi.org/10.1111/j.1749-6632.2010.05938.x (2011).
Capobianco, A. & Rovere Querini, P. Endometriosis, a disease of the macrophage. Front. Immunol. 4. (2013).
Giuliani, E., Parkin, K. L., Lessey, B. A., Young, S. L. & Fazleabas, A. T. Characterization of uterine NK cells in women with infertility or recurrent pregnancy loss and Associated Endometriosis. Am. J. Reprod. Immunol. 72(3), 262–269. https://doi.org/10.1111/aji.12259 (2014).
Odukoya, O., Bansal, A. & Cooke, I. Serum endometrial IgG antibodies and soluble CD23 concentrations in patients with endometriosis. Acta Obstet. Gynecol. Scand. 75(10), 927–931. https://doi.org/10.3109/00016349609055030 (1996).
White, H. D. et al. CD3 + CD8 + CTL activity within the human female reproductive tract: Infuence of stage of the menstrual cycle and menopause. J. Immunol. 158(6), 3017 (1997).
Witz, C. A., Montoya, I. A., Dey, T. D. & Schenken, R. S. Characterization of lymphocyte subpopulations and T cell activation in endometriosis. Am. J. Reprod. Immunol. 32(3), 173–179. https://doi.org/10.1111/j.1600-0897.1994.tb01110.x (1994).
Bulmer, J. N., Jones, R. K. & Searle, R. F. Intraepithelial leukocytes in endometriosis and adenomyosis: Comparison of eutopic and ectopic endometrium with normal endometrium. Hum. Reprod. 13(10), 2910–2915. https://doi.org/10.1093/humrep/13.10.2910 (1998).
Antsiferova, Y. S., Sotnikova, N. Y., Posiseeva, L. V. & Shor, A. L. Changes in the T-helper cytokine profile and in lymphocyte activation at the systemic and local levels in women with endometriosis. Fertil. Steril. 84(6), 1705–1711 (2005).
Zhang, X., Xu, H., Lin, J., Qian, Y. & Deng, L. Peritoneal fluid concentrations of interleukin-17 correlate with the severity of endometriosis and infertility of this disorder. BJOG: Int. J. Obstet. Gynecol. 112(8), 1153–1155. https://doi.org/10.1111/j.1471-0528.2005.00639.x (2005).
Capalbo, L. et al. Coordinated regulation of the ESCRT-III component CHMP4C by the chromosomal passenger complex and centralspindlin during cytokinesis. Open Biol. 6(10). (2016).
Petsalaki, E., Dandoulaki, M. & Zachos, G. Chmp4c is required for stable kinetochore-microtubule attachments. Chromosoma 127(4), 461–473. https://doi.org/10.1007/s00412-018-0675-8 (2018).
Carlton, J. G., Caballe, A., Agromayor, M., Kloc, M. & Martin-Serrano, J. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Sci. (New York N Y). 336(6078), 220–225. https://doi.org/10.1126/science.1217180 (2012).
Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2(2), a001057. https://doi.org/10.1101/cshperspect.a001057 (2010).
Antsiferova, Y. S., Sotnikova, N. Y., Posiseeva, L. V. & Shor, A. L. Changes in the T-helper cytokine profile and in lymphocyte activation at the systemic and local levels in women with endometriosis. Fertil. Steril. 84(6), 1705–1711. https://doi.org/10.1016/j.fertnstert.2005.05.066 (2005).
Scheerer, C. et al. Characterization of endometriosis-associated immune cell infiltrates (Emaici). Arch. Gynecol. Obstet. 294(3), 657–664. https://doi.org/10.1007/s00404-016-4142-6 (2016).
Riccio, L. G. C. et al. B lymphocytes inactivation by ibrutinib limits endometriosis progression in mice. Hum. Reprod. 34(7), 1225–1234. https://doi.org/10.1093/humrep/dez071 (2019).
Wild, R. A. & Shivers, C. A. Antiendometrial antibodies in patients with endometriosis. Am. J. Reprod. Immunol. Microbiol. 8(3), 84–86. https://doi.org/10.1111/j.1600-0897.1985.tb00314.x (1985).
Shen, P. & Fillatreau, S. Antibody-independent functions of b cells: A focus on cytokines. Nat. Rev. Immunol. 15(7), 441–451. https://doi.org/10.1038/nri3857 (2015).
Fournier, M. et al. KAT2A/KAT2B-targeted acetylome reveals a role for PLK4 acetylation in preventing centrosome amplification. Nat. Commun. 7, 13227. https://doi.org/10.1038/ncomms13227 (2016).
Zhang, G. et al. MicroRNA-200c and microRNA- 141 are regulated by a FOXP3-KAT2B axis and associated with tumor metastasis in breast cancer. Breast Cancer Research: BCR. 19 (1), 73. https://doi.org/10.1186/s13058-017-0858-x (2017).
Ma, J. et al. hMSCs-derived exosome circCDK13 inhibits liver fibrosis by regulating the expression of MFGE8 through miR-17-5p/KAT2B. Cell Biol. Toxicol. https://doi.org/10.1007/s10565-022-09714-4 (2022).
Qi, X. et al. miR-30b-5p releases HMGB1 via UBE2D2/KAT2B/HMGB1 pathway to promote pro-inflammatory polarization and recruitment of macrophages. Atherosclerosis 324, 38–45. https://doi.org/10.1016/j.atherosclerosis.2021.02.016 (2021).
Bai, A. H. C. et al. Dysregulated lysine acetyltransferase 2B promotes inflammatory Bowel Disease Pathogenesis through Transcriptional repression of Interleukin-10. J. Crohn’s Colitis. 10(6), 726–734. https://doi.org/10.1093/ecco-jcc/jjw020 (2016).
Li, Y. et al. Type I IFN operates pyroptosis and necroptosis during multidrug-resistant A. baumannii infection. Cell Death Differ. 25(7), 1304–1318. https://doi.org/10.1038/s41418-017-0041-z (2018).
Yang, G., Zhang, Y. & Yang, J. A Five-microRNA signature as Prognostic Biomarker in Colorectal Cancer by Bioinformatics Analysis. Front. Oncol. 9, 1207. https://doi.org/10.3389/fonc.2019.01207 (2019).
Zhou, X., Wang, N., Zhang, Y., Yu, H. & Wu, Q. KAT2B is an immune infiltration-associated biomarker predicting prognosis and response to immunotherapy in non-small cell lung cancer. Investig. New Drugs. 40(1), 43–57. https://doi.org/10.1007/s10637-021-01159-6 (2022).
Kapoor, R., Stratopoulou, C. A. & Dolmans, M. M. Pathogenesis of endometriosis: New insights into prospective therapies. Int. J. Mol. Sci. 22(21), 11700. https://doi.org/10.3390/ijms222111700 (2021).
Li, W. N. et al. Extracellular vesicle-associated VEGF-C promotes lymphangiogenesis and immune cells infiltration in endometriosis. Proc. Natl. Acad. Sci. U.S.A. 117(41), 25859–25868. https://doi.org/10.1073/pnas.1920037117 (2020).
Berbic, M. & Fraser, I. S. Regulatory T cells and other leukocytes in the pathogenesis of endometriosis. J. Reprod. Immunol. 88(2), 149–155 (2011).
Jeljeli, M. et al. Macrophage immune memory controls endometriosis in mice and humans. Cell. Rep. 33(5), 108325. https://doi.org/10.1016/j.celrep.2020.108325 (2020).
Guo, X. et al. NLRP3 inflammasome activation of mast cells by estrogen the nuclear-initiated signaling pathway contributes to the development of endometriosis. Front. Immunol. 12, 749979. https://doi.org/10.3389/fimmu.2021.749979 (2021).
De Felice, F. et al. Immune check-point in cervical cancer. Crit. Rev. Oncol. Hematol. 129, 40–43. https://doi.org/10.1016/j.critrevonc.2018.06.006 (2018).
Fournel, L. et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 464 https://doi.org/10.1016/j.canlet.2019.08.005 (2019).
Funding
This study was supported by National Natural Science Foundation of China (Grant No. 81672861), Science and Technology Development Plan of Shandong Province (Grant No. 2017GSF218029).
Author information
Authors and Affiliations
Contributions
KL, XL and YL contributed to the conception and design of the study. KL and JW wrote the manuscript, YD, KW, ML and XZ helped to the writing, review, and/or revision of the manuscript. XL performed the qRT-PCR. All authors contributed the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
This study was approved by the Institutional Research Ethics Committees of Shandong Provincial Hospital Affiliated to Shandong University. All clinical samples patients were informed of the purpose of the study and signed the consent form.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, K., Wang, J., Liu, X. et al. Identification of hub biomarkers and immune cell infiltrations participating in the pathogenesis of endometriosis. Sci Rep 15, 2802 (2025). https://doi.org/10.1038/s41598-025-86164-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86164-y
