
Abstract
Cuproptosis, a recently discovered form of cell death, has emerged as a crucial player in tumor development, although its role in uterine corpus endometrial carcinoma (UCEC) remains inadequately explored. This study aims to identify prognostically relevant cuproptosis-related genes in endometrial cancer. Cuproptosis-related genes were sourced from previously published studies and the FerrDb database. UCEC gene expression profiles and clinical data were obtained from the TCGA database. Differential gene expression was determined using LIMMA analysis, and functional enrichment analysis was conducted on identified cuproptosis-related genes. A prognostic model for UCEC was developed using LASSO Cox regression analysis and a Nomogram, integrating survival data, status, and gene signatures. TIMER analysis assessed the impact of crucial cuproptosis-related genes on immune cell infiltration in UCEC. Validation of the selected genes, CDKN2A, GLS, and PPAT, was performed at both mRNA and protein levels. A total of 27 cuproptosis-related genes were identified, with 19 upregulated and 6 downregulated in UCEC. These genes were associated with key signaling pathways, including the TCA cycle, Pyruvate metabolism, Glycolysis/Gluconeogenesis, and Platinum drug resistance. The LASSO regression and Nomogram models demonstrated robust predictive performance for UCEC prognosis, identifying CDKN2A, GLS, and PPAT as critical prognostic genes. Furthermore, these genes played essential roles in immune cell infiltration in UCEC, confirming their significance. Validation at both mRNA and protein levels solidified the role of CDKN2A, GLS, and PPAT. The identified signature of CDKN2A, GLS, and PPAT demonstrates significant predictive value for UCEC prognosis, suggesting their potential as therapeutic targets, including their application in immunotherapy strategies.
Similar content being viewed by others
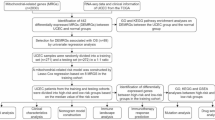

Introduction
Endometrial cancer, one of the most prevalent malignancies in the female reproductive system, ranks second in incidence among such tumors in China and first in the United States and other Western developed nations1,2,3. While incidence rates have either increased or stabilized globally over the past decade, notable surges are evident in South Africa and several Asian countries4. Despite these trends, patients with poorly differentiated and locally advanced tumors face inevitable metastasis and postoperative recurrence, often leading to mortality5,6. This highlights an urgent need for a deeper exploration of the molecular mechanisms underlying endometrial cancer development, particularly in the context of cell death. The identification of novel candidate genes is imperative to enhance early diagnosis and inform treatment decisions.
An increasing number of novel non-apoptotic forms of regulated cell death (RCD) have been identified, including necroptosis, pyroptosis, ferroptosis, endocytic cell death, reticulocyte death, parthanatos, lysosome-dependent Sexual cell death, autophagy-dependent cell death, alkaline cell death, and hypoxia7. Understanding and applying these RCD mechanisms could lead to discovering new therapeutic targets and provide rich means for diagnosing and treating tumors.
In March 2022, a landmark publication in SCIENCE described a newly characterized mechanism of cell death, distinct from previously known RCD pathways. This mechanism, termed “cuproptosis,” was elucidated by Tsvetkov et al. Through their analysis of phenomena, mechanisms, and disease models, the researchers found that copper directly binds to the fatty acylation components of the tricarboxylic acid cycle (TCA), leading to the aggregation of fatty acylated proteins and the loss of iron-sulfur cluster proteins, which in turn trigger proteotoxicity stress and eventually lead to cell death8.
There is increasing evidence that copper is involved in tumor cell initiation and progression. However, research on the role of cuproptosis-related genes in the progression and prognosis of UCEC remains limited. Therefore, this study aims to explore the role of cuproptosis-related genes in UCEC by constructing a predictive model, with the goal of identifying potential prognostic markers for UCEC.
Materials and methods
UCEC gene expression data and clinical data acquisition
The TCGA-UCEC data set and clinical information were downloaded from the Cancer Genome Atlas (TCGA) database. The Sangerbox online analysis tool was used for data standardization and transformation. Samples with absent, repeated, missing, or severely incorrect gene expression information were excluded. A summary of the clinical information is provided in Table 1.
Acquisition of cuproptosis-related genes
Cuproptosis-related genes were identified from a seminal article published in SCIENCE on cuproptosis. Using the FerrDb database (http://www.zhounan.org/ferrdb), 27 cuproptosis-related genes were curated. Unlike previous studies, our research incorporated an additional 17 cuproptosis-related genes, establishing a robust foundation for subsequent analysis (Table S1).
Identification of UCEC differentially expressed genes and cuproptosis-related genes
Differentially expressed genes (DEGs) between UCEC and normal endometrial tissue samples were identified using the LIMMA R package. Based on these results, expression profiles of 27 cuproptosis-related genes were analyzed.
Functional enrichment analysis of cuproptosis-related genes
Gene Ontology (GO) annotations and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were conducted using online tools (www.kegg.jp/kegg/kegg1.html). Permission from the Kanehisa laboratory was obtained for tool usage.
Construction of LASSO model
The Least Absolute Shrinkage and Selection Operator (LASSO)-Cox regression model was constructed using the R package “glmnet.” Survival time, survival status, and expression levels of 27 cuproptosis-related genes were integrated into the model.
Multivariate survival analysis and Kaplan-Meier survival curves
Survival data were analyzed using the R package “survival”. The prognostic significance of individual genes was assessed through Cox regression. The optimal RiskScore cutoff value was determined using the “maxstat” package, setting minimum and maximum grouping sample proportions to > 25% and < 75%, respectively. Patients were stratified into high-risk and low-risk groups. Prognostic differences between groups were evaluated using the log-rank test.
Construction of the Nomogram model
To predict outcome risks, a Nomogram was constructed using the R package “rms.” Data from survival time, survival status, and eight features (age, BMI, stage, grade, essential cuproptosis-related genes, and risk score) were integrated, and prognostic significance was assessed across 536 samples.
Immune infiltration analysis of cuproptosis-related genes
Unified and standardized pan-cancer datasets from UCSC Xena (https://xenabrowser.net/) were utilized. Three cuproptosis-related genes’ expression data were extracted, and their relationships with six immune cell infiltration scores were analyzed using the TIMER database. Additionally, Pearson’s correlation coefficients between these genes and immune infiltration scores in UCEC were calculated using the R package “psych” (version 2.1.6).
Validation of cuproptosis-related gene expression
To validate mRNA and protein expression of CDKN2A, GLS, and PPAT, quantitative real-time PCR (qPCR) and Western blot analyses were conducted on 30 UCEC and normal endometrial tissue pairs.
Sample size estimation is based on predetermined effect size (d), significance level (α = 0.05), and statistical power (β = 0.80). The sample size of 60 (30 per group) exceeds these requirements9,10.
-
n: Required sample size for each group.
-
Δ: Expected mean difference between groups.
-
σ: Standard deviation.
-
Zα/2 and Zβ: Z-scores corresponding to the significance level and statistical power (1.96 and 0.84, respectively).
qPCR procedure
Total RNA was extracted using Trizol reagent, followed by reverse transcription with the Revert Aid First-Strand cDNA Synthesis Kit. qPCR was performed using the SYBR Premix Ex Taq II Kit. GAPDH served as the endogenous control, and relative mRNA expression was calculated using the 2 − ΔΔCq method. Each sample was analyzed in triplicate.
Western blot procedure
Protein supernatants were separated via 10% SDS-PAGE and transferred to PVDF membranes. Membranes were blocked with 5% milk and incubated with primary antibodies (CDKN2A, GLS, PPAT, and GAPDH) overnight at 4 °C. Horseradish peroxidase-conjugated secondary antibodies were applied, and visualization was achieved with ECL chromogenic substrate reagent. Semi-quantitative analysis was performed using Image J, with GAPDH as the reference.
Results
Expression profiles of 27 cuproptosis-related genes derived from UCEC differentially expressed genes
From 536 cases of UCEC and 11 cases of normal endometrium tissue, a total of 2,516 differentially expressed genes were identified by LIMMA analysis, including 1,107 upregulated genes and 1,409 downregulated genes (Fig. 1A and B). Further analysis of these results yielded the expression profiles of 27 cuproptosis-related genes. Among these, 19 genes were upregulated (FDX1, DLAT, PDHA1, PDHB, SLC31A1, CDKN2A, DNA2, NTHL1, POLE, POLD1, CISD1, GLRX5, PPAT, ELP3, CDK5RAP1, ISCA2, ETFDH, HSPA1B, and ATP7B), while 6 genes were down-regulated (LIAS, LIPT1, MTF1, GLS, HSPA1A, and ATP7A). Two genes, DLD and RTEL1, showed no significant differences in expression (Fig. 1C).

Expression profiles of differentially expressed genes and cuproptosis-related genes in UCEC. (A). The UCEC differentially expressed gene volcano map, calculated by 2-fold differential expression, showed 1 107 up-regulated genes and 1 409 down-regulated genes. (B). Heat map of differentially expressed genes in UCEC. (C). Expression profiles of 27 cuproptosis-related genes in UCEC, including 19 up-regulated genes, 6 down-regulated genes, and 2 genes with no difference.
Functional enrichment analysis of 27 cuproptosis-related genes
Using KEGG Pathway annotations and GO annotations, we identified significant enrichment in pathways such as the TCA cycle, Pyruvate metabolism, Glycolysis/Gluconeogenesis, Platinum drug resistance, and Carbon metabolism(Fig. 2A). Additionally, these genes were involved in functions including iron-sulfur cluster binding, metal cluster binding, copper ion transmembrane transporter activity, and copper ion import(Fig. 2B).

Functional enrichment analysis of 27 cuproptosis-related genes. (A). KEGG Pathway of 27 cuproptosis-related genes. (B). GO annotation of 27 cuproptosis-related genes.
Construction of the LASSO-COX model
A LASSO-COX model was developed using the R package “glmnet” by integrating survival time, survival status, and expression data of 27 cuproptosis-related genes(Fig. 3A). The optimal Lambda value of 0.023 was used, resulting in the selection of six genes. The model formula is as follows (Fig. 3C):

Lasso-cox regression analysis results. (A). The lasso-cox optimal model obtained 6 cuproptosis-related genes, namely GLS, CDKN2A, PPAT, PDHA1, CDK5RAP1, and ATP7B. (B). ROC analysis to obtain the AUC curve. (C). Lasso-cox regression analysis sets the Lambda value to 0.0234. (D). The prognosis of the high RiskScore group and the low RiskScore group showed a significant difference (P = 7.0e−10), and the best cut-off value of RiskScore was 1.5187.
RiskScore = 0.0099*PDHA1.
+ 0.1906*GLS.
+ 0.1684*CDKN2A.
+ 0.0673*PPAT.
+ 0.0543*CDK5RAP1.
+ 0.0527*ATP7B.
The final area under the curve (AUC) values obtained via ROC analysis were 0.61 at 365 days, 0.70 at 1095 days, and 0.71 at 1825 days, demonstrating the model’s reliability (Fig. 3B). Using an optimal cutoff value of 1.5187, patients were classified into high-risk and low-risk groups. Significant differences in prognosis between these groups were identified (P = 7.0e−10) using the log-rank test (Fig. 3D).
LASSO heatmap and multivariate analysis
Risk scores were analyzed alongside patients’ follow-up times, events, and gene expression profiles. Increasing risk scores corresponded with decreased survival rates in UCEC patients (Fig. 4A). A heatmap of OS-related CRGs expression was generated. Multivariate Cox regression identified six key cuproptosis-related genes (GLS, CDKN2A, PPAT, PDHA1, CDK5RAP1, and ATP7B) significantly associated with survival (Fig. 4B).

Lasso-cox regression analysis expression heat map and multivariate analysis. (A). Lasso-cox regression analysis expression heat map. (B). Multivariate analysis of forest plot. Multivariate Cox regression showing 6 cuproptosis-related genes (GLS, CDKN2A, PPAT, PDHA1, CDK5RAP1, and ATP7B) associated with OS in UCEC.
KM survival curves of six cuproptosis-related genes
Kaplan-Meier survival curves indicated that three genes (CDKN2A: P = 2.4e−4, GLS: P = 2.5e−3, and PPAT: P = 2.5e−3) were significantly associated with prognosis (Fig. 5A-F). Among these, high-risk groups consistently showed worse prognoses. CDKN2A, GLS, and PPAT were identified as potential prognostic biomarkers for UCEC. Subgroup analyses across age and BMI categories revealed consistent trends (Fig. 6).

Survival rates of UCEC patients with high-risk and low-risk patients with UCEC in 6 cuproptosis-related genes. (A). KM curve of CDKN2A, P = 2.4*10−4. (B). KM curve of GLS, P = 2.5*10−3. (C). KM curve of ATP7B, P = 0.08. (D). KM curve of PPAT, P = 2.5*10−3. (E). KM curve for DHA1, P = 0.10. (F). KM curves of CDK5RAP1, P = 0.10.

Survival rates of UCEC patients with high and low-risk patients with UCEC in the subgroups based on clinicopathological characteristics. (A) The subgroup of age ≥ 60 years. (B) The subgroup of age < 60 years. (C) The subgroup of BMI ≥ 30. (D) The subgroup of BMI < 30.
Construction and assessment of the Nomogram
A Nomogram was constructed to predict 1-, 3-, and 5-year survival probabilities based on age and risk scores (Fig. 7A). The model exhibited strong performance, with a concordance index of 0.785 (95% CI: 0.735–0.835, P = 4.10e−29). Calibration curves indicated high consistency between predicted and observed survival times (Fig. 7B).

Establishment of the nomogram prediction model. (A). A prognostic nomogram based on the risk score using the age, BMI, stage, grade, and prognostic cuproptosis-related genes signature (CDKN2A, GLS, and PPAT) to predict 1-, 3-, and 5-year survival in patients with UCEC. (B). Calibration curves corrected for deviations in agreement between the predicted and observed survival rates at 1-, 3-, and 5 years.
Functional enrichment analysis of three key cuproptosis-related genes
Functional analysis of CDKN2A, GLS, and PPAT revealed their involvement in pathways such as the TCA cycle, Pyruvate metabolism, and Platinum drug resistance. Their roles in molecular functions like iron-sulfur cluster binding and metal cluster binding were also noted (Fig. 8). Protein-protein interaction analysis demonstrated a co-expression network among the three genes.

6 cuproptosis-related key genes co-expression network and functional enrichment. (A). KEGG Pathway of 6 cuproptosis-related key genes. (B). GO annotation of 6 cuproptosis-related key genes. (C). Protein-protein Interaction Enrichment Analysis shows the 6 cuproptosis-related key genes co-expression network.
Immune infiltration analysis of three key cuproptosis-related genes
Immune infiltration analysis indicated a negative correlation between CDKN2A expression and CD8 + T cell invasion scores, but positive correlations with CD4 + T cells and dendritic cells. GLS expression correlated positively with neutrophils and dendritic cells and negatively with macrophages. PPAT expression correlated positively with dendritic cells and negatively with B cells and macrophages (Fig. 9A-C). Overall, the three genes were significantly associated with immune infiltration levels in UCEC (Fig. 9D-F).

Immune cell infiltration score and immune infiltration score for 3 cuproptosis-related genes. (A-C) B cell, T cell CD4, T cell CD8, Neutrophil, Macrophage, and DC infiltration scores of 3 cuproptosis-related genes. (D-F) Immune infiltration score of 3 cuproptosis-related genes.
Validation of mRNA and protein expression levels of three key cuproptosis-related genes
Quantitative PCR and Western blot analyses were conducted on 30 pairs of UCEC and normal endometrial tissues. The mRNA levels of CDKN2A, GLS, and PPAT were significantly different between UCEC and normal tissues (Fig. 10A). Similarly, their protein levels showed significant differences, confirming their potential as prognostic markers.(Fig. 10B-E).

The mRNA and protein level of the 3 cuproptosis-related genes in UCEC tissues and normal tissues. (A) mRNA levels of the 3 cuproptosis-related genes in 30 UCEC and normal endometrial tissues; (B-E) Protein level of 3 cuproptosis-related genes in 30 UCEC and normal endometrial tissues.
Discussion
Cuproptosis, a newly identified form of regulated cell death characterized by the accumulation of copper ions and their interaction with mitochondrial components, has emerged as a compelling focus in cancer research. Studies have confirmed the role of copper in promoting angiogenesis, a critical process in tumor progression and metastasis, through the activation of factors such as angiopoietin, VEGF, FGF-1, and IL-1. Copper also stabilizes HIF-1α, enhancing the expression of pro-angiogenic molecules, thereby promoting tumor proliferation and invasion11,12,13,14. These findings highlight Copper’s integral role in cancer biology, making it a potential therapeutic target for cancer treatment and prevention15.
First introduced in Science, cuproptosis delineates its unique mechanisms from other cell death pathways and highlights a set of associated genes, offering a novel paradigm for cancer research8. Building on this foundation, Li et al. revealed an association between a high cuproptosis index and advanced hepatocellular carcinoma, emphasizing its role in tumor aggressiveness16. Song et al. successfully applied a cuproptosis scoring system to predict bladder cancer prognosis and immune responses, indicating its broader utility in oncology17.
Gene expression profiles of TCGA-UCEC were analyzed in combination with 27 cuproptosis-related genes from the FerrDb database, providing a robust framework for studying endometrial cancer. Functional enrichment analysis revealed that these genes are involved in crucial pathways such as the TCA cycle, glycolysis/gluconeogenesis, and metal ion transmembrane transporter activity. These pathways underscore the critical role of Copper and its related genes in endometrial cancer development. By constructing a LASSO Cox regression model, six genes were identified as prognostic markers, and three key genes—CDKN2A, GLS, and PPAT—were further validated at both mRNA and protein levels, affirming their significance in UCEC prognosis.
While CDKN2A is established as a regulator of cell cycle checkpoints and autophagy18,19, GLS plays a crucial role in glutamine metabolism, supporting tumor growth20,21. However, the role of PPAT in UCEC remains unexplored, representing an exciting avenue for future studies. Additionally, our findings align with prior work demonstrating the prognostic significance of CDKN2A and GLS but uniquely emphasize their potential interplay within the context of cuproptosis.
Notably, this study’s approach integrates advanced bioinformatics tools to provide a comprehensive analysis of cuproptosis-related pathways and their impact on UCEC. The combination of KEGG pathway analysis, gene ontology annotations, and prognostic modeling offers novel insights into the role of cuproptosis in cancer.
Unlike previous studies relying on limited gene sets, our approach utilized a broader dataset from FerrDb, encompassing 27 genes to improve model accuracy. Previous studies have utilized limited gene sets, such as the 13 cuproptosis-related genes directly derived from the SCIENCE article22,23, which may constrain the scope of research. In contrast, our use of the FerrDb database expanded the gene set to 27, enhancing model precision and allowing for a broader exploration of cuproptosis-related mechanisms. Notably, while some studies identified different genes like LIPT1 as relevant to UCEC22, the absence of LIPT1 in our analysis may reflect disparities in methodological frameworks or gene set definitions. This highlights the need for standardized datasets and cross-study validations to ensure reproducibility. Another notable aspect of this study is its methodological rigor. Unlike some prior analyses that relied solely on WGCNA for differential gene selection23, we incorporated functional enrichment analyses and Nomogram modeling to ensure a robust prognostic model.
The Nomogram model offers several advantages over WGCNA in bioinformatics analysis. Nomograms integrate gene features with clinical variables (e.g., age, tumor stage) to create a quantifiable and intuitive prediction tool. Using methods like LASSO regression, Nomograms efficiently select key variables, avoiding multicollinearity and overfitting. Moreover, the predictive performance of Nomograms can be validated using metrics like the concordance index (C-index) and calibration curves, providing results with higher clinical applicability. In contrast, WGCNA primarily identifies gene co-expression modules and their correlation with phenotypes. While it reveals potential biological mechanisms, it lacks direct predictive capabilities and often requires additional analysis for clinical application. Previous studies show that WGCNA’s complex networks and module analyses are better suited for exploratory research. In comparison, Nomograms, with their intuitive risk scoring and robust validation systems, provide a rapid and efficient tool for clinical translation. Thus, Nomograms are widely used in cancer prognosis model construction, contributing significantly to precision medicine. Comparative evaluations reveal that Nomogram modeling provides quantifiable and clinically applicable results, outperforming WGCNA in predictive precision and risk stratification24,25.
Nevertheless, despite the strengths of our bioinformatics approach, we noted that the racial and regional distribution of TCGA-UCEC database samples might contribute to variability in gene expression results. And we acknowledge the absence of in vivo and in vitro validation experiments, which are crucial for translating our findings into clinical applications. Future studies should incorporate functional validation of identified biomarkers to strengthen their clinical applicability.
In conclusion, this study advances the understanding of cuproptosis in UCEC by identifying novel prognostic biomarkers and providing a framework for further research. However, the biological mechanisms underlying cuproptosis in UCEC, particularly involving PPAT, require additional experimental exploration to uncover potential therapeutic strategies.
Data availability
The datasets generated and analysed during the current study are available in the TCGA database repository, https://portal.gdc.cancer.gov/.
References
Chen, W. et al. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016 Mar-Apr ;66(2):115–132. https://doi.org/10.3322/caac.21338
Siegel, R. L., Miller, K. D., Fuchs, H. E., Jemal, A. & Cancer statistics CA Cancer J Clin. 2022;72(1):7–33. (2022). https://doi.org/10.3322/caac.21708
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249. https://doi.org/10.3322/caac.21660 (2021).
Lortet-Tieulent, J., Ferlay, J., Bray, F. & Jemal, A. International patterns and trends in Endometrial Cancer incidence, 1978–2013. J. Natl. Cancer Inst. 110 (4), 354–361. https://doi.org/10.1093/jnci/djx214 (2018).
Han, C. et al. Novel targeted therapies in ovarian and uterine carcinosarcomas. Discov Med. 25 (140), 309–319 (2018).
Travaglino, A. et al. Impact of endometrial carcinoma histotype on the prognostic value of the TCGA molecular subgroups. Arch. Gynecol. Obstet. 301 (6), 1355–1363. https://doi.org/10.1007/s00404-020-05542-1 (2020).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell. Res. 29 (5), 347–364. https://doi.org/10.1038/s41422-019-0164-5 (2019).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375 (6586), 1254–1261. https://doi.org/10.1126/science.abf0529 (2022).
Vittinghoff, E. & McCulloch, C. E. Relaxing the rule of ten events per variable in logistic and Cox regression. Am. J. Epidemiol. 165 (6), 710–718. https://doi.org/10.1093/aje/kwk052 (2007).
Biau, D. J., Kernéis, S. & Porcher, R. Statistics in brief: the importance of sample size in the planning and interpretation of medical research. Clin. Orthop. Relat. Res. 466 (9), 2282–2288. https://doi.org/10.1007/s11999-008-0346-9 (2008).
Oliveri, V. Selective targeting of Cancer cells by copper ionophores: an overview. Front. Mol. Biosci. 9, 841814. https://doi.org/10.3389/fmolb.2022.841814 (2022).
Shao, S., Si, J. & Shen, Y. Copper as the target for anticancer nanomedicine[J]. Adv. Ther. 2 (5), 1800147 (2019).
Lelièvre, P., Sancey, L., Coll, J. L., Deniaud, A. & Busser, B. The multifaceted roles of copper in Cancer: a Trace Metal element with Dysregulated Metabolism, but also a target or a bullet for Therapy. Cancers (Basel). 12 (12), 3594. https://doi.org/10.3390/cancers12123594 (2020).
Li, Y. Copper homeostasis: an emerging target for cancer treatment. IUBMB Life. 72 (9), 1900–1908. https://doi.org/10.1002/iub.2341 (2020).
Shanbhag, V. C. et al. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell. Res. 1868 (2), 118893. https://doi.org/10.1016/j.bbamcr.2020.118893 (2021).
Li, X. et al. Analysis of cuproptosis in hepatocellular carcinoma using multi-omics reveals a comprehensive HCC landscape and the immune patterns of cuproptosis. Front. Oncol. 12, 1009036. https://doi.org/10.3389/fonc.2022.1009036 (2022).
Song, Q., Zhou, R., Shu, F. & Fu, W. Cuproptosis scoring system to predict the clinical outcome and immune response in bladder cancer. Front. Immunol. 13, 958368. https://doi.org/10.3389/fimmu.2022.958368 (2022).
Liu, Q., Yu, M. & Zhang, T. Construction of oxidative stress-related genes risk Model predicts the prognosis of Uterine Corpus Endometrial Cancer patients. Cancers (Basel). 14 (22), 5572. https://doi.org/10.3390/cancers14225572 (2022).
Zhang, J. et al. An integrated autophagy-related gene signature predicts prognosis in human endometrial cancer. BMC Cancer. 20 (1), 1030. https://doi.org/10.1186/s12885-020-07535-4 (2020).
Zhou, W. J. et al. Estrogen inhibits autophagy and promotes the growth of endometrial cancer by promoting glutamine metabolism. Cell. Commun. Signal. 17 (1), 99. https://doi.org/10.1186/s12964-019-0412-9 (2019).
Devis-Jauregui, L., Eritja, N., Davis, M. L., Matias-Guiu, X. & Llobet-Navàs, D. Autophagy in the physiological endometrium and cancer. Autophagy 17 (5), 1077–1095. https://doi.org/10.1080/15548627.2020.1752548 (2021).
Chen, Y. Identification and Validation of Cuproptosis-Related Prognostic Signature and Associated Regulatory Axis in Uterine Corpus Endometrial Carcinoma. Front. Genet. 13, 912037. https://doi.org/10.3389/fgene.2022.912037 (2022).
Wu, Q. et al. A comprehensive analysis focusing on cuproptosis to investigate its clinical and biological relevance in uterine corpus endometrial carcinoma and its potential in indicating prognosis. Front. Mol. Biosci. 9, 1048356. https://doi.org/10.3389/fmolb.2022.1048356 (2022).
Iasonos, A., Schrag, D., Raj, G. V. & Panageas, K. S. How to build and interpret a nomogram for cancer prognosis. J. Clin. Oncol. 26 (8), 1364–1370. https://doi.org/10.1200/JCO.2007.12.9791 (2008).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Acknowledgements
The authors thank the developers of Sanger box tools, a free online platform for data analysis (http://vip.sangerbox.com/).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conceptualization: Jianqi Li; Formal analysis: Jianqi Li; Funding acquisition: Jianqi Li; Methodology: Yanfang Huang, Guoxian Luo and Xiujie Sheng; Software: Guoxian Luo; Writing – original draft: Yanfang Huang, Guoxian Luo, Xiujie Sheng and Jianqi Li; Writing – review & editing: Yanfang Huang, Guoxian Luo and Jianqi Li.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not Applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Y., Luo, G., Sheng, X. et al. Construction of cuproptosis-related genes risk model predicts the prognosis of Uterine Corpus Endometrial Carcinoma. Sci Rep 15, 2210 (2025). https://doi.org/10.1038/s41598-025-86756-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86756-8
Keywords
This article is cited by
-
Targeting cuproptosis for treatment and prognostic assessment in endometrial cancer
Cancer Cell International (2025)
-
Novel cuproptosis-related lncRNAs risk model to predicting prognosis and guiding immunotherapy for OSCC patients
Discover Oncology (2025)
