
Abstract
Lethal alleles are mutations in the genome that cause embryonic losses in affected homozygous embryos and, therefore, can negatively influence reproduction rates in commercial populations. Thus, this study aimed to identify genomic regions containing potential lethal haplotypes in Nellore breed; identify candidate genes located within these regions; and investigate the reproductive performance of heterozygous carriers of lethal haplotypes in Nellore cattle. Forty-five genomic regions harboring putative lethal haplotypes were identified, which overlap with 360 genes. Gene ontology analyses of these genes revealed biological processes associated with the development of sexual traits in males and females, key functions of the immune system, energy homeostasis, and embryonic development. The gene networks were involved in metabolic pathways including ovarian steroidogenesis, oocyte meiosis, and insulin secretion. Matings between carrier dam and carrier sire led to a reduction of up to -203.46% in pregnancy success probability, an increase of 275.15% in probability of pregnancy loss, 295.03% for stillbirth occurrence, and 301.40% for pre-weaning mortality when compared to non-carrier dam and sire matings. The results highlight the importance of identifying animals that are carriers of lethal haplotypes to avoid the propagation of these haplotypes in the population.
Similar content being viewed by others

Introduction
Most mutations in diploid organisms typically manifest as recessive alleles, resulting in a harmful effect when homozygous1. Lethal alleles have been identified to significantly impact the profitability and efficiency of livestock industry. These genetic variants have the potential to reduce the expression of economically important traits2, cause embryonic losses3, reduce pregnancy and rebreeding rates4,5, and increase post-natal mortality5.
Lethal alleles can be tracked using a haplotype-based approach, where the harmful region in the genome can be inferred based on observed and expected frequencies of homozygous haplotypes in the population6. This approach uses data from living animals, instead of relying on information from affected embryos (that may not have survived). Its efficacy depends on having a sufficient number of genotyped animals in a population, as well as accurate genealogical records for tracing inheritance patterns5,6,7. If the number of genotyped animals is sufficiently high, the complete absence of homozygous carriers of some haplotypes in different segments of the genome may not be a completely random event6.
In populations under direct selection for economically important traits, the presence of a lethal allele conferring an advantage to heterozygous individuals in their phenotypic performance, may facilitate the propagation of this deleterious genetic variant across subsequent generations2,7. This event hides the antagonistic effects associated with the homozygous recessive state of the allele, which results in harmful effects or mortality among affected animals6. Furthermore, higher linkage disequilibrium between the lethal allele and alleles with favorable effects on the target traits in breeding programs may also maintain the lethal allele in the population due to selection practices2.
Most studies aimed at identifying lethal genetic variants in bovine species have been focused on taurine breeds (Bos taurus taurus), with few reports in animals of Zebu origin (Bos taurus indicus). Schmidt et al.5 reported 30 potentially lethal haplotypes in the Nellore breed (Bos taurus indicus), distinct from those identified in taurine breeds. Similarly, Id-Lahoucine et al.4 identified 19 potentially lethal haplotypes in the Angus breed based on a transmission ratio distortion (TRD) approach and observed that heterozygous carriers of these haplotypes showed a reduction of 15% in pregnancy rates. These results support the hypothesis that genomic regions harboring putative lethal haplotypes may have undesirable effects even in heterozygous carriers. Furthermore, identifying carriers of lethal haplotypes to guide mating decisions may be an effective strategy to prevent the spread of lethal alleles to future generations, potentially enhancing reproductive efficiency.
In this study, we used data belonging to a closed experimental Nellore breeding program selecting animals for stabilizing and directional selection, based on YW for 44 years8,9. These closed selection lines differ from each other on phenotypic performance, such as average body weight at different ages, body measurements and carcass trait. This distinctive breeding design can offer valuable insights into the implications of the harmful effect of recessive alleles on reproductive aspects within a closed Nellore population under selection. Hence, the objectives of this study were to identify potential genomic regions containing lethal haplotypes in a Nellore (Bos taurus indicus) cattle population; to find genes and functional processes linked to the gene network within specified genomic regions harboring putative candidates for lethal haplotypes; and to investigate the reproductive performance of carriers of lethal haplotypes, with a specific focus on the probabilities of pregnancy success, pregnancy losses, stillbirth, and pre-weaning mortality in at-risk matings.
Materials and methods
Ethical statement
The production system evaluated followed all animal welfare guidelines established by Law No. 11.977 of the State of São Paulo, Brazil. Data were obtained from an existing database, and therefore, approval from the Ethics Committee was not required.
Population
The Nellore dataset belonged to an experimental selection design from the Beef Cattle Research Center (Institute of Animal Science – IZ, Sertãozinho, SP, Brazil; Fig. 1). In 1980, the Nellore IZ experimental breeding program was established with the aim of selecting animals based on YW, measured at 378 days of age in young bulls and 550 days of age in heifers. Three distinct selection lines were designed: Line 1 - Nellore control (NeC), with animals selected for YW close to the average of the contemporary group (stablishing selection), within birth year x herd; Line 2 - Nellore Selection (NeS), with animals selected for the YW with a maximum selection differential, within birth year x herd; and Line 3 - Nellore Traditional (NeT) with animals selected for higher YW, and since 2008 has been also selected for lower residual feed intake (RFI), within birth year x herd.

Animals of the Nellore breed from the experimental breeding program of the Institute of Animal Science—Beef Cattle Research Center (Sertãozinho, SP, Brazil).
The selection lines are closed, which means that only bulls and dams born in the respective selection line are used for breeding. The inbreeding levels of all three herds are controlled with planned mattings considering the co-ancestry coefficient. The bulls are utilized in mating seasons for two consecutive years, beginning at two years of age with 15 cows and at three years of age with 30 cows. Annually, on average, 4, 6, and 8 bulls are utilized for breeding in the NeC, NeS, and NeT selection lines, respectively.
Pedigree and genomic databases
The pedigree dataset included genealogical information of 12,843 individuals born from 1950 to 2022, involving 478 sires and 2927 dams. We used the RelaX2 software10 for pedigree evaluation and it was identified a total of 13 founders for NeC, 27 for NeS, and 29 for NeT. The estimated effective population size was 107 ± 15 animals, considering the three selection lines, based on the methodology outlined by Gutiérrez et al.11. Additionally, the number of equivalent complete generations (ECG) was 5.02, while the number of discrete generations (DGE) was 6.50 for the three selection lines. The mean inbreeding was 2.81% for NeC, 2.23% for NeS, and 2.15% for NeT, with the highest inbred animal exhibiting 25.00% of inbreeding.
The genomic dataset included 3,226 genotyped animals, with 1,741 males and 1,485 females genotyped with different Bead chip assays (Supplementary Table S1). Before imputation, were removed monomorphic markers, markers with the same coordinate, located on non-autosomal chromosomes and with GenCall score lower than 0.90. After this quality control (QC), the animals genotyped with medium-density panels (50 K and 75 K SNP panels) were imputed to the Illumina BovineHD panel (770 K) using the FImpute v.3 software12. The expected accuracy from the imputation process was higher than 0.9713,14. After the imputation, 3,226 animals and 612,154 SNP markers remained in the genomic dataset for further analyses. Possible genotype inconsistencies between parents and progeny were adjusted using the seekparentf90 software15.
Reproductive records
A total of 5,093 reproductive records (pregnancy success, pregnancy loss, stillbirth and pre-weaning mortality) from 1,258 cows and 190 sires genotyped were used for the analyses. Annually, cows and heifers were exposed to natural or artificial insemination at a fixed time during 90-day breeding season. After approximately 30 days from the end of the breeding season, an ultrasound evaluation was performed to diagnose pregnancy, and females were classified as pregnant (1, success) or non-pregnant (0, failure). The pregnancy loss was determined by attributing a value of 1 to females with a positive pregnancy diagnosis but did not calve and 0 otherwise. For non-pregnant cows, no information about pregnancy loss was given.
The stillbirths were considered as calves that died within the first 48 h after birth being assigned as 1 (stillbirth), and calves that survived beyond this period were assigned as 0 (non-stillborn). Calves were weaned at around 7 months and pre-weaning mortality was determined by assigning a value of 1 (mortality) for calves that did not reach weaning age and 0 (survived) for animals that survived until weaning.
Contemporary groups (CG) for pregnancy success, pregnancy loss, and stillbirth were based on the combination of the breeding year (1980 to 2022) and selection line (NeC, NeS, and NeT). We did not include the season information in CG because all cows were bred in a single season of the year. For pre-weaning mortality, CG consisted of calves’ birth year (1981 to 2023), sex (male or female), and selection line (NeC, NeS, and NeT). CGs with less than three animals and no phenotypic variability within CG were excluded from further analyses.
Identification of candidates for lethal haplotypes
To identify potential candidates for lethal haplotypes this study followed the methods proposed by VanRaden et al.6 and applied by Schmidt et al.5. The findhap.f90 software v.316 was used to obtain the haplotypes using the sliding windows method17,18. The haplotype construction involved three-step iterative processes: initially, haplotypes consisting of 2,000 markers on the same chromosome were identified; next, haplotypes comprising 632 markers; and, finally, it identified haplotypes containing up to 200 markers, which were subsequently used and considered for further analyses. These numbers were defined based on the recommendations of findhap.f90 software considering the SNP panel density used for this study16.
The haplotypes that showed frequencies higher than 2% in the population but were not observed in a homozygous state were selected for further evaluation as putative lethal haplotypes. We established this threshold value to select only the haplotypes present in a significant number of genotyped animals. This was done with the aim of identifying genetic variants with the highest lethal potential within the population. This threshold value was also applied by Hoff et al.7 and Schmidt et al.2.
In accordance with the methods described by VanRaden et al.6, two approaches were used to estimate the expected number of homozygous individuals for each haplotype: (1) the Simple method – assumes random mating across the population over time. The calculation involves dividing the number of genotyped individuals by 4 and then multiplying it by the square of the carrier frequency of the haplotypes; and (2) the Mating method – accounts for the actual mating patterns observed in the population that generated the genotyped individuals. In this approach, the expected number of homozygous individuals is calculated by dividing the number of carrier mating sire × carrier maternal grandsire pairs by 4. This approach assumes that the allele frequencies for maternal granddams and maternal grandsires are equal.
The probabilities of observing zero homozygotes when n > 0 is expected were obtained using analogous formulas applied by VanRaden et al.6, Jenko et al.2, and Schmidt et al.5. For the Simple method, this probability (Phh) was calculated as (1 – C2/4) × N, where the absence of homozygous animals depends on the carrier frequency of heterozygous animals (C) and the number of genotyped animals (N). In contrast, for the Mating method, the probability follows a Bernoulli distribution and is equal to 0.75 raised to the power of the observed number of carrier mating sire × carrier maternal grandsire pairs.
To identify a putative recessive lethal haplotype region among the thousands of haplotypes that could present zero homozygous individuals by chance, specific conditions were set following Wu et al.3: (1) the haplotype carrier frequency had to exceed 2%; (2) the number of expected homozygous individuals for the haplotype had to be greater than 1 and (3) the probabilities of observing zero homozygotes had to be less than 0.6. All haplotypes satisfying these conditions were selected for further analyses.
Functional and gene set enrichment
The genomic regions identified as potential hotspots for lethal haplotype candidates were mapped to the genes located within these regions using the BioMart tool from the ENSEMBL (https://www.ensembl.org/biomart/martview/), considering the ARS-UCD 1.2 Bos taurus assembly19. Functional classification of genes for biological mechanisms (Gene Ontology - GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were identified using the cluster- Profiler R Package20.
Statistical analyses
After identifying all potential lethal haplotypes and their respective carriers, four mating classes were defined according to the carrier status of each lethal haplotype: (1) noncarriers (\(\:{\text{N}\text{C}}_{\text{D}\text{a}\text{m}}\:\times\:\:{\text{N}\text{C}}_{\text{S}\text{i}\text{r}\text{e}}\)); (2) carrier dam with noncarrier sire (\(\:{\text{C}}_{\text{D}\text{a}\text{m}}\:\times\:\:{\text{N}\text{C}}_{\text{S}\text{i}\text{r}\text{e}}\)); (3) noncarrier dam with carrier sire (\(\:{\text{N}\text{C}}_{\text{D}\text{a}\text{m}}\:\times\:\:{\text{C}}_{\text{S}\text{i}\text{r}\text{e}}\)); and (4) carrier dam with carrier sire (\(\:{\text{C}}_{\text{D}\text{a}\text{m}}\:\times\:\:{\text{C}}_{\text{S}\text{i}\text{r}\text{e}}\)). Following these mating classes, a logistic regression model considering a logit link function, was used to describe the effects of the four mating classes on the probabilities of pregnancy success, pregnancy loss, stillbirth, and pre-weaning mortality:
where \(\:\varvec{\upeta\:}\) was the logit transformation of the phenotypic records (θ) with \(\:{{\upeta\:}}_{i}=\text{l}\text{n}\left(\frac{{{\uptheta\:}}_{i}}{1-{{\uptheta\:}}_{i}\:}\right)\), assumed as y ~ bin(1, θ); y is the vector of phenotypic records (pregnancy, pregnancy loss, stillbirth, and pre-weaning mortality), in which ith element in y followed a Bernoulli distribution \(\:{\text{y}}_{\text{i}}\)~ Be(\(\:{{\uptheta\:}}_{\text{i}}\)); m is a vector with the fixed effects of mating class; Xm is an incidence matrix relating phenotypic records to mating class effect for each lethal haplotype; CG is a vector of fixed effects of CG; XCG is an incidence matrix relating phenotypic records to the CG effect; \(\:\mathbf{c}\mathbf{a}\) is a vector with the linear and quadratic effects of cow’s age at breeding as a covariate; \(\:{\mathbf{X}}_{\mathbf{c}\mathbf{a}}\) is an incidence matrix relating phenotypic records to the cow’s age effects; \(\:\mathbf{e}\) is the residual vector assumed as e ~ N(0, \(\:{\sigma\:}_{e}^{2}\)), where \(\:{\sigma\:}_{e}^{2}\) is the variance of the residuals.
The effects of mating type were expressed as an odds ratio probability, comparing the elements m1 with the other elements (m2, m3, and m4) of m corresponding mating classes. All the analyses described in this section were performed using Rstudio version 4.3.2 with the package stats21.
Results
Candidates for lethal haplotypes
A total of 45 genomic regions located on 18 chromosomes exhibited lethal haplotypes with an observed frequency higher than 2% and with no observed homozygous haplotypes in the genotyped animals (Table 1).
The BTAs that exhibited at least 2 potential candidate lethal haplotypes were identified on: BTA1 (7 candidates), BTA2 (4 candidates), BTA7 (3 candidates), BTA10 (2 candidates), BTA13 (4 candidates), BTA15 (3 candidates), BTA17 (4 candidates), BTA18 (4 candidates), BTA26 (3 candidates), BTA27 (2 candidates), and BTA29 (2 candidates). A total of 45 lethal candidate haplotypes presented probabilities lower than 0.60 using both, the simple and mating detection methods. Among these 45 putative lethal haplotypes, 42 had not been reported in other studies with cattle as potential carriers of lethal alleles. Only the haplotypes 998.14 in BTA7 (segment 998, haplotype 14), 2213.8 in BTA17 (segment 2213, haplotype 8), and 2620.22 in BTA 22 (segment 2620, haplotype 22) were previously identified as lethal haplotypes5.
The lethal haplotype with the highest number of carriers (735 animals) was the 1947.12 haplotype in BTA15 (segment 1947, haplotype 12), with size of 0.74 Mb and a frequency of 11.39% in the population. For this haplotype, 26 homozygous individuals were expected considering the Simple method (random mating) and 43 homozygous individuals considering the Mating method (observed mating pattern). The lethal haplotype with the lowest number of carriers was located in BTA26 (2850.17; segment 2850, haplotype 17), which has a size of 1.06 Mb, 224 carriers and a frequency of 3.48% in the evaluated population.
The lethal haplotypes exhibited an average size of 0.91 ± 0.43 Mb, with the largest haplotype identified on BTA10 (1387.10; segment 1387, haplotype 10), with a size of 3.42 Mb, 367 carriers and a frequency of 5.69% in genotyped the population. The smallest was haplotype 1637.10 located in BTA12 (segment 1637, haplotype 10), with a size of 0.57 Mb, 245 carriers and a frequency of 3.80% in the population.
Identified genes and functional enrichment analyses
A total of 360 genes were identified within the genomic regions indicated as a putative lethal haplotype (Supplementary Table S2). Enrichment analysis identified biological processes associated with the development of sexual traits in males and females, key functions of the immune system, energy homeostasis, and embryonic development (Table 2).
Specifically, the zinc finger protein (ZFP42), GATA binding protein 3 (GATA3), luteinizing hormone subunit beta (LHB), LDL receptor related protein 2 (LRP2), BCL2 associated X, apoptosis regulator (BAX), and aldo-keto reductase family 1 member C3 (AKR1C3) genes, were annotated and associated with the development of primary male sexual traits and male gonad development (GO:0046546; GO:0008584). Furthermore, the LRP2, BAX, and retinol binding protein 4 (RBP4) genes were identified to influence female genitalia development (GO:0030540).
Regarding the immune system, dehydrogenase/reductase 9 (DHRS9), aldo-keto reductase family 1 member C4 (AKR1C4), cytochrome P450 Family 26 subfamily A Member 1 (CYP26A1), GATA3, cytochrome P450 Family 26 subfamily C Member 1 (CYP26C1), LHB, RBP4, beta-carotene oxygenase 2 (BCO2), and AKR1C3 genes were associated with the hormone metabolic process (GO:0042445).
Concerning energy homeostasis, the glucose homeostasis process (GO:0042593) was identified in the GO analyses, with the fibroblast growth factor 21 (FGF21), forkhead box A3 (FOXA3), insulin (INS), transient receptor potential cation channel subfamily M member 4 (TRPM4), glucose-6-phosphatase catalytic subunit 2 (G6PC2), RBP4, transformer 2 beta homolog (TRA2B), SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 (SMARCB1), and tyrosine hydroxylase (TH) genes annotated and associated with this function. The glycogen synthase 1 (GYS1), insulin like growth factor 2 (IGF2), INS, glycogen phosphorylase B (PYGB), and pleckstrin homology like domain family A member 2 (PHLDA2) genes influenced the energy reserve metabolic process (GO:00006112). For the regulation of embryonic development (GO:0045995) there were also identified genes such as RuvB like AAAATPase 2 (RUVBL2), NLR family pyrin domain containing 5 (NLRP5), PHLDA2, and actin-related protein 8 (ACTR8).
Gene enrichment analysis identified metabolic pathways affecting the ovarian steroidogenesis by the action of the gene set (INS, LHB, and AKR1C3), oocyte meiosis (INS, PPP2R1B, YWHAH, and CALM3); and insulin secretion (INS, TRPM4 and CACNA1D) (Table 2).
Reproductive performance of matings involving carriers of lethal haplotypes
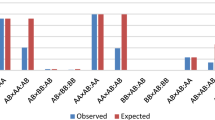
Among the 1,258 genotyped cows with mating records, 1,209 of them were carriers of at least one candidate lethal haplotype in a heterozygous state. Regarding the sires, 186 out of 190 were carriers of at least one putative lethal haplotype. Overall, 2,564 combinations between the mating type CDam × CSire were observed in the reproductive records (Supplementary Table S3). The number of matings for each lethal haplotype involving NCDam × NCSire, CDam × NCSire, NCDam × CSire and CDam × CSire are presented in Supplementary Table S4.
Only the haplotypes 42.4 (BTA1), 935.15 (BTA6), and 2214.4 (BTA17) exhibited associations with the four traits: pregnancy success, pregnancy loss, stillbirth and pre-weaning mortality (P-value < 0.05). On the other hand, the haplotypes 1736.1 (BTA13), 1756.27 (BTA13), 1774.2 (BTA13), 1892.3 (BTA14), 1947.12 (BTA15), 1948.12 (BTA15), 2213.8 (BTA17), 2219.3 (BTA17), 2260.18 (BTA18), 2300.15 (BTA18), 2787.1 (BTA25), 2850.17 (BTA26), and 3072.6 (BTA29) were not associated with pregnancy success, pregnancy loss, stillbirth and pre-weaning mortality occurrences (P-value > 0.05).
Twenty-one lethal haplotypes had significant effects (P-value < 0.05) on pregnancy success (Table 3). Matings involving the combination of CDam × NCSire exhibited reductions of up to -86.83% in pregnancy success probability when compared with NCDam × NCSire mating, as observed with haplotype 223.10 (BTA2). Similarly, mating NCDam × CSire showed reductions of up to -88.02% in pregnancy success when compared to NCDam × NCSire mating, detected with haplotype 193.7 (BTA2). Furthermore, mating CDam × CSire exhibited reductions of up to -203.46% in pregnancy success probability when compared to NCDam × NCSire mating considering haplotype 998.14 (BTA7).
For pregnancy loss, 11 haplotypes exhibited significant effects on its occurrence (P < 0.05) (Table 4). The matings CDam × NCSire increased the probability of pregnancy loss by 78.36% when compared to NCDam × NCSire, as observed in haplotype 2923.7 (BTA27). Similarly, mating NCDam × CSire had up to 105.23% probability of experiencing pregnancy loss, observed in carriers of haplotype 2923.7 (BTA27). Regarding matings of CDam × CSire, the probabilities of pregnancy loss occurrence ranged from 39.04%, observed in haplotype 10.14 (BTA1), to 275.15% when considering haplotype 1739.3 (BTA13).
Thirteen candidate lethal haplotypes significantly influenced stillbirth occurrence (P < 0.05), with 6 of them being located on BTA1 (Table 4). In matings between CDam × NCSire and NCDam × CSire of haplotype 2027.5 (BTA16), the probabilities of observing a stillbirth were of 112.50% and 181.13%, respectively. Furthermore, matings between carriers of haplotype 935.15 in the CDam × CSire class showed the highest probabilities for stillbirth occurrence, with a 295.03% increase in its likelihood.
Considering pre-weaning mortality, 14 candidate lethal haplotypes had significant effects on the pre-weaning mortality of calves (P < 0.05) (Table 4). The probabilities of observing pre-weaning mortality ranged from 14.56% (haplotype 935.15, BTA6) to 85.43% (haplotype 2027.5, BTA16) in CDam × NCSire matings, 14.88% (haplotype 1110.12, BTA8) to 80.19% (haplotype 2852.58, BTA26) in NCDam × CSire matings, and 44.03% (haplotype 42.4, BTA1) to 301.40% (haplotype 1110.12, BTA8) in CDam × CSire matings.
Discussion
Candidates for lethal haplotypes
Lethal haplotypes have been identified in livestock species, including horses22,23, sheep24, dairy cattle3,25,26, and beef cattle2,5,27. The identification of lethal haplotypes represents a strategy for guiding mating decisions aimed at mitigating the dissemination of lethal alleles to subsequent generations and compromising herd productive efficiency. However, there is a lack of research on the Nellore breed (Bos taurus indicus), having only one study to date that identified potential lethal haplotypes candidates5, despite its significant contribution to global meat production. The study of Nellore cattle is particularly relevant due to their prominent role in Brazilian beef production, as they are resilient to environmental challenges of tropical climates9. Furthermore, the experimental breeding program that provided the data for this study is one of the pioneering initiatives in genetic improvement of the Nellore breed in Brazil. Genetic material from elite animals is supplied to herds across the entire country, playing an important role in establishing diverse herds throughout Brazilian territory, and some Latin American countries. This emphasizes the importance of investigating genetic factors that may influence the productivity and sustainability of Nellore cattle, such as the occurrence of lethal haplotypes.
In this study, 45 chromosomal regions containing putative lethal haplotypes were identified (Table 1). These haplotypes exhibited frequencies exceeding 2% in the population with no homozygous individuals observed among genotyped animals. Furthermore, the probabilities of observing zero homozygous individuals were lower than 0.60, as determined by the two detection tests used to search for lethal alleles (simple and mating methods) based on the expected frequency of homozygous haplotype carriers, as described by VanRaden et al.6.
Out of the 45 candidates, 42 represent novel genomic regions that have not been previously reported in other cattle studies as hotspots for lethal alleles. This finding reinforces the hypothesis that the Nellore breed may harbor distinct genomic segments containing previously unreported deleterious mutations. Furthermore, it is important to note that the population evaluated in this study originates from a selection experiment initiated in the 1980s and has since been maintained as closed selection lines. A closed population can make high frequency haplotypes easier to find, but then they may have low frequency in the general Nellore population. Besides, populations of closed breeding programs may also encounter challenges in maintaining genetic variability across generations, leading to increased levels of inbreeding resulting from mating between genetically related animals28. In this study, the highest inbreed animal was observed in the selection line NeC and carried 5 potential lethal haplotypes. Increased inbreeding can lead to an increased frequency of deleterious alleles29,30.
The haplotypes 998.14 (BTA7), 2213.8 (BTA17), and 2620.22 (BTA22), previously identified by Schmidt et al.5, were also found in this study as potential candidates for lethal haplotypes in the Nellore breed (Table 1). This result suggests that these haplotypes may be specific to the Nellore cattle breed and highlights potential fixation of these genomic regions in Nellore animals. Schmidt et al.5 conducted their study using data from 276 commercial herds located in different Brazilian regions. One possible explanation for the recurrence of these three lethal haplotypes in our study is that all Nellore animals trace back to a few common ancestors, which may contribute to the identification of these haplotypes across studies.
The genomic regions containing putative lethal haplotypes were identified on 18 chromosomes (1, 2, 6, 7, 8, 10, 12, 13, 14, 15, 16, 17, 18, 22, 25, 26, 27, and 29), with heterozygous carrier frequencies ranging from 3.48 to 11.39% in the population (Table 1). Schmidt et al.5 reported frequencies of lethal haplotypes ranging from 2.98 to 12.21% in Nellore cattle considering 30 haplotypes harboring potential lethal alleles, while Wu et al.26 in a Nordic Red Dairy cattle population observed frequencies ranging from 3.82 to 12.91% on 18 putative recessive lethal regions.
The haplotypes exhibiting high frequencies within the population may be propagated through the population via the use of popular elite sires for breeding26 or pleiotropic effects linked with enhanced performance in target traits of breeding programs2. In this study, the sire with the largest number of offspring (82 animals) carried 4 potential lethal haplotypes, which may have contributed to spreading these haplotypes across generations. Kadri et al.31 reported the effects of a pleiotropic recessive allele in Nordic Red Dairy cattle that cause embryonic death but is also associated with greater milk yield.
Some of the identified haplotypes are located in adjacent segments with a similar number of carriers, such as haplotypes 41.4, 42.4, 43.4, 44.4, and 46.4 on BTA1; 223.10, 224.9, 225.11 on BTA2; 1736.1 and 1739.3 on BTA13; 1946.13, 1947.12, and 1948.12 on BTA15; 2213.8, 2214.9, and 2214.4 on BTA17; 2291.5 and 2293.2 on BTA18; 2850.17, 2852.5, and 2852.58 on BTA26; and 2922.8 and 2923.7 on BTA27 (Table 1). This result suggests that these hotspots may be influenced by the same lethal variant, reinforcing the potential lethality of these regions. This trend was also observed by Wu et al.26 and Schmidt et al.5, where potential lethal haplotypes located in nearby segments were also found.
These findings provide insights into genomic regions that may harbor putative haplotypes containing lethal alleles. Understanding the distribution of these haplotypes within the Nellore breed, as well as identifying their carriers, is of paramount importance to prevent mating between animals carrying the same copy of a lethal allele in a specific locus. This strategy may enhance genetic gain in populations undergoing artificial selection towards specific breeding goals and contribute to the long-term sustainability of breeding programs.
Identified genes and functional enrichment
Considering all genomic regions harboring putative lethal haplotypes, 360 candidate genes were identified (Supplementary Table S2). The functional enrichment analysis revealed biological processes associated with the development of sexual traits in both males and females, immune system essential functions, energy metabolism, and embryonic development (Table 2). The annotated metabolic pathways included ovarian steroidogenesis, oocyte meiosis, and insulin secretion (Table 2). In this discussion, the main focus was to understand how the identified biological and metabolic processes could be linked to occurrences of embryonic lethality or reproductive inefficiency, which may help in elucidating the effects associated with lethal haplotypes.
The development of primary sexual traits in males and females play a significant role in the cattle reproductive performance, as they can affect fertility in both sexes. The genes ZFP42 (BTA27), GATA3 (BTA3), LHB (BTA18), LRP2 (BTA2), BAX (BTA18), and AKR1C3 (BTA13) were identified as regulators of these processes, and some studies have reported their association with reproductive traits in pigs32, humans33, zebrafish34, and cattle35. Sires with underdeveloped gonads can exhibit inferior sperm quality, primarily due to lower concentrations of serum follicle-stimulating hormone (FSH), which in turn affects the endocrine regulation of testicular functions and the number of Sertoli cells36. On the other hand, heifers with genital developmental issues during the pubertal period may have lower leptin secretion, leading to decreased ovarian follicle development, and lower ovulation rates37. This can also be reflected in progesterone concentrations during gestation, increasing the likelihood of embryo loss38,39,40. These processes can impact reproduction in both sexes and consequently influence the expected frequencies of homozygous haplotypes in live animals due to reproductive inefficiency.
The protein-coding gene AKR1C4 (BTA13) was found to play a role in the hormone metabolic process, converting PGH2 to PGF2α41. In cattle, this conversion was already associated with embryo development and immune function at the fetal-maternal interface during early pregnancy42,43. Uterine production of PGF2α has a negative impact on ongoing embryonic development from the morula to the blastocyst stage, slowing down embryo growth42,44.
The INS (BTA29) and IGF2 (BTA29) genes have been identified and linked to glucose homeostasis and energy reserve metabolic process. The function of the INS gene is significantly influenced by the hormone metabolism, being crucial for glucose absorption and maintaining glucose balance45,46. In cows, the INS gene also controls insulin receptors in embryos from the zygote to the blastocyst stage. Elevated insulin levels during this period can impair the developmental potential of the embryo, resulting in embryo loss45. The IGF2 gene is known to regulate insulin concentrations and facilitate fetal growth by ensuring the supply of oxygen and nutrients to the fetus through placental circulation, contributing to the process of energy homeostasis47,48,49. Studies have documented a mutation in this gene that results in fetal death, attributable to disruptions in the delivery of oxygen and nutrients to the fetus49,50.
The regulation of embryonic development was also reported in the enrichment analysis with the genes RUVBL2 (BTA18), NLRP5 (BTA18), PHLDA2 (BTA29) and ACTR8 (BTA22) being responsible for this process. During embryonic development in cattle, several factors can influence and lead to embryonic loss51, including incorrect chromosome segregation during cell division in meiosis52, genetic incompatibility between the maternal immune system and the embryo53, hormonal disorders54, and failures during embryo implantation in the maternal uterus55. These findings support the hypothesis that the main effect of lethal alleles may be on the occurrence of embryonic loss in affected homozygous embryos6, possibly due to alterations in the regulation of embryonic development.
The RUVBL2 gene directly controls transcription through chromatin remodeling in multi-protein complexes56, and it has been linked to the differentiation of neuroectoderm during early embryogenesis in mouse57. Furthermore, the NLRP5 gene is a protein coding that has been associated with impaired fertility58 and age at first calving59 in cattle, regulation of embryo development via mitochondrial functions in mouse60, and embryo progression in the early stages in ovine species61. The expression of the PHLDA2 gene has been associated with fetal growth restrictions in cattle62,63, leading to adverse perinatal outcomes such as embryo death49 and placental inefficiency64. The ACTR8 gene is involved in chromatin remodeling, transcription regulation and DNA recombination65. ACTR8 has been associated with Stayability in Nellore cattle5 and signatures of selection in Holstein cattle66.
The metabolic pathways identified in the KEGG enrichment analysis for the gene network were ovarian steroidogenesis (genes INS, LHB and AKR1C3), oocyte meiosis (genes INS, PPP2R1B, YWHAH and CALM3), and insulin secretion (genes INS, TRPM4 and CACNA1D). These processes are associated with reproductive efficiency in cattle, which supports the hypothesis that the chromosomal regions harboring putative lethal haplotypes may have an impact on the reproductive performance of their carriers.
Ovarian steroidogenesis is a critical process for normal uterine function, as well as the establishment and maintenance of pregnancy67. Challenges in this process can also result in the development of nonviable oocytes67,68,69. Conversely, oocyte meiosis in mammals initiates before birth during embryonic development and resumes during puberty in response to luteinizing hormones during estrus70,71. Issues with the oocyte meiosis mechanism can hinder follicle maturation and delay reproduction71. Furthermore, insulin secretion plays a pivotal role in body metabolism and can influence cattle reproduction68,72. Adequate insulin concentrations are necessary for normal follicular steroidogenesis and secretion of estradiol73.
The INS gene was found to be involved in all three pathways described. Some studies have indicated that higher insulin and glucose levels are associated with anestrus in Holstein cows74, while other authors have reported that hyperinsulinemia and increased plasma insulin concentrations impair oocyte quality and subsequent embryo development in cattle68. Regarding the other genes associated with metabolic pathways, few studies linking their expression on reproductive traits in cattle were found. For example, the LHB gene, involved in the ovarian steroidogenesis mechanism, did not affected the development of ovarian follicular growth or the number of follicles in crossbreed Holstein-Gyr female fetuses, which are crucial factors in ovarian reserve and fertility75. On the other hand, the PPP2R1B gene was identified in a co-expression network analysis of the metabolome and transcriptome to influence fertility in beef heifers76.
Reproductive performance of mating involving carriers of lethal haplotypes
In this study, 5093 reproductive records were analyzed, and 45 potential lethal haplotypes were identified, totaling 229,185 possible results for matings between noncarriers or carriers of lethal haplotypes (Supplementary Table S4). A total of 2564 combinations between the mating class CDam × CSire were effectively observed in the reproductive records (Supplementary Table 3). Among the 1258 genotyped cows that entered reproduction stage, 1209 were identified as carriers of at least one candidate lethal haplotype, while 186 out of 190 genotyped bulls were also found to carry at least one candidate lethal haplotype (Supplementary Table S3).
This finding is crucial in demonstrating that despite the low probability of deleterious mutations emerging in populations and being considered rare2,26, the lethal haplotypes are prevalent among the animals engaged in reproduction within the evaluated population. This result highlights the importance of implementing targeted mating schemes to prevent combinations between animals harboring identical copies of a candidate lethal haplotype. Furthermore, it is imperative to emphasize that the effective population size is small (107 ± 15 animals), have an estimated founder population of 69 animals, and the breeding program is closed for selecting progenitors exclusively from within each selection line. This scenario difficult the management of the dissemination of these haplotypes within the population, particularly if a bull deemed superior for target traits is a carrier of a candidate haplotype.
Three haplotypes (42.4 – BTA1, 935.15 – BTA6, and 2214.4 – BTA17) had significant effects (P-value < 0.05) on the probabilities of pregnancy success, pregnancy loss, stillbirth, and pre-weaning mortality (Tables 3 and 4). In the mating class CDam × CSire, the odds ratio of pregnancy success was 55.14%, 46.18%, and 56.54% lower to NCDam × NCSire considering the haplotypes 42.4, 935.15, and 2214.4, respectively (Table 3). The probabilities of observing pregnancy loss in CDam × CSire were 50.91% higher when considering the haplotype 42.4, 210.81% higher for haplotype 935.15, and 84.72% higher for haplotype 2214.4 (Table 4). The stillbirth and pre-weaning mortality occurrences were also higher for the CDam × CSire mating category (Table 4), with probabilities of 141.02% (stillbirth) and 44.03% (pre-weaning mortality) for haplotype 42.4, 295.03% (stillbirth), and 189.56% (pre-weaning mortality) for haplotype 935.15, and 54.01% (stillbirth) and 101.14% (pre-weaning mortality) for haplotype 2214.4.
These results demonstrate that even in heterozygous carriers of potential candidates for lethal haplotypes, there is a significant decline in their reproductive performance. Similar results were observed in mating categories CDam× NCSire and NCDam × CSire, where a loss in reproductive performance was evident for certain haplotypes (Tables 3 and 4). This trend has also been reported in other studies. For instance, Id-Lahoucine et al.4,26 found in Angus cattle that the probability of pregnancy decreased by up to 15% in carriers of specific lethal haplotypes. Wu et al.26 observed two candidate lethal haplotypes in Nordic Holsteins that increased the return-to-estrus rate in previously identified pregnant cows, suggesting early embryonic lethality. Similarly, Id-Lahoucine et al.77 noted that the occurrence of certain genomic regions with evidence of transmission ratio distortion phenomenon, potentially harboring lethal alleles, increased the likelihood of stillbirth by up to 254%. Likewise, Schmidt et al.5 identified 15 candidate lethal haplotypes in Nellore cattle that exerted significant effects on post-natal mortality occurrence.
Specifically, considering pregnancy success probabilities, reductions of up to 203.46% in the odds ratio were observed in the mating category CDam × CSire compared to the scenario NCDam × NCSire (Table 3), considering haplotype 998.14 (BTA7). This haplotype was also identified by Schmidt et al.5, who reported significant negative effects of its occurrence on the probability of heifer rebreeding and stayability. This result supports the finding that this genomic region likely harbors a specific lethal allele in Nellore cattle that influences the reproductive efficiency of its carriers. No annotated genes were overlapped with this haplotype region (36.55–37.25 Mb, BTA7).
The haplotype 1739.3 was responsible for the higher increase in the occurrence of pregnancy loss (275.15%) in the mating category CDam × CSire compared to the category NCDam × NCSire (Table 4). The genomic region harboring haplotype 1739.3 (15.76–16.49 Mb, BTA13) had not been previously reported as a potential carrier of lethal haplotype candidates in cattle breeds. The genes GATA3, TAF3, ATP5F1C, KIN, ITIH2, ITIH5, and SFMBT2 were found in this region. It is important to highlight the GATA3 gene, which was observed in the functional enrichment analysis of the gene network and was associated with biological processes related to development of primary male sexual characteristics, male gonad development, and hormone metabolic processes (Table 2). In cattle, the GATA3 gene has been linked to regulating lineage specification during early embryonic development, and its deletion disrupts transcription in bovine blastocysts78.
Mating involving the CDam × CSire category, considering carriers of haplotype 935.15, experienced a 295.03% increase probability in stillbirth occurrence (Table 4). This haplotype is located on BTA6 between 111.03 and 111.68 Mb, encompassing the genes CD38, FGFBP1, PROM1, and TAPT1. This genomic region had not been previously reported as a potential host of putative lethal haplotypes. Among the identified genes, CD38 was the only gene associated with postnatal survival traits in livestock species, with a significant effect reported on the occurrence of stillborn piglets in purebred Yorkshire pigs79.
Regarding pre-weaning mortality, a 301.40% increase in the probability of its occurrence was observed among matings in the CDam × CSire category, considering carriers of the haplotype 1110.12 (Table 4), compared to the non-carrier category (NCDam × NCSire). Among the genes identified in the region of this putative lethal haplotype (21.62–22.74 Mb, BTA8), the CDKN2A gene was found. This gene has been associated with cell cycle regulation in humans80 and ribeye area in Nellore cattle81, providing limited information on the mechanisms of this genomic region that may be influencing the occurrence of pre-weaning mortality.
Taken together, these findings support the argument that lethal alleles in Nellore cattle breeding should be traced within the population to prevent reductions in pregnancy rates, pregnancy loss, stillbirth, and pre-weaning mortality occurrences. Individuals selected for reproduction should be tested for the lethal haplotypes they carry, and mating between individuals carrying the same copy of a lethal haplotype should be avoided. These practices can prevent the spread of lethal alleles to future generations and potentially increase reproductive efficiency, as demonstrated by the results of this study in matings within the CDam × CSire category when compared to the NCDam × NCSire category.
Further studies on this topic involving Nellore cattle are still needed, given the impact of this breed on the socioeconomic landscape and the production of beef for the global market. It is worth noting that among the 45 candidate lethal haplotypes found in this study, only 3 had been reported in previous studies5. Although the population evaluated in this study belongs to an experimental herd, there may still be genomic regions containing lethal alleles that have not yet been identified in Nellore cattle. Additional studies utilizing larger databases and whole-genome sequence data would be beneficial to confirm the potential lethality of these haplotypes and reinforcing that their complete absence is not merely a random event. This comprehensive approach would provide more robust evidence regarding the association between these haplotypes, lethal outcomes and reproductive inefficiency, enhancing our understanding of their genetic effects and informing breeding practices for improved livestock management.
Conclusions
A total of 45 candidate lethal haplotypes were identified in closed experimental Nellore cattle lines. Within the genomic regions of these candidate lethal haplotypes, 360 genes were found, and the enrichment analyses revealed associations with reproductive, hormonal, and metabolic aspects. Carriers of the potential lethal haplotypes exhibited reduced reproductive efficiency compared to noncarriers. This resulted in decreased pregnancy success rates, increased pregnancy loss, stillbirth occurrence, and pre-weaning mortality. These results highlight the importance of monitoring lethal genetic variants in Nellore cattle that exert negative effects on reproduction and production efficiency, avoiding their propagation in the population.
Data availability
The haplotypes results obtained in this study have been deposited in the Harvard Dataverse and can be accessed via the following link: https://doi.org/10.7910/DVN/XBAHMZ. The parameters and guidelines used for haplotype identification can be found in the findhap.f90 manual provided by the Agricultural Research Service (USDA, https://www.ars.usda.gov/northeast-area/beltsville-md-barc/beltsville-agricultural-research-center/agil/aip/software/findhap/). Phenotypic and genomic information can be required for academic use contacting MEZM (email: mezmercadante@gmail.com).
References
Gebreyesus, G., Sahana, G., Christian Sørensen, A., Lund, M. S. & Su, G. Novel approach to incorporate information about recessive lethal genes increases the accuracy of genomic prediction for mortality traits. Hered 125, 155–166 (2020).
Jenko, J. et al. Analysis of a large dataset reveals haplotypes carrying putatively recessive lethal and semi-lethal alleles with pleiotropic effects on economically important traits in beef cattle. Genet. Sel. Evol. 51, 1–14 (2019).
Wu, X., Mesbah-Uddin, M., Guldbrandtsen, B., Lund, M. S. & Sahana, G. Haplotypes responsible for early embryonic lethality detected in nordic holsteins. J. Dairy. Sci. 102, 11116–11123 (2019).
Id-Lahoucine, S. et al. Distortion of Mendelian segregation across the Angus cattle genome uncovering regions affecting reproduction. Sci Rep 2023;13:1–10. (2023) 13:1.
Schmidt, P. I. et al. Identification of candidate lethal haplotypes and genomic association with post-natal mortality and reproductive traits in Nellore cattle. Sci. Rep. 13, 1–12 (2023).
VanRaden, P. M., Olson, K. M., Null, D. J. & Hutchison, J. L. Harmful recessive effects on fertility detected by absence of homozygous haplotypes. J. Dairy. Sci. 94, 6153–6161 (2011).
Hoff, J. L., Decker, J. E., Schnabel, R. D. & Taylor, J. F. Candidate lethal haplotypes and causal mutations in Angus cattle. BMC Genom. 18, 1–11 (2017).
Benfica, L. F. et al. Detection and characterization of copy number variation in three differentially-selected Nellore cattle populations. Front. Genet. 15, 1–14 (2024).
Rodrigues, G. R. D. et al. Animal growth models as a tool to estimate resilience indicators in Bos indicus and Bos taurus heifers: selection effects and genetics parameters. Livest. Sci. 282, 1–9 (2024).
Strandén, I. RelaX2 program for pedigree analysis, User’s guide for version 1.65. (2014).
Gutiérrez, J. P., Cervantes, I., Molina, A., Valera, M. & Goyache, F. Individual increase in inbreeding allows estimating effective sizes from pedigrees. Genet. Sel. Evol. 40, 359–378 (2008).
Sargolzaei, M., Chesnais, J. P. & Schenkel, F. S. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 15, 1–12 (2014).
Mota, L. F. M. et al. Meta-analysis across Nellore cattle populations identifies common metabolic mechanisms that regulate feed efficiency-related traits. BMC Genom. 23, 1–12 (2022).
Mota, L. F. M. et al. Benchmarking machine learning and parametric methods for genomic prediction of feed efficiency-related traits in Nellore cattle. Sci. Rep. 14, 1–14 (2024).
Misztal, I. et al. Manual for BLUPF90 family of programs. 1st edition. Athens: University of Georgia; (2014).
VanRaden, P. M., Sun, C. & O’Connell, J. R. Fast imputation using medium or low-coverage sequence data. BMC Genet. 16, 1–12 (2015).
Li, Y., Sung, W. K. & Liu, J. J. Association mapping via regularized regression analysis of single-nucleotide-polymorphism haplotypes in variable-sized sliding windows. Am. J. Hum. Genet. 80, 705–715 (2007).
Guo, Y., Li, J., Bonham, A. J., Wang, Y. & Deng, H. Gains in power for exhaustive analyses of haplotypes using variable-sized sliding window strategy: a comparison of association-mapping strategies. Eur. J. Hum. Genet. 17, 785–792 (2009).
Rosen, B. D. et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 9, 1–9 (2020).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 1–9 (2021).
R Core Team. R: The R Project for Statistical Computing. (2022). https://www.r-project.org/
McFadden, A. et al. Population Analysis identifies 15 Multi-variant Dominant White haplotypes in horses. Animals 14, 1–12 (2024).
Todd, E. T. et al. A genome-wide scan for candidate lethal variants in Thoroughbred horses. Sci. Rep. 10, 1–10 (2020).
Ben Braiek, M., Fabre, S., Hozé, C., Astruc, J. M. & Moreno-Romieux, C. Identification of homozygous haplotypes carrying putative recessive lethal mutations that compromise fertility traits in French lacaune dairy sheep. Genet. Sel. Evol. 53, 1–13 (2021).
Häfliger, I. M., Spengeler, M., Seefried, F. R. & Drögemüller, C. Four novel candidate causal variants for deficient homozygous haplotypes in Holstein cattle. Sci. Rep. 12, 1–13 (2022).
Wu, X., Mesbah-Uddin, M., Guldbrandtsen, B., Lund, M. S. & Sahana, G. Novel haplotypes responsible for prenatal death in nordic red and Danish Jersey cattle. J. Dairy. Sci. 103, 4570–4578 (2020).
Sasaki, S. et al. Identification of deleterious recessive haplotypes and candidate deleterious recessive mutations in Japanese black cattle. Sci. Rep. 11, 1–13 (2021).
Howard, J. T., Pryce, J. E., Baes, C. & Maltecca, C. Invited review: inbreeding in the genomics era: inbreeding, inbreeding depression, and management of genomic variability. J. Dairy. Sci. 100, 6009–6024 (2017).
Bosse, M., Megens, H. J., Derks, M. F. L., de Cara, Á. M. R. & Groenen, M. A. M. Deleterious alleles in the context of domestication, inbreeding, and selection. Evol. Appl. 12, 6–17 (2019).
Pausch, H. et al. Homozygous haplotype deficiency reveals deleterious mutations compromising reproductive and rearing success in cattle. BMC Genom. 16, 1–13 (2015).
Kadri, N. K. et al. A 660-Kb deletion with antagonistic effects on fertility and milk production segregates at high frequency in nordic red cattle: additional evidence for the common occurrence of balancing selection in Livestock. PLoS Genet. 10, 1–11 (2014).
Liu, T. et al. High expression of ZFP42 improves Early Development of Pig embryos produced by Handmade Cloning. Cell. Reprogram. 26, 57–67 (2024).
Beiraghdar, M., Beiraghdar, M. & Khosravi, S. The methylation status of GATA3 potentially predicts the outcomes of assisted reproductive technologies. Hum. Fertil. 26, 1279–1285 (2023).
Chu, L., Li, J., Liu, Y., Hu, W. & Cheng, C. H. K. Targeted gene disruption in zebrafish reveals noncanonical functions of lh signaling in reproduction. Mol. Endocrinol. 28, 1785–1795 (2014).
Yang, M. Y. & Rajamahendran, R. Expression of Bcl-2 and Bax proteins in relation to quality of bovine oocytes and embryos produced in vitro. Anim. Reprod. Sci. 70, 159–169 (2002).
Burns, B. M., Gazzola, C., Holroyd, R. G., Crisp, J. & Mcgowan, M. R. Male reproductive traits and their relationship to reproductive traits in their female progeny: a systematic review. Reprod. Domest. Anim. 46, 534–553 (2011).
Williams, G. L. et al. Leptin and its role in the central regulation of reproduction in cattle. Domest. Anim. Endocrinol. 23, 339–349 (2002).
Carter, F. et al. Effect of increasing progesterone concentration from Day 3 of pregnancy on subsequent embryo survival and development in beef heifers. Reprod. Fertil. Dev. 20, 368–375 (2008).
Kasimanickam, R. K., Kasimanickam, V. R., Oldham, J. & Whitmore, M. Cyclicity, estrus expression and pregnancy rates in beef heifers with different reproductive tract scores following progesterone supplementation. Theriogenology 145, 39–47 (2020).
Martins, K. R. et al. Regulation and function of leptin during ovarian follicular development in cows. Anim. Reprod. Sci. 227, 1–12 (2021).
Parent, M., Madore, E., MacLaren, L. A. & Fortier, M. A. 15-hydroxyprostaglandin dehydrogenase in the bovine endometrium during the oestrous cycle and early pregnancy. Reproduction 131, 573–582 (2006).
Emond, V., Fortier, M. A. & Murphy, B. D. Lambert2 RD. prostaglandin E 2 regulates both Interleukin-2 and granulocyte-macrophage colony-stimulating factor gene expression in bovine lymphocytes’. Biol. Reprod. 58, 143–151 (1998).
Scenna, F. N. et al. Detrimental effects of prostaglandin F2α on preimplantation bovine embryos. Prostaglandins Other Lipid Mediat. 73, 215–226 (2004).
Sponchiado, M. et al. Pre-hatching embryo-dependent and -independent programming of endometrial function in cattle. PLoS One. 12, 1–23 (2017).
Laskowski, D. et al. The functional role of insulin in fertility and embryonic development-what can we learn from the bovine model? Theriogenology 86, 457–464 (2016).
D’Occhio, M. J. et al. Pleomorphic adenoma gene (PLAG1) in reproduction and implication for embryonic survival in cattle: a review. J. Anim. Sci. 102, 1–12 (2024).
Thatcher’, W. W. et al. Guzeloglu1 A, Meikle2 A, Kamimura3 S, Regulation of embryo survival in cattle. Reproduction. ;61:253–66. (2003).
Gobikrushanth, M. et al. The relationship between serum insulin-like growth factor-1 (IGF-1) concentration and reproductive performance, and genome-wide associations for serum IGF-1 in Holstein cows. J. Dairy. Sci. 101, 9154–9167 (2018).
Sigdel, A., Bisinotto, R. S. & Peñagaricano, F. Genes and pathways associated with pregnancy loss in dairy cattle. Sci. Rep. 11, 1–11 (2021).
Bergman, D., Halje, M., Nordin, M. & Engström, W. Insulin-like growth factor 2 in development and disease: a mini-review. Gerontology 59, 240–249 (2013).
Lonergan, P., Fair, T., Forde, N. & Rizos, D. Embryo development in dairy cattle. Theriogenology 86, 270–277 (2016).
De Coster, T. et al. Parental genomes segregate into distinct blastomeres during multipolar zygotic divisions leading to mixoploid and chimeric blastocysts. Genome Biol. 23, 1–29 (2022).
Fair, T. Embryo maternal immune interactions in cattle. Anim. Reprod. 13, 346–354 (2016).
Ribeiro, E. S. & Carvalho, M. R. Impact and mechanisms of inflammatory diseases on embryonic development and fertility in cattle. Anim. Reprod. 14, 589–600 (2017).
Lonergan, P. & Forde, N. Maternal-embryo interaction leading up to the initiation of implantation of pregnancy in cattle. Animal 8, 64–69 (2014).
Gorynia, S. et al. Structural and functional insights into a dodecameric molecular machine - the RuvBL1/RuvBL2 complex. J. Struct. Biol. 176, 279–291 (2011).
Hong, S. et al. RuvB-Like protein 2 (Ruvbl2) has a role in directing the neuroectodermal differentiation of mouse embryonic stem cells. Stem Cells Dev. 25, 1376–1385 (2016).
Pennetier, S. et al. MATER protein expression and intracellular localization throughout folliculogenesis and preimplantation embryo development in the bovine. BMC Dev. Biol. 6, 1–9 (2006).
Alves, A. A. C. et al. A Random Forest-based genome-wide scan reveals fertility-related candidate genes and potential inter-chromosomal epistatic regions Associated with Age at First Calving in Nellore cattle. Front. Genet. 13, 1–18 (2022).
Fernandes, R. et al. NLRP5 mediates mitochondrial function in mouse oocytes and embryos. Biol. Reprod. 86, 1–10 (2012).
Bebbere, D. et al. Expression of maternally derived KHDC3, NLRP5, OOEP and TLE6 is associated with oocyte developmental competence in the ovine species. BMC Dev. Biol. 14, 1–13 (2014).
Guillomot, M. et al. Abnormal expression of the imprinted gene Phlda2 in cloned Bovine Placenta. Placenta 31, 482–490 (2010).
Wang, M. et al. Biallelic expression of Tssc4, Nap1l4, Phlda2 and Osbpl5 in adult cattle. J. Genet. 94, 391–395 (2015).
Chavatte-Palmer, P. et al. Review: placental perturbations induce the developmental abnormalities often observed in bovine somatic cell nuclear transfer. Placenta 26, 99–104 (2012).
Choe, S. H. et al. A single mutation in the ACTR8 gene associated with lineage-specific expression in primates. BMC Evol. Biol. 20, 1–12 (2020).
Naderi, S. et al. Assessing selection signatures within and between selected lines of dual-purpose black and white and German holstein cattle. Anim. Genet. 51, 391–408 (2020).
Wood, J. R. & Strauss, J. F. III Multiple Signal Transduction pathways regulate ovarian steroidogenesis. Rev. Endocr. Metab. Disord. 3, 33–46 (2002).
Sartori, R. et al. Metabolic hormones and reproductive function in cattle. Anim. Reprod. 10, 199–205 (2013).
Gareis, N. C. et al. Impaired insulin signaling pathways affect ovarian steroidogenesis in cows with COD. Anim. Reprod. Sci. 192, 298–312 (2018).
Von Stetina, J. R. & Orr-Weaver, T. L. Developmental control of oocyte maturation and egg activation in metazoan models. Cold Spring Harb Perspect. Biol. 3, 1–19 (2011).
He, M., Zhang, T., Yang, Y. & Wang, C. Mechanisms of oocyte maturation and related epigenetic regulation. Front. Cell. Dev. Biol. 9, 1–18 (2021).
Baruselli, P. S. et al. Intrinsic and extrinsic factors that influence ovarian environment and efficiency of reproduction in cattle. Anim. Reprod. 14, 48–60 (2017).
Silva, J. M. & Price, C. A. Insulin and IGF-I are necessary for FSH-induced cytochrome P450 aromatase but not cytochrome P450 side-chain cleavage gene expression in oestrogenic bovine granulosa cells in vitro. J. Endocrinol. 174, 499–507 (2002).
Azarbayejani, R. & Mohammadsadegh, M. Glucose, insulin, and cortisol concentrations and glucose tolerance test in Holstein cows with inactive ovaries. Trop. Anim. Health Prod. 53, 3–9 (2021).
Weller, M. M. D. C. A. et al. Effect of maternal nutrition and days of gestation on pituitary gland and gonadal gene expression in cattle. J. Dairy. Sci. 99, 3056–3071 (2016).
Banerjee, P., Rodning, S. P., Diniz, W. J. S. & Dyce, P. W. Co-expression Network and Integrative Analysis of Metabolome and Transcriptome uncovers Biological pathways for Fertility in Beef heifers. Metabolites 12, 1–18 (2022).
Id-Lahoucine, S., Cánovas, A., Legarra, A. & Casellas, J. Transmission ratio distortion regions in the context of genomic evaluation and their effects on reproductive traits in cattle. J. Dairy. Sci. 106, 7786–7798 (2023).
Shi, Y. et al. Functional role of GATA3 and CDX2 in lineage specification during bovine early embryonic development. Reproduction 165, 325–333 (2023).
Jonas, E. & Rydhmer, L. Effect of candidate genes for maternal ability on piglet survival and growth. Livest. Sci. 207, 83–90 (2018).
Nielsen, G. P., Burns, K. L., Rosenberg, A. E. & Louis, D. N. CDKN2A gene deletions and loss of p16 expression occur in Osteosarcomas that Lack RB alterations. (1998).
Fernandes Júnior, G. A. et al. Genome scan for postmortem carcass traits in Nellore cattle. J. Anim. Sci. 94, 4087–4095 (2016).
Acknowledgements
The authors thank Dr. Paul M. VanRaden (USDA) for generously providing the codes for obtaining the lethal haplotypes, and the staff of the Institute of Animal Science (Sertãozinho, SP, Brazil) for their efforts in collecting the data used in this study.
Funding
São Paulo Research Foundation (FAPESP) for financial support (grants 2017/10630-2, 2018/20026-8, and 2017/50339-5), and scholarships to GRDR (grants 2023/11176-4 and 2024/01909-7), LFMM (grant 2022/11852-7), PIS (2021/09942-5), and JPSV (2024/05697-4). Coordination for the Improvement of Higher Education Personnel (CAPES, Finance Code 001) for providing scholarship to ESO. National Council for Science and Technological Development (CNPq) for providing grants to LGA and MEZM.
Author information
Authors and Affiliations
Contributions
MEZM coordinated the team and supervised all the stages of the study. GRDR, JNSGC, LFMM, and MEZM conceived and designed the study. GRDR, JNSGC, LFMM, PIS, and MEZM conducted the data analyses. LGA and MEZM obtained the resources for this research. GRDR, JNSGC, LFMM, PIS, JPSV, ESO, LGA, LFB, and MEZM contributed to the data acquisition and interpretation of the results. GRDR, JNSGC, LFMM, LFB, and MEZM wrote and edited the manuscript. All authors reviewed and contributed to the editing of the manuscript and approved its final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Animal Care and Use Committee approval was not obtained for this study because all the analyses were performed using pre-existing datasets.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rodrigues, G.R.D., Cyrillo, J.N.S.G., Mota, L.F.M. et al. Effect of genomic regions harboring putative lethal haplotypes on reproductive performance in closed experimental selection lines of Nellore cattle. Sci Rep 15, 4113 (2025). https://doi.org/10.1038/s41598-025-88501-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88501-7
