
Abstract
Benign prostatic hyperplasia (BPH) is a prevalent urinary system disorder. Despite evidence of a significant genetic component from previous studies, the specific pathogenic genes and biological mechanisms are still largely unknown. The study utilized the FinnGen R10 dataset, encompassing 177,901 individuals (36,601 cases and 141,300 controls), and the GTEx v8 EQTLs files to conduct single-tissue and cross-tissue transcriptome-wide association studies (TWAS). FUSION method was utilized to validate the findings in specific tissues. Gene Analysis and Multi-marker Analysis of Genomic Annotation (MAGMA) were used to identify potential susceptibility genes. The intersection genes of the above results were analyzed by Mendelian randomization (MR), summary data-based MR (SMR) and colocalization studies. Fine-mapping of causal gene sets (FOCUS) software was employed to accurately locate risk genes. Gene Expression Omnibus (GEO) analysis explores the differential expression of genes. Finally, The GeneMANIA tool was utilized to further understand the functional roles of these susceptibility genes. The cross-tissue TWAS analysis revealed 28 genes associated with BPH susceptibility. Single-tissue TWAS and MAGMA further refined eight genes, and subsequent MR, SMR, FOCUS and colocalization analysis pinpointed INO80B as the key gene. The differential expression of this result was verified by GEO. INO80B may help prevent excessive prostatic cell proliferation by regulating cell cycle-related gene expression. Our research identified the INO80B gene, whose predicted expression is associated with BPH risk and hence provided a new insight into the genetic basis of this disease. However, further functional studies are necessary to elucidate the potential biological activity of these significant signals.
Similar content being viewed by others

Introduction
Benign prostatic hyperplasia (BPH) is a prevalent chronic urological condition marked by nonmalignant prostate enlargement, resulting in lower urinary tract symptoms (LUTS) that considerably impact patients’ quality of life and daily activities1. BPH is of vital concern for world health due to its high prevalence and the significant resources it requires from health services. According to estimates from the Global Burden of Disease Study 2019 in Lancet, BPH is among the leading causes of DALYs among elderly males2. The incidence of BPH increases with age, affecting roughly 50% of men between 50 and 60 years and nearly 90% of those men over 80 years of age3,4. The condition imposes considerable economic costs due to the need for medical treatment and surgical interventions, and it also impacts patients’ psychological well-being and social interactions, further underlining the importance of effective management and therapeutic strategies5.
A study on the genetic susceptibility of benign prostatic hyperplasia (BPH) revealed that the lifetime cumulative risk of male relatives of early-onset BPH patients undergoing prostatectomy due to BPH is as high as 66%.The autosomal dominant model indicates that 7% of men with early-onset BPH possess a gene with an 89% lifetime penetrance6. The polygenic effect is considered due to many genetic variants carrying individually small effects, which sum up to influence the disorder. Recent genome-wide association studies (GWAS)estimated that about 60% of variation in phenotypes related to BPH risk is accounted for by genetic factors7. Whereas specific mutations may account for rare hereditary forms of BPH, most cases are mediated by the interplay of many genetic factors. Recent GWAS identified 14 risk loci associated with BPH8. However, many of the identified loci reside in non-coding regions; this implication certainly makes the determination of functional roles of these regions very complicated9. Furthermore, complex LD patterns may mask the identification of causal variants responsible for those associations10.
Transcriptome-wide association studies (TWAS) incorporate eQTL information with summary statistics from GWAS to provide candidate genes and probe gene phenotype associations11. A most recent significant development in this study is the Unified Test for Molecular Signature (UTMOST), a novel gene-level association analysis across multiple tissues12. UTMOST provides a unique solution not captured by other single-tissue approaches and utilizes a “group-lasso penalty” in improving the precision and efficiency of imputation models. This model allows shared eQTL effects across tissues while also identifying significant tissue-specific eQTL effects. In this study, besides the major effect attributed to prostate tissue in BPH, there are associations with other tissue types. (Fine-mapping of causal gene sets) FOCUS is a statistical approach that identifies putative causal genes by leveraging eQTL weights, LD patterns, and GWAS summary statistics through null model prioritization. This method demonstrates particular efficacy in detecting disease-associated genes when regional genetic elements influence downstream phenotypes13.
Herein, we conducted a cross-tissue TWAS in which the summary statistics of the BPH GWAS were combined with eQTL data sourced from the Genotype-Tissue Expression Project (GTEx) v8. In addition, we conducted tissue-specific association tests using FUSION14, FOCUS and further replication was conducted with Multi-marker Analysis of Genomic Annotation (MAGMA)15. Bioinformatic investigations of candidate genes followed MR, after colocalization.
Materials and methods
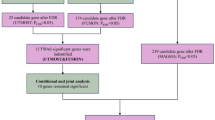
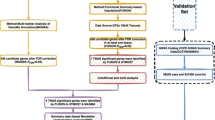
The study flow is illustrated schematically in Fig. 1.

The flowchart of this study.
Data sources of BPH
The summary statistic GWAS data for BPH were retrieved from the FinnGen R11 dataset (https://r11.finngen.fi/) on of genes across 49 distinguishable tissues16. To verify the stability of the results, the study of Jiang et al. was used as a validation set17.
Cross-tissue TWAS analyses
UTMOST analyses were applied to multi-tissue data to estimate the overall gene-trait associations at an organismal level. This approach allows more gene discoveries in tissues enriched for trait heritability and/or higher imputation accuracy of underlying signal12. For tissue-wise integrated association, we applied the generalized Berk-Jones (GBJ) test which uses covariance obtained from single tissue summary statistics18. Significance was taken as FDR < 0.05 after applying FDR correction.
Single-tissue TWAS analyses
To interrogate disease-gene associations in BPH, we performed a comprehensive TWAS using the FUSION method, where we integrated BPH GWAS with the eQTL data from 49 different tissues in GTEx V819. We first characterized the linkage disequilibrium (LD) between single nucleotide polymorphisms (SNPs) of the prediction model in respect to each GWAS locus. with 1,000 Genomes Project European population data. For each tissue and gene, we combined gene expression predictions based on the following multiple statistical models: BLUP, BSLMM, and LASSO, and Elastic Net models. In the final model, we also used the best 1 model for each tissue and gene. To compute gene expression weights, we used the best weight model in terms of prediction performance. Z-scores from the BPH GWAS and weights derived from the gene expression predictions were used to perform the TWAS. We also tried to find any genetic associations to migraine and conducted the TWAS to this disease. Candidate genes were allowed to pass the significance threshold if they had an FDR < 0.05 in the cross-tissue TWAS and at least one single-tissue TWAS. Candidate genes in such a multi-step process amazing from the cross-fertilization of genes associated with different diseases that are previously done and hence provide a new insight into the genetic base of complex diseases.
Conditional and joint analysis
In FUSION, identifying multiple features within a genetic locus necessitates determining their conditional independence. We employed the COJO analysis module in FUSION for post-processing to isolate independent genetic signals19. The COJO analysis improves our comprehension of genetic architecture by considering LD among markers, providing a more detailed perspective on trait variation20. After conducting this analysis, genes that maintained their significance were classified as jointly significant, indicating strong independent associations. Conversely, genes that lost significance were deemed marginally significant, reflecting their conditional dependency on other genetic factors.
FOCUS for precise gene location
FOCUS software was employed to fine-map transcriptome-wide associations between genetic variants and disease risk regions. The platform integrates GWAS summary statistics with multi-tissue eQTL weights, including GTExv8 PrediXcan data, to identify probable causal genes13. Risk genes were identified using dual criteria: marginal posterior inclusion probability > 0.8 and P < 5 × 10−8.
MAGMA for gene annotation
We used MAGMA with default parameters to aggregate the SNP-level association statistics into gene scores in all our gene analyses. In this way, the extent of association of each separate gene with the investigated phenotype could be appraised21,22. For detailed parameter settings and methodology, follow the original MAGMA documentation15.
Bayesian colocalization and MR
Key genes identified from the convergence of MAGMA, UTMOST, and FUSION analyses underwent further investigation through MR, SMR, FOCUS and Bayesian colocalization. MR study employed cis-eQTL SNPs as instrumental variables (IVs) under the conditions of p < 5 × 10−8, R2< 0.001, and a 10,000 kb window size23. When only a single IV was available, the causal effect was estimated using the Wald ratio method, with significance determined at p < 0.05. it’s worth noting that summary data-based Mendelian randomization (SMR) offers greater statistical power compared to traditional MR analyses by leveraging top-ranked cis-QTLs. We selected the optimal cis-QTL within a 1,000 kb window centered on the gene of interest, using a stringent significance threshold of p < 5.0 × 10−8. Furthermore, to mitigate the potential confounding effects of pleiotropy and linkage, we excluded SNPs with an allele frequency difference greater than 0.2 in paired datasets and employed the (heterogeneity in dependent instrument) HEIDI test. A p-value less than 0.01 was considered indicative of pleiotropic associations24. Bayesian co-localization analysis was performed to verify the posterior probability (PP) of the five possible relationships25, A posterior probability (PPH4) exceeding 0.7 suggests a shared causal variant between GWAS and eQTL25,26. All analyses were carried out using the “TwoSampleMR” and “coloc” R packages27.
Gene expression omnibus (GEO) analysis
To identify differentially expressed genes (DEGs) between BPH and normal prostate tissues, we analyzed the GSE3868 dataset from the Gene Expression Omnibus (GEO) database. The analysis included two BPH samples and two matched normal prostate samples. Following data normalization, DEGs were determined using the GEO2R online tools(https://www.ncbi.nlm.nih.gov/geo/geo2r/). The cut off criterion was set as adj.pval < 0.05 and (log2 fold change [FC]) > 1.
GeneMANIA analysis
The GeneMANIA platform28 (https://genemania.org/) integrates various datasets, including genetic interactions, pathways, and co-expression data, to provide insights into target genes and their functions. By incorporating these diverse gene-function relationships, GeneMANIA enhances our understanding of the biological roles and interactions of the target genes29.
Results
TWAS analyses results of BPH
Of those, a total of 314 genes reached significance in the cross-tissue TWAS analysis at P < 0.05 (Supplemental Table 1), of which 28 genes remained significant after FDR correction at PFDR < 0.05 (Table 1). In the validation dataset, the cross-tissue analysis screened a total of 4 important genes after FDR correction (PFDR < 0.05) (Supplemental Table 2). In the single tissue TWAS analysis, 493 genes across at least one tissue reached PFDR < 0.05 (Supplemental Table 3). In contrast, in the single-tissue analysis of the validation dataset, a total of 54 significant genes were identified after FDR correction (Supplemental Table 4). Given that there was no intersecting genes in the validation set, subsequent analyses were further explored using the discovery cohort. Thus 12 candidate genes passed strict screening thresholds in both cross-tissue and single-tissue analyses in discovery set. The genes included eleven protein-coding genes (FGFR3, INO80B, TACC3, PTPN13, OXER1, SPARCL1, SMIM43, MTMR3, BCL11A, FEZ2, NT5C1B) and one non-coding gene (CHKB-DT) (Supplementary Table 5).
COJO analysis
COJO analysis was performed on the 12 candidate genes, mainly found on chromosomes 2, 4, and 22, in their respective tissues to eliminate false positives caused by linkage disequilibrium (LD) (Supplementary Table 6). in Adipose_Subcutaneous tissue, the regulation of FGFR3 expression resulted in a significant reduction of TWAS signaling in TACC3, a result that was validated in Artery_Coronary tissue. Meanwhile, BCL11A, SMIM43, OXER1, CHKB-DT, INO80B, and SPARCL1 did not exhibit false-positive results (Supplementary Fig. 1). due to their significance in TWAS results from only a single tissue and the potential impact of LD, NT5C1B, PTPN13, FEZ2, and MTMR3 were excluded from further analyses.
Gene analysis of MAGMA
The gene-based scan by MAGMA gave 443 genes associated with the BPH phenotype after FDR correction (p < 0.05) (Supplemental Table 7). Next, the ten most significant genes in the Bonferroni correction were labeled on the Manhattan map (Fig. 2A). In terms of tissue specific enrichment, after FDR correction, a total of 6 tissues showed positive results (p < 0.05), including bladder, prostate and urethra. MAGMA pathway enrichment analysis revealed significant enrichment in developmental processes (prostate gland branching and mammary gland formation), transcriptional regulation (RNA biosynthesis and nucleobase metabolism), and cellular processes (Golgi vesicle tethering and biosynthetic regulation) (Supplementary Fig. 2).

(A) Manhattan plot of the MAGMA results for BPH; (B) Venn diagram.
MR and colocalization results
The intersection of nominally significant genes from three analytical approaches identified eight key genes: BCL11A, FGFR3, NT5C1B, INO80B, MTMR3, SMIM43, PTPN13 and TACC3 (Fig. 2B). MR analyses demonstrated a causal relationship between the eight genes associated with BPH (Fig. 3 and Supplemental Table S8), in addition, we conducted SMR analysis on the potential eight genes and corresponding tissues, and HEIDI test showed no obvious heterogeneity and pleiotropy, and the results of SMR analysis were consistent with those of MR (Supplemental Table S9). while co-localization provided stronger evidence of causality for the INO80B gene (posterior probability PPH4 > 0.7) (Fig. 4 and Supplemental Table S10). the INO80B gene is located on human chromosome 2, specifically at the 2q14.2 position. The rs11695896 variant was identified as the most significant co-localization locus for BPH across 18 tissues.

The forest map of MR results.

The colocalization results were obtain in Adipose_Subcutaneous.
The results of FOCUS precision positioning
Using FOCUS software, we performed fine-mapped TWAS analysis on European ancestry data across 49 tissues. With threshold criteria of model credible parameter = 1 and PIP > 0.8, we identified 321 candidate genes (Supplementary Table S11). FOCUS generated regional plots showing predicted expression correlations, with TWAS statistics and PIPs for each gene illustrated in Supplementary Fig. 3.
DEGs and GeneMANIA analysis
After normalizing the dataset GSE3868, we found that all eight previously identified genes were down-regulated in BPH tissues, with significant differences in INO80B (Supplemental Table S12 and Figure S4). Figure 5 illustrates the gene interaction network with INO80B as the central linker. INO80B is mainly associated with the formation of the INO80-type complex, DNA helicase complex, and SWI/SNF superfamily-type complex within the gene network (Supplementary Table 13).

GeneMANIA gene network.
Discussion
We integrated the GWAS summary statistics for BPH with eQTL summary data obtained from GTEx V8 to investigate how genetic susceptibility influences gene expression in the development of BPH. TWAS analysis, followed by MR and co-localization results, suggested that INO80B is a susceptibility gene for BPH.
Giri et al.30 explored the genetic links between metabolism-related traits and benign prostatic hyperplasia (BPH).They used the Illumina Cardio-MetaboChip platform for genotyping and constructed genetic risk scores (GRS) for height, BMI, and waist-to-hip ratio (WHR).This research highlighted how metabolic disorders might influence BPH development. Additionally, a separate GWAS employed the Sequenom iPLEX system to genotype specific SNPs. following Bonferroni correction, a significant association was established between GATA3 (rs17144046) and the mechanisms driving BPH31. This study utilizes UTMOST’s cross-tissue TWAS to improve the identification of genes associated with complex traits. Integrating gene expression data from multiple tissues provides a comprehensive view of gene-trait relationships, enhancing the detection of associations that single-tissue analyses might overlook. Importantly, the study identifies INO80B as a gene linked to BPH risk, a novel association not previously documented.
The INO80 protein complex is integral to chromatin remodeling, where it regulates chromatin structure by repositioning or reorganizing nucleosomes. This action directly influences gene expression by modulating the accessibility of chromatin32. Furthermore, INO80 is essential for repairing DNA double-strand breaks (DSBs).it facilitates the recruitment and localization of repair proteins to the site of damage, thereby aiding the homologous recombination repair (HRR) process. Additionally, the INO80 complex is vital in managing replication stress at the replication fork, ensuring the continuity of the replication process and preventing the accumulation of DNA damage33,34. Previous studies have found that certain INO80 subunits are overexpressed in various cancers, including breast cancer35, neuroendocrine prostate cancer36, and melanoma37. Moreover, elevated expression levels of these subunits are positively correlated with poor prognosis38.
On the other hand, INO80B is a crucial subunit of the INO80 chromatin remodeling complex, which plays essential roles in transcription regulation, DNA replication, and DNA repair39. The INO80 complex has been implicated in various cellular processes, including cell growth and proliferation control, making its potential involvement in prostatic hyperplasia biologically plausible. The negative correlation observed suggests that decreased INO80B expression might contribute to BPH development. This aligns with previous studies showing that chromatin remodeling complexes can act as regulatory factors in prostate tissue homeostasis. For instance, the SWI/SNF chromatin remodeling complex has been shown to influence prostate development and disease40.
Interestingly, we discovered that INO80B is involved in the inhibition of benign prostatic hyperplasia (BPH). The development of BPH is typically associated with various factors, including abnormal androgen signaling, inflammatory responses, and overactivation of cell proliferation signals41. BPH is characterized by the excessive proliferation of prostate cells and the abnormal accumulation of extracellular matrix42. The INO80 complex potentially curbs excessive cell proliferation by modulating the expression of cell cycle-related genes, especially those linked to the androgen receptor43. Further research is needed to validate these findings.
Protein-protein interaction network analysis demonstrates that INO80B functions within a complex molecular framework, involving multiple cellular processes relevant to prostatic tissue regulation. The INO80B interaction network reveals several key functional modules that may explain its protective effect against BPH. At the core of this network, INO80B strongly interacts with other INO80 complex components (INO80, INO80E) and the ATP-dependent DNA helicases RUVBL1 and RUVBL2, suggesting its fundamental role in chromatin remodeling activities39. The presence of YY1, a crucial transcription factor, indicates INO80B’s involvement in promoter regulation and enhancer-promoter interactions, potentially influencing prostate-specific gene expression patterns.
The network also reveals connections to growth factor signaling through IGFBP2, suggesting a role in cellular proliferation control, which is particularly relevant to BPH pathogenesis. The interaction with ZNHIT family proteins (ZNHIT1, ZNHIT2, ZNHIT3, ZNHIT6) implies involvement in histone modification and nucleosome positioning, potentially affecting global gene expression patterns in prostatic tissue. These interactions align with previous observations of chromatin remodeling complexes influencing prostate development and disease40. The presence of DDX59 and ACTR5 in the network suggests INO80B’s involvement in RNA metabolism, DNA repair, and nuclear organization, processes crucial for maintaining tissue homeostasis. This comprehensive interaction network supports the observed negative correlation with BPH by demonstrating INO80B’s role as a master regulator of multiple cellular processes relevant to prostate tissue maintenance. The interaction with the SWI/SNF chromatin remodeling complex components further supports its role in prostate tissue regulation, as these complexes have been shown to influence prostate development and disease progression44. The complex also interacts with androgen receptor (AR) signaling pathways, as demonstrated by previous studies showing chromatin remodeling complexes modulating AR-dependent transcription45. The inflammatory response regulation through chromatin remodeling, as suggested by Wu et al.46, may represent another mechanism by which INO80B influences BPH development.
Several limitations are to be pointed out concerning our study. First, the samples obtained from European populations may reduce the generalizability of our conclusions. Of course, such discrepant results for the discovery and validation sets should be interpreted with caution. there are genetic background differences between the UKB and Finnish populations, the pattern of LD may be different in different populations, and allele frequencies may be significantly different in different populations. Second, although we employed FDR correction to reduce false positive rates, the lack of independent validation sets presents a limitation in verifying our findings. Remaining studies are also needed in the future to involve more in vitro and in vivo experiments, which would explain the mechanisms at work. Our study provides new perspectives and insights into the pathophysiological mechanisms of BPH, despite its limitations.
Conclusion
In summary, our cross-tissue TWAS analyses revealed a correlation between INO80B expression and the risk of BPH, offering new insights into the genetic architecture of this condition. However, this is only a preliminary exploratory finding, further functional studies are necessary to elucidate the potential biological activity of these significant signals.
Data availability
This study used R version 4.4.1 for data collation and analysis. The S-PrediXcan package (https://github.com/hakyimlab/MetaXcan) was used to analyze TWAS, while TWAS fine mapping was conducted using the FOCUS package (https://github.com/bogdanlab/focus/). The FUSION analysis of relative weights and sample download from (http://gusevlab.org/projects/fusion/), MAGMA analysis reference FUMA generated online website (https://fuma.ctglab.nl/snp2gene), The analysis of the SMR reference from (https://yanglab.westlake.edu.cn/software/smr/). The Code via the following link: https://github.com/wangli19971118/xTWAS. All datasets in this study are available for download in the online dataset/supplementary file and further contact the corresponding author if necessary.
References
Roehrborn, C. G. Benign prostatic hyperplasia: an overview. Rev. Urol. 7(Suppl 9), S3–S14 (2005).
Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396(10258), 1204–1222 (2020).
Barry, M. J. et al. The natural history of patients with benign prostatic hyperplasia as diagnosed by North American urologists. J. Urol. 157(1), 10–14 (1997).
Martin, S., Lange, K., Haren, M. T., Taylor, A. W. & Wittert, G. Risk factors for progression or improvement of lower urinary tract symptoms in a prospective cohort of men. J. Urol. 191(1), 130–137 (2014).
Welliver, C. et al. Trends in Lower Urinary Tract Symptoms Associated with Benign Prostatic Hyperplasia, 2004 to 2013: the Urologic Diseases in America Project. J. Urol. 203(1), 171–178 (2020).
Sanda, M. G., Beaty, T. H., Stutzman, R. E., Childs, B. & Walsh, P. C. Genetic susceptibility of benign prostatic hyperplasia. J. Urol. 152(1), 115–119 (1994).
Hellwege, J. N. et al. Heritability and genome-wide association study of benign prostatic hyperplasia (BPH) in the eMERGE network. Sci. Rep. 9(1), 6077 (2019).
Gudmundsson, J. et al. Genome-wide associations for benign prostatic hyperplasia reveal a genetic correlation with serum levels of PSA. Nat. Commun. 9(1), 4568 (2018).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337(6099), 1190–1195 (2012).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20(8), 467–484 (2019).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47(9), 1091–1098 (2015).
Hu, Y. et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 51(3), 568–576 (2019).
Mancuso, N. et al. Pasaniuc BJNg: Probabilistic fine-mapping of transcriptome-wide association studies. 51(4), 675–682 (2019).
Gui, J. et al. Identification of novel proteins for sleep apnea by integrating genome-wide association data and human brain proteomes. Sleep Med. 114, 92–99 (2024).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11(4), e1004219 (2015).
The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45(6), 580–585 (2013).
Jiang, L., Zheng, Z., Fang, H. & Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 53(11), 1616–1621 (2021).
Sun, R., Hui, S., Bader, G. D., Lin, X. & Kraft, P. Powerful gene set analysis in GWAS with the Generalized Berk-Jones statistic. PLoS Genet. 15(3), e1007530 (2019).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48(3), 245–252 (2016).
Liao, C. et al. Transcriptome-wide association study of attention deficit hyperactivity disorder identifies associated genes and phenotypes. Nat. Commun. 10(1), 4450 (2019).
de Leeuw, C. A., Neale, B. M., Heskes, T. & Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 17(6), 353–364 (2016).
de Leeuw, C. A., Stringer, S., Dekkers, I. A., Heskes, T. & Posthuma, D. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat. Commun. 9(1), 3768 (2018).
Wang, L., Deng, J. Y., Li, K. P., Shan, Y. & Zhu, P. Y. Inflammatory bowel disease and bladder cancer risk: based on a Mendelian randomization study. BMC Urol. 23(1), 195 (2023).
Zhu, T. et al. Mitochondrial FIS1 As a Novel Drug Target for the Treatment of Erectile Dysfunction: A Multi-Omic and Epigenomic Association Study. 42 (2024).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10(5), e1004383 (2014).
Zhu, M. et al. A cross-tissue transcriptome-wide association study identifies novel susceptibility genes for lung cancer in Chinese populations. Hum. Mol. Genet. 30(17), 1666–1676 (2021).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7 (2018).
Mostafavi, S. et al. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 9(Suppl 1), S4 (2008).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38(Web Server issue), W214–220 (2010).
Giri, A., Edwards, T. L., Motley, S. S., Byerly, S. H. & Fowke, J. H. Genetic Determinants of Metabolism and Benign Prostate Enlargement: Associations with Prostate Volume. PLoS One 10(7), e0132028 (2015).
Na, R. et al. A genetic variant near GATA3 implicated in inherited susceptibility and etiology of benign prostatic hyperplasia (BPH) and lower urinary tract symptoms (LUTS). Prostate 77(11), 1213–1220 (2017).
Poli, J., Gasser, S. M., & Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. Lond. Ser. B, Biol. Sci. 372(1731) (2017).
Lafon, A. et al. INO80 Chromatin Remodeler Facilitates Release of RNA Polymerase II from Chromatin for Ubiquitin-Mediated Proteasomal Degradation. Mol. Cell 60(5), 784–796 (2015).
Poli, J. et al. Mec 1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev. 30(3), 337–354 (2016).
Eirew, P. et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518(7539), 422–426 (2015).
Beltran, H. et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 22(3), 298–305 (2016).
Zhou, B. et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 30(12), 1440–1453 (2016).
Hu, J. et al. Ino80 promotes cervical cancer tumorigenesis by activating Nanog expression. Oncotarget 7(44), 72250–72262 (2016).
Watanabe, S. et al. Structural analyses of the chromatin remodelling enzymes INO80-C and SWR-C. 6(1), 7108 (2015).
Muthuswami, R. et al. BRG1 is a prognostic indicator and a potential therapeutic target for prostate cancer. 234(9), 15194–15205 (2019).
Wilson, J. D. The pathogenesis of benign prostatic hyperplasia. Am. J. Med. 68(5), 745–756 (1980).
Wang, L., Chen, X., Li, K., Yin, S. & Zhu, P. Perioperative and functional outcomes of salvage versus primary Holmium laser enucleation of the prostate: Evidence-based on controlled studies. Asian J. Surg. 46(12), 5411–5420 (2023).
Jafari, H., Hussain, S. & Campbell, M. J. Nuclear Receptor Coregulators in Hormone-Dependent. Cancers. 14(10), 2402 (2022).
Zhou, B. et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. 30(12), 1440–1453 (2016).
Zhao, D. et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. 542(7642), 484–488 (2017).
Wu, Q. et al. The BRG1 chromatin remodeling enzyme links cancer cell metabolism and proliferation. 7(25), 38270 (2016).
Acknowledgements
We are grateful to the consortium that provided all the public GWAS data.
Funding
This study was supported by The Key Research and Development Program of Science and Technology Department of Gansu Province (24YFFA049).
Author information
Authors and Affiliations
Contributions
Conceptualization: WL, SY, XR. Search and evaluation: KP, WL, JW, KY. Data Analysis: WL, SW. Writing—original draft and Visualization: WL, LY. Writing—Review and Revision: LY. Super-vision and Project Management: WL, SY, KP, LY.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Chen, Sy., Yang, Jw. et al. A cross-tissue transcriptome-wide association study identifies new susceptibility genes for benign prostatic hyperplasia. Sci Rep 15, 3186 (2025). https://doi.org/10.1038/s41598-025-87651-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87651-y
