
Abstract
Expanding the genetic toolkit for Leptospira spp. is a crucial step toward advancing our understanding of the biology and virulence of these atypical bacteria. Pathogenic Leptospira are responsible for over 1 million human leptospirosis cases annually and significantly impact domestic animals. Bovine leptospirosis causes substantial financial losses due to abortion, stillbirths, and suboptimal reproductive performance. The advent of the CRISPR/Cas9 system has marked a turning point in genetic manipulation, with applications across multiple Leptospira species. However, incorporating controlled protein expression into existing genetic tools could further expand their utility. We developed and demonstrated the functionality of IPTG-inducible heterologous protein expression in Leptospira spp. This system was applied for regulated expression of dead Cas9 (dCas9) to generate knockdown mutants, and Cas9 to produce knockout mutants by inducing double-strand breaks (DSB) into desired targets. IPTG-induced dCas9 expression enabled validation of essential genes and non-coding RNAs. Additionally, IPTG-controlled Cas9 expression combined with a constitutive non-homologous end-joining (NHEJ) system allowed for successful recovery of knockout mutants, even in the absence of IPTG. These newly controlled protein expression systems will advance studies on the basic biology and virulence of Leptospira, as well as facilitate knockout mutant generation for improved veterinary vaccines.
Similar content being viewed by others

Introduction
Pathogenic Leptospiraspp. are the causative agent of leptospirosis1,2,3, a worldwide neglected zoonosis responsible for more than 1 million human cases and approximately 60,000 deaths annually4. Leptospires are versatile bacteria with genome plasticity5 capable of expressing a distinct arsenal of proteins in response to environmental and host cues, including osmolarity, temperature and serum6,7,8,9,10,11. These bacteria can survive in wet environments and infect mammalian hosts through penetration of abraded skin or mucosa, rapidly disseminating in the bloodstream, where they overcome several host immune defenses, reaching immune privileged target organs12.
Recent advances in functional genomics13,14,15, plasmid delivery by E. coli conjugation16 and mutagenesis tools for Leptospira spp.17,18,19, are enabling the elucidation of the biology of Leptospira, and their pathophysiology and virulence factors. The generation of mutants is a pivotal step to confirm the roles of expressed genes, either by in vitro assays or evaluation of virulence in animal models20,21,22,23. In past years, the application of the “memory immunity” bacterial CRISPR/Cas9 system from Streptococcus pyogenes has advanced the generation of knockdown and knockout mutants in several species and serovars of Leptospira18,19,24,25,26.
The CRISPR/Cas9 system relies on the expression of a programmable RNA-guided nuclease, called Cas9, and a single-guide RNA (sgRNA), comprising a 5′ 20-nt sequence termed protospacer, that dictates Cas9 targets by base-pairing, and a scaffold region responsible for Cas9 binding27,28. Upon recognition of the protospacer adjacent motif (PAM) 5’ NGG 3’ and sgRNA:DNA base pairing, Cas9 utilizes its two nuclease domains to promote double-strand breaks (DSBs) in the target sequence, which needs to be repaired so cells can retain their viability29,30. Even though eukaryotes present with multi-protein non-homologous end-joining (NHEJ) pathways to re-ligate DNA ends in the absence of a template for recombination31, most bacterial cells lack such machinery, with a few exceptions32.
It was experimentally demonstrated for Leptospira spp. that DSBs caused by Cas9 are lethal to the cells17,24, and the first approach to overcome this obstacle was to employ a catalytically inactive Cas9 (dCas9). Mechanistically, target recognition by dCas9 is similar to that of Cas9, however, dCas9 is not capable of cutting the DNA, and acts merely as a steric barrier preventing RNA polymerase elongation33. This physical hindrance results in gene silencing, in a strategy termed CRISPR interference (CRISPRi), which was successfully used to generate several mutants in Leptospira spp.21,34,35,26. In its current state, CRISPRi with constitutively expressed dCas9 cannot differentiate a failed conjugation experiment from a lethality caused by silencing of an essential gene.
Lethal DSBs caused by Cas9 were overcome by co-expression of this nuclease with a prokaryotic version of the NHEJ from Mycobacteria smegmatis, composed of sequences coding for two proteins, LigD and Ku. This strategy allowed the recovery of knockout markerless mutants with target specificity but variable deletion lengths from the Cas9 cutting site24, since NHEJ is an error prone process. The success of the Cas9-NHEJ approach is dependent on the efficiency of conjugation, which tends to be low for certain species and serovars of Leptospira, and on the efficiency of DSB repair. At present, these efficiencies are interdependent events resulting in low mutant recovery rates for pathogenic Leptospira. Nonetheless, mutant recovery by Cas9-NHEJ appears to be higher than allelic exchange20, which was, for several years, the only technique available for targeted mutagenesis in Leptospira.
It is noteworthy that a system for controlled protein expression would open new horizons for both CRISPRi and Cas9-NHEJ methodologies, expanding the toolbox for Leptospira mutagenesis. In the case of CRISPRi, an inducible dCas9 expression would favor the validation of essential genes, as the same conjugation reaction could be evaluated in both the presence and absence of an inducer for dCas9 expression. For Cas9-NHEJ, repressed Cas9 expression would be beneficial for colony recovery since hypothetically, the frequency of this first step would be determined by the efficiency of conjugation. After colony recovery and growth of leptospires to high cell densities, Cas9 can then be expressed by the addition of an inducer; thus, the frequency of DNA repair by the NHEJ proteins would be acting in this subsequent step only, and thus increasing the chances of knockout mutant recovery.
In this study, we developed a cassette for heterologous protein expression in Leptospira spp. based on the lactose operon from E. coli. Protein expression induced by IPTG was demonstrated and the inducible cassette was used to control expression of dCas9 and Cas9 in Leptospira. The applicability of inducible expression of dCas9 for validating essential genes, and Cas9 for improved knockout mutant recovery were evaluated in distinct species and serovars of Leptospira.
Methods
Bacterial strains and media
Pathogenic L. interrogans serovar Copenhageni strain FIOCRUZ L1-13036 and serovar Canicola strain LAD-125 were grown in HAN medium37 at 29 °C. Saprophytic L. biflexa serovar Patoc strain Patoc1 was cultured in EMJH medium (Difco, BD, Franklin Lakes, NJ) or HAN medium, according to the experimental design. For agar plates, media was supplemented with 1.2% noble agar (Difco). To select for recombinant cells, spectinomycin was added at 40 µg/mL. E. coli strain β216338, auxotrophic for diaminopimelic acid (DAP), was grown in Lysogeny Broth (LB, Difco) medium supplemented with DAP (0.3 mM, Sigma).
Development of an IPTG-inducible cassette for Leptospira spp
The promoter for the highly expressed gene lipL2139 from L. interrogans serovar Copenhageni strain Fiocruz L1-130 was used to express the codon optimized Lac repressor (LacI), followed by a Borrelia bmpB gene transcription terminator40. For inducible protein expression, the promoter for the highly expressed gene lipL41 was used and included 253 pb upstream from the transcription start site (TSS)41 immediately after the TSS, the Lac operator (lacO) sequence was included, followed by the 5’ UTR from lipL41, which includes the putative Shine-Delgarno (SD) sequence, and the CDS for lipL41 (Fig. 1A). The entire sequence was synthetized by GeneArt and included the restriction sites XbaI and NotI at the 5’ and 3’ends, respectively. The cassette was ligated into the pMaOri plasmid42, previously digested with the same enzymes, in a T4 ligase-mediated reaction. The final plasmid was named pMaOri.Inducible:LipL41. A plasmid named pMaOri:LipL41, without the IPTG-inducible cassette, was made by cloning the lipL41 wild-type cassette into the pMaOri backbone by Gibson assembly using amplicons generated by PCR with primers for lipL41 as listed in Supplementary Table 1.

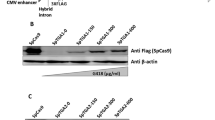
Validation of an IPTG-inducible cassette in L. biflexa.(A) An inducible cassette developed for Leptospira spp. comprising the lipL21 promoter directing the transcription of codon optimized lac repressor LacI, followed by a B. burgdorferi transcription terminator. The constitutive lipL41 promoter was used to transcribe the reporter gene (either lipL41 or lipL32), with inclusion of the lacO sequence (red) after TSS. Shine-Dalgarno sequence (SD, blue) is responsible for ribosome binding. L. biflexa cells containing the plasmid for IPTG-controlled expression of LipL41 (pMaOri.Inducible:LipL41) (B) or LipL32 (pMaOri.Inducible:LipL32) (C) were grown in liquid media in the absence (-) or presence ( +) of IPTG and protein expression evaluated by immunoblot. Controlled LipL32 expression was also evaluated in different concentrations of IPTG (D) and times of induction (E). Cell lysates from L. interrogans serovar Copenhageni strain Fiocruz L1-130 were used as a control.
The plasmid pMaOri.Inducible:LipL32 was prepared by substituting the lipL41 CDS with that of the lipL32 CDS. The plasmid backbone and inducible cassette, excluding the lipL41 CDS, were amplified from pMaOri.Inducible:LipL41 using Q5 DNA polymerase (New England Biolabs) with primers pMaOriInducibleF and InducibleCassetteR. The lipL32 CDS was amplified from genomic DNA of L. interrogans serovar Copenhageni strain Fiocruz L1-130 with primers LipL32 F and R. Both fragments were ligated by Gibson Assembly according to manufacturer’s specifications.
Conjugation of Leptospira spp. and transconjugant recovery
The conjugative E. coli strain β2163 was transformed by heat-shock with each of the recombinant plasmids described herein. Individual colonies were selected from agar plates, confirmed by colony PCR, and propagated in liquid media. E. coli to Leptospira spp. conjugation protocols were performed as previously described18,19. Transconjugant Leptospira were recovered from filter membranes after 24 h conjugation and plated onto EMJH (for saprophytic strains) or HAN (pathogenic strains) solid media containing 40 µg/mL spectinomycin, in the presence or absence of 1 mM IPTG. Plates were incubated at 29 °C until colonies were visible.
Inclusion of the inducible cassette in pMaOri.dCas9 and pMaOriCas9NHEJ plasmids
The S. pyogenes constitutive cas9 promoter within pMaOri.dCas917 and pMaOriCas9NHEJsmegmatis24 was substituted with the IPTG-inducible cassette by Gibson Assembly. Briefly, PCR amplification of plasmid pMaOri.dCas9 with primers pdCas9 F and R or amplification of plasmid pMaOriCas9NHEJsmegmatis with primer pdCas9 F and pNHEJ R resulted in amplicons without the S. pyogenes promoter. The inducible cassette was amplified from plasmid pMaOri.Inducible:LipL41 with primers cassette_dCas9 F and R for ligation into the dCas9 plasmid, or primers cassette_NHEJ F and cassette_dCas9 R for ligation into the Cas9NHEJ plasmid. Appropriate amplicons were then used for Gibson Assembly (New England BioLabs) reactions according to manufacturer’s instructions. Ligated reactions were used to transform E. coli β2163 strain as described above and recovered colonies were evaluated by colony PCR to confirm inclusion of the inducible cassette with primers InducibleCassette F and R. The final plasmids pMaOri.Inducible:dCas9 and pMaOriNHEJ.Inducible:Cas9 were confirmed by whole sequencing with Nanopore long-read technology (Plasmidsaurus Inc, Arcadia, California).
Inclusion of sgRNA cassettes in the inducible plasmids
sgRNA cassettes previously made for L. biflexa dnaK (LEPBI_RS16560) and ompL1 (LEPBI_RS04010), or for L. interrogans lipL21 (LIC_RS00055), lipL41 (LIC_RS15260) and ompL1 (LIC_RS05025) were amplified from plasmids with primers sgRNA F and R, and ligated to XmaI digested plasmids pMaOri.Inducible:dCas9 or pMaOriNHEJ.Inducible:Cas9 by Gibson Assembly at a 1(plasmid):10(amplicon) molar ratio. For sgRNA cassettes targeting the non-coding RNase P (LEPBI_RS18770), pMaOri.dCas9sgRNAlipL21 was used as template for PCR amplification using primers RNaseP_sense F and R and RNaseP_antisense F and R to substitute the protospacer within the plasmid for the ones targeting the sense and antisense strands of RNase P, respectively. Amplicons were circularized by Gibson Assembly. Recombinant plasmids were then used for sgRNA amplification, following ligation into XmaI digested pMaOri.Inducible:dCas9, as above mentioned.
The conjugative E. coli β2163 was transformed with the ligation reactions by heat-shock and cells were seeded onto LB plates containing 0.3 mM DAP and 40 µg/mL spectinomycin. Positive clones containing the recombinant plasmids were validated by PCR with primers Inducible cassette F and R, and pMaOri2 F and R. Targets and selected protospacers are listed in Table 1.
Validation of IPTG-inducible protein expression
To validate the IPTG-inducible cassette for controlled protein expression in Leptospira, colonies were recovered from agar plates after conjugation and grown in liquid medium without or with IPTG (as indicated) until mid-late log phase (2–5 × 108/mL). Cells were harvested by centrifugation (10,000 × g, 15 min), washed twice with PBS and cell lysates prepared by boiling at 95 °C for 10 min in SDS-PAGE loading buffer for gel electrophoresis on either 12% or 4–15% polyacrylamide gels (BioRad). Protein visualization was achieved by staining with Sypro Ruby (Invitrogen, CA) according to the manufacturer’s instructions. For immunoblotting, proteins were electrotransferred onto polyvinylidene difluoride (PVDF) membranes (BioRad) using a semidry transfer method. Membranes were then blocked with SuperBlock (PBS) Blocking Buffer (Thermo) for 1 h, followed by incubation with primary rabbit antibodies (anti-LipL32, anti-LipL21 and anti-LipL41 at 1:4,000, OmpL1 at 1:1,000, anti-Cas9 at 1:2,000 dilution) in blocking buffer for 1 h at room temperature. After three washes with PBS containing 0.1% Tween 20 (PBS-T), membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:4,000 dilution) in blocking buffer for 1 h at room temperature. Following six washes, antigen reactivity was detected using Clarity Max ECL (BioRad), and luminescence was captured with a ChemiDoc MP Imaging System (BioRad).
Validation of IPTG-induced dCas9 function to silence gene expression
E. coli β2163 containing the plasmids for IPTG-induced dCas9 expression and with sgRNA targeting the putative leptospiral genes dnaK (LEPBI_RS16560), ompL1 (LEPBI_RS04010) or the non-coding RNaseP (LEPBI_RS18770) were used for conjugation with wild-type L. biflexa. After recovery of transconjugants from filters, suspensions were spread onto EMJH plates containing 40 µg/mL spectinomycin, with or without 1 mM IPTG. The presence of IPTG induces expression of dCas9 and thus the subsequent silencing of genes targeted by the sgRNA acts as a first indication as to whether a targeted gene provides an essential function as evidenced by no, or a drastically reduced number of, recovered colonies compared to plating in the absence of IPTG. To further validate the essentiality of the gene of interest, individual colonies were then selected from plates without IPTG and inoculated (105/mL) into liquid medium plus spectinomycin, with or without IPTG. To confirm that the addition of IPTG to plates was not impairing colony formation, a control containing pMaOri.Inducible:dCas9 with no sgRNA was included in each experiment, and cells were plated in the presence or absence of IPTG. When growth was observed in the presence of IPTG, cells were evaluated for expression of dCas9. IPTG-induced gene silencing by dCas9 targeting ompL1 and lipL41 was also performed in pathogenic L. interrogans, using conjugation methods for pathogenic species as described above.
Recovery of knockout mutants by IPTG-induced Cas9 expression in conjunction with constitutively expressed NHEJ machinery from M. smegmatis.
The plasmid pMaOriNHEJ.Inducible:Cas9 was designed for IPTG-inducible expression of Cas9 nuclease and constitutive expression of two M. smegmatis proteins, LigD and Ku, responsible for DSB repair after Cas9 DNA target cleavage. This plasmid, with or without a sgRNA targeting lipL21, was delivered by conjugation to pathogenic Leptospira. Recovered cells were plated onto HAN media containing 40 µg/mL spectinomycin, with or without 1 mM IPTG. Plates were incubated at 29 °C until colonies were visible, and colonies were selected from plates without IPTG for growth in liquid media, with or without IPTG. Cultures were monitored daily to account for any delayed growth in the presence of IPTG. Cell lysates were evaluated by immunoblotting, and upon confirmation of knockout phenotypes, mutant cultures were diluted to 103 cells/mL and plated onto HAN plates for individual colony isolation. Individual colonies were selected, grown in liquid media for extraction of DNA, PCR amplification of mutated sites, and amplicon sequencing. The lipL21 gene was amplified by PCR with primers spanning approximately 200 bp from the Cas9 cutting site (Table 1). PCR products were then cleaned up with ExoSAP-IT (Applied Biosystems, Foster City, California, USA) and labeled using the Big Dye Terminator v3.1 cycle sequencing reagent (Applied Biosystems). Sequencing was performed using the ABI 3130XL Genetic Analyzer.
Results
Validation of IPTG-induced expression in the surrogate L. biflexa
The lipL41 gene was initially selected to validate expression of the IPTG-inducible cassette in L. biflexa. For that, the lipL41 CDS, from start codon ATG to 128 nucleotides downstream of the stop codon, was utilized. The expression of codon optimized lac repressor, LacI, is driven by the constitutive and strong lipL21 promoter. To ensure proper termination, the B. burgdorferi bmpB gene intrinsic terminator downstream of the lacI coding sequence was included (Fig. 1A). Next, the lipL41 promoter spanning 254 nucleotides from the experimentally defined transcription start site (TSS), as reported by Zhukova et al. in (2017), was used, followed by the lacO sequence and the lipL41 5’UTR, including the putative Shine-Dalgarno sequence (Fig. 1A).
Plasmid pMaOri containing the inducible cassette for lip41 expression was delivered to L. biflexa by conjugation with E. coli cells, and recovered colonies grown in liquid media to generate whole-cell lysates for immunoblotting. No LipL41 protein could be detected in the recombinant L. biflexa cells, indicating that this protein is not expressed in the saprophytic strain (Fig. 1B). As an alternative method to demonstrate IPTG-controlled expression, the lip41 CDS was substituted by the lipL32 CDS (Fig. 1A), since LipL32 protein has been successfully expressed in L. biflexa previously17. Colonies were randomly selected from plates and grown in liquid media with or without IPTG. Subsequent analysis by immunoblotting demonstrated that expression of LipL32 was detected only in the presence of the inducer, indicating controlled gene expression by the inducible cassette (Fig. 1C). No LipL32 was detected in the absence of IPTG even after overexposure of immunoblots (data not shown). L. biflexa cells containing the inducible cassette for lipL32 were used to determine an optimal concentration of 1 mM IPTG for expression of the inducible cassette (Fig. 1D). LipL32 was faintly detected at 6 h post IPTG induction, with peak expression apparent after 1 day (Fig. 1E).
IPTG-induced expression of dCas9 and Cas9 proteins in L. biflexa
The native S. pyogenes promoter within the pMaOri.dCas9 and pMaOri.Cas9NHEJsmegmatis was substituted for the IPTG-inducible cassette by PCR, resulting in the generation of plasmids pMaOri.Inducible:dCas9 and pMaOriNHEJ.Inducible:Cas9, respectively. Final plasmids were confirmed by sequencing and their genetic elements and complete sequence are presented in Supplementary Fig. 1 and Supplementary Table 2, respectively. Recombinant L. biflexa colonies were grown in media with or without IPTG for validation by SDS-PAGE and immunoblotting with anti-Cas9 antibodies. An ~ 160 kDa band was detected in whole cell lysates of cells containing the control plasmid pMaOri.dCas9, in which dCas9 is constitutively expressed, as well as in the clones containing the inducible cassette for dCas9 in the presence of IPTG (Fig. 2A and upper panel). The same profile was observed in the cells containing the inducible cassette for Cas9 (Fig. 2B and upper panel). Clones containing each construct were evaluated by immunoblotting, corroborating the IPTG-induced expression of dCas9 and Cas9, with no bands being detected in the absence of the inducer. These encouraging results prompted further experiments to target genes considered essential for Leptospira by using IPTG-induced gene silencing with dCas9, and IPTG-induced gene knockout and recovery with Cas9 expression in conjunction with NHEJ proteins.

Evaluation of IPTG-induced expression of dCas9 and Cas9 in L. biflexa. Total protein from L. biflexa cells containing the plasmids pMaOri.Inducible:dCas9 (A) and pMaOriNHEJ.Inducible:Cas9 (B) and grown without (-) or with ( +) IPTG were evaluated by Sypro Ruby staining. Upper panel highlights the appearance of dCas9 or Cas9 protein in the presence of IPTG. Wild-type (WT) and L. biflexa containing the plasmid pMaOri.dCas9 were used as controls. dCas9 and Cas9 controlled expression was validated by immunoblotting with anti-Cas9 antibodies (C).
IPTG-induced gene silencing to identify essential genes
Cassettes containing sgRNA targeting expression of the dnaK gene in L. biflexa were obtained by PCR and ligated into pMaOri.Inducible:dCas9. After conjugation, cell mixtures were plated onto EMJH plates containing spectinomycin, with or without 1 mM IPTG. After 6 days of incubation, colonies began to be apparent in all groups in the absence of IPTG. For those cells containing the control plasmid with no sgRNA cassette, the presence of IPTG, and therefore the expression of dCas9, delayed colony formation, since colonies were apparent at 8 days (Fig. 3A, one representative experiment out of four). When sgRNA targeting dnak was included in the plasmid, the presence of IPTG abrogated or drastically reduced colony formation, indicating that the absence of the chaperone DnaK impaired leptospiral growth on plates (Fig. 3A and B). Figure 3B depicts the number of colonies recovered after each conjugation experiment, in both the absence and presence of IPTG. These results agree with previous attempts to recover dnak knockdown mutants by CRISPRi17.

Evaluation of conditional dnaK knockdown mutants inL. biflexa. (A) L. biflexa cells containing the plasmids pMaOri.Inducible dCas9 alone or with a sgRNA targeting the dnaK gene were seeded onto EMJH media containing spectinomycin without (-) or with ( +) 1 mM IPTG. Colony formation was monitored and recorded at different time points. (B) Colonies observed across four independent experiments. Number of colonies recovered in the absence or presence of IPTG was compared by t-test (*p < 0.05). Colonies from plates without IPTG (n = 3) were picked, cells released from the agar and inoculated in liquid HAN media containing spectinomycin at 105 cells/mL, in the absence or presence of IPTG, and growth was monitored daily at both 29 (C) and 37 °C (D).
To further confirm the essential role of the DnaK protein on leptospiral physiology, individual colonies were selected from plates without IPTG, and cells inoculated into liquid HAN media, which supports the growth of leptospires at both 29 and 37 °C, supplemented with spectinomycin with or without IPTG. At 29 °C, cells containing plasmid with no sgRNA displayed similar growth curves in the presence or absence of IPTG (Fig. 3C); however, in the presence of IPTG, cells containing pMOri.Inducible:dCas9sgRNAdnaK displayed abnormal morphology, and an early stationary phase with lower cell densities (~ 107/mL) (Fig. 3C, Supplementary Fig. 1).
Incubation of cells with IPTG at 37 °C completely abolished growth in the conditional dnaK knockdown mutant, indicating that the chaperone DnaK is essential for cells to cope with higher temperatures (Fig. 3D). Even though leptospires with an unusual morphology were observed at day 1 (Supplementary Fig. 2), no cells could be detected beyond day 2 in the presence of IPTG, and in contrast to growth in the absence of IPTG, or control cells with no sgRNA (Fig. 3D).
IPTG-inducible gene silencing of non-coding RNA
To further corroborate the applicability of IPTG-inducible gene silencing to establish whether targets are essential, and to demonstrate the feasibility of silencing non-coding RNA in Leptospira, a sgRNA cassette targeting RNAseP was constructed. Both IPTG-inducible and constitutively expressed dCas9 strategies were tested and included a sgRNA targeting the antisense strand of RNAseP for partial silencing43,17.
When using the plasmids for constitutive expression of dCas9, colonies could be recovered when cells contained the pMaOri.dCas9 plasmid alone or with the sgRNA targeting the antisense strand of RNAseP. In contrast, a reduced number or no colonies were obtained using the sgRNA targeting the sense strand for RNAseP (Fig. 4A). Since reduced colony recovery could also be due to reduced conjugation efficiencies, the IPTG-inducible plasmid was used for complete silencing of RNAseP. Plating of transconjugants onto EMJH plates with or without IPTG determined that indeed, silencing expression of RNAseP affects cell viability since colony formation was drastically impaired in the presence of IPTG (Fig. 4B). Two clones of E. coli β2163 containing the pMaOri.Inducible:dCas9sgRNasePsense were tested. Results corroborate that inducible silencing is a feasible strategy to validate the essential requirement of genes as well as non-coding RNA, and represents the first demonstration of silencing of this class of macromolecules in Leptospira spp.

Silencing of the ncRNA RNase P in L. biflexa. (A) L. biflexa cells containing plasmids pMaOri.dCas9 alone, or with sgRNA cassettes for pairing to the sense (complete silencing) or antisense (incomplete) strand of the RNaseP sequence, were plated onto EMJH media containing spectinomycin. Colonies obtained from two independent experiments were counted. (B)Validation of essentiality of ncRNA RNaseP was demonstrated by inducible silencing. Cells harboring the pMaOri.Inducible:dCas9 alone or with the sgRNasePsense were seeded onto EMJH plates with spectinomycin without (-) or with ( +) IPTG. Two independent experiments were performed and at each, two clones of conjugative E. coli were used. Number of colonies recovered in the absence or presence of IPTG was compared by t-test (*p < 0.05).
Reassessing the essential role for the OmpL1 porin in Leptospira spp.
A sgRNA cassette targeting the ompL1 gene of L. biflexa was generated and the final plasmid was delivered to leptospiral cells by conjugation. Cells were plated onto EMJH media containing spectinomycin without (-) or with ( +) IPTG, and colony growth was monitored daily. As seen in Fig. 5A, colonies were observed in both control (no sgRNA) and gene knockdown groups (pMaOri.Inducible:dCas9sgRNAompL1biflexa), in either the absence or presence of IPTG, suggesting that OmpL1 silencing is tolerated by L. biflexa. Colonies were enumerated and results of three independent experiments are presented in Fig. 5B. Results corroborate that cells remain viable in the absence of OmpL1 porin, and as confirmed by cell growth in liquid media (Fig. 5C). Since antibodies specifically targeting OmpL1 in the saprophyte L. biflexa are unavailable, gene silencing of ompL1 in the pathogen L. interrogans serovar Copenhageni strain Fiocruz L1-130 was performed, not only to demonstrate silencing by probing the target protein with specific antibodies, but also to confirm that L. interrogans cells were viable in the absence of the porin. Colonies of L. interrogans could be recovered after conjugation from HAN plates without or with IPTG across 3 independent experiments (Fig. 5D).

OmpL1 porin silencing in L. biflexa and L. interrogans. (A) L. biflexa cells containing plasmid pMaOri.Inducible:dCas9 alone, or with a sgRNA cassette targeting the ompL1 gene, were seeded onto EMJH media containing spectinomycin without (-) or with ( +) 1 mM IPTG. Colony formation was monitored and recorded at different time points. (B) Three independent experiments were performed, and numbers of colonies recovered in the absence or presence of IPTG were recorded. (C) Cell viability was also demonstrated in liquid media in the absence (-) or presence ( +) of IPTG. (D-F) Silencing was also performed in the ompL1 ortholog in L. interrogans strain Fiocruz L1-130. (D) Three independent conjugation experiments were performed and for each, two clones of conjugative E. coli containing the plasmid for ompL1 silencing were used. (E) Colonies from plates without IPTG (n = 2) were picked, cells released from the agar and inoculated in liquid HAN media containing spectinomycin in the absence (-) or presence ( +) of IPTG. Cells at mid-log phase were recovered and cell lysates evaluated by immunoblotting. (F) Viability was also confirmed by growth curves in liquid EMJH containing spectinomycin, in the presence or absence of IPTG.
Colonies were recovered from agar plates without IPTG and propagated in liquid media containing spectinomycin without (-) or with ( +) IPTG. Mid-log phase leptospires were collected and whole-cell lysates were evaluated by immunoblotting with anti-OmpL1 and anti-Cas9 antibodies (Fig. 5E). As expected, dCas9 was detected only in cells grown in the presence of IPTG; no dCas9 expression was observed in the absence of IPTG even after overexposure of membranes (not shown). However, diminished expression of OmpL1 was observed in transconjugants with the plasmid pMaOri.Inducible:dCas9sgRNAompL1interrogans in the absence of IPTG when compared with those without the sgRNA for ompL1, suggesting that basal “leaky”, yet undetectable, levels of dCas9 are present. Nevertheless, the presence of IPTG completely abolished expression of OmpL1, as confirmed by immunoblot (Fig. 5E). Nor was expression of OmpL1 required for growth in liquid media (Fig. 5F). “Leaky” silencing was also observed when L. interrogans cells contained the plasmid pMaOri.Inducible:dCas9sgLipL41 targeting expression of LipL41 (Supplementary Fig. 3).
Recovery of knockout mutants for lipL21 in pathogenic Leptospira
A sgRNA cassette targeting the lipL21 gene of L. interrogans strain Fiocruz L1-130 was included into the pMaOriNHEJ.Inducible:Cas9 plasmid. Conjugation reactions for L. interrogans serogroup Canicola strain LAD-1 or serovar Copenhageni strain Fiocruz L1-130 were plated in HAN media without (-) or with ( +) IPTG, and colonies were enumerated (Fig. 6A and B). For strain LAD-1, which displayed overall higher conjugation efficiencies than strain L1-130, the absence of IPTG favored colony recovery, despite the variability in conjugation efficiencies observed across three independent experiments. Nevertheless, a reduced number of colonies were retrieved in groups containing the sgRNA compared to those without (pMaOriNHEJ.Inducible:Cas9), even in the absence of IPTG, implying that to some extent, lethal DSB were occurring due to a basal “leaky” protein expression of Cas9, and as observed for the IPTG-induced dCas9 cassette.

lipL21 knockout by inducible Cas9 in different strains of L. interrogans. Conjugation reactions for L. interrogans serogroup Canicola strains LAD-1 (A) and serovar Copenhageni strain Fiocruz L1-130 (B) were seeded onto HAN plates with spectinomycin, without (-) or with ( +) 1 mM IPTG. Numbers of recovered colonies after 3 independent experiments were recorded, utilizing two clones of E. coli with the plasmid containing the sgRNA targeting lipL21 in each experiment. Control plasmid with no sgRNA, therefore no DSB, was also employed. Distinct colonies from the plates without IPTG were selected, grown in liquid media without or with IPTG, and whole cell lysates were by SDS-PAGE followed by Sypro Ruby staining (C, LAD-1; D, L1-130) and immunoblotting (E, LAD-1; F, L1-130). Cultures (a-c for LAD-1; a and b for L1-130) were plated for individual colonies, which were selected and lipL21 mutations were assessed by sequencing. (G) Mutation in distinct colonies (mutation/total colonies) from original colonies “a-c” in LAD-1, and (H) from original colonies “a and b” in L1-130. Sequence in green denotes the protospacer contained in the sgRNA, including a mismatch (underlined) for LAD-1, followed by the PAM (purple, not contained in the sgRNA sequence). Cas9 cleavage sites are indicated by the red triangles.
Colonies from agar plates without IPTG were selected, grown in liquid HAN without or with IPTG and whole cell lysates prepared for evaluation of total protein profiles (Fig. 6C and D) and immunoblotting (Fig. 6E and F). For both strains LAD-1 and L1-130, Cas9 expression could be readily observed in total protein profiles only in the presence of IPTG, with no detectable expression of LipL21 in proteins from cells grown in the absence or presence of IPTG. Immunoblotting confirmed the absence of LipL21 protein for LAD-1 cells, even without IPTG, confirming a basal, yet undetectable, expression of Cas9; for L1-130 cells, reduced levels of LipL21 were observed, suggesting that the mutation inflicted into lipL21 gene affected protein expression.
Populations derived from each selected colony (“a-c” for strain LAD-1; “a” and “b” for strain L1-130) were plated and individual colonies were selected to confirm mutation by gene sequencing (Fig. 6G). For strain LAD-1, all colonies from population “a” and “b” presented with a 1 and 10 nucleotide deletion, respectively, starting from the Cas9 cleavage site. For colony “c”, and out of 8 clonal isolates evaluated, 5 presented with a deletion of 27 nucleotides, and 3 presented with a 1 nucleotide insertion; this suggests that cells underwent DSB after at least one generation, resulting in a mixed population of mutant cells within the same colony. Since the sgRNA used in this study targets the 3’ end of lipL21, it was unexpected that the slight truncation of the final protein product would so drastically affect protein expression. Of note, the sgRNA for lipL21 was designed based on the genome of strain L1-130 and is mismatched with the target sequence of strain LAD-1 (underlined nucleotide in the green sequence). The full-length LipL21 protein, excluding the signal peptide, has a predicted molecular mass of 17.82 kDa. The predicted molecular mass for LipL21 is 15.26 kDa in clone “a” (containing a 1 nucleotide deletion), 13.53 kDa in clone “b” (containing 10 nucleotides deletion), and either 16.90 kDa (27 nucleotides deletion, in frame mutation) or 14.51 kDa (1 nucleotide insertion) for clone “c”.
Analysis of mutations for lipL21 in colonies of strain L1-130 showed that all those derived from clone “a” displayed a 12-nucleotide deletion (resulting in a protein of 17.45 kDa) and those derived from clone “b” had a 15-nucleotide deletion (resulting in a truncated LipL21 of 17.33 kDa) (Fig. 6H). Despite this minor reduction on the protein’s molecular mass, it was again observed that protein expression was affected.
Discussion
The negative regulation of the lactose operon in E. coli is achieved by a DNA binding repressor called LacI44,45,46. The lac repressor acts as a homotetramer, consisting of two functional homodimers of LacI polypeptides44,46, capable of binding to a DNA sequence termed the lac operator (lacO), located upstream of the structural genes of the lac operon. When bound to allolactose, or its analog IPTG, LacI undergoes allosteric modification which in turn changes its ability to bind DNA, derepressing transcription of the operon. The regulatory elements of the lac operon have been extensively applied to molecular and system biology, including the spirochete Borrelia burgdorferi47,48.
It has been previously shown that the lac system is functional in Leptospira, being used to express heterologous gfp49. In this early version of the IPTG-inducible cassette, the B. burgdorferi promoter flaB was used to transcribe the lacI gene, codon optimized for Borrelia, followed by the hsp10 promoter and lacO sequence. These results indicated that Leptospira cells are permeable to IPTG, most probably via passive diffusion50, since Leptospira spp. lack a lactose permease51. These results prompted us to develop an optimized inducible cassette, comprising promoters with high transcriptional activity and a lacI sequence codon optimized for Leptospira spp.
When LipL41 protein was used to validated conditional protein expression in the surrogate L. biflexa, no protein band corresponding to the heterologous protein could be observed; we further used the wild type lipL41 cassette from L. interrogans, without any exogenous genetic elements, and recombinant L. biflexa cells were still incapable of expressing LipL41. This is probably due to the absence of the lepcoding sequence in the constructs, since Lep is a molecular chaperone essential for the stable expression of LipL4152. Nevertheless, inclusion of lipL32 CDS to the IPTG-inducible cassette validated the successful protein expression only in the presence of the inducer, whereas no band was observed in its absence, suggesting a well-regulated system. However, it is well established that transcription from the lac promoter is never completely abolished, and some basal “leaky” levels of mRNA can be expected53.
When the IPTG-inducible cassette was used for controlled dCas9 expression, the “leaky” basal, yet undetectable, expression of dCas9 was indirectly demonstrated. Even though no dCas9 protein could be detected when L. biflexa and L. interrogans cells grew in the absence of IPTG, a reduced expression of the target proteins was observed when L. interrogans contained plasmids for IPTG-induced expression of dCas9 and sgRNA targeting either OmpL1 or LipL41, compared to the control cells harboring the plasmids with no sgRNA, even in the absence of the inducer. However, the addition of IPTG and consequently observable dCas9 expression resulted in complete silencing of the target genes. Basal expression of dCas9 was also observed in the IPTG-inducible system for B. burgdorferi47, more pronouncedly when the strains carried the system in a shuttle vector which displays a 5:1 ratio to the chromosome. Leakage was also observed in the previously constructed IPTG-inducible cassette for L. biflexa49.
Interestingly, even with target genes affected by basal dCas9 expression, our system could be successfully used to validate essential genes in the model L. biflexa,with no deleterious phenotypes being observed in the absence of IPTG. It is worth mentioning that the selected targets for silencing in this work code for highly abundant products, since DnaK is expressed in over 3,000 copies/cell39 and RNase P is a major ncRNA in Leptospira41,54. Nonetheless, when working with less abundant targets for essentiality assessments, the basal leakage of dCas9 and its effect target silencing in the absence of IPTG could be more impactful to the phenotype. Accordingly, when Takacs et al.47 used an all-in-one shuttle vector for controlled silencing for rodA, mreB or ftsI, they observed phenotypes consistent with significant basal CRISPRi activity, where they were unable to generate clones containing the plasmid targeting mreB, even in the absence of IPTG. The authors also observed a delayed colony formation for rodAplasmid-containing transformants, with cells often displaying a phenotype in line with a RodA depletion phenotype, even without IPTG. The lack of observable phenotype in the absence of IPTG in our study could also be related to the 1:1 ratio of pMaOri plasmid, the backbone of our all-in-one plasmid for IPTG-inducible silencing, regarding the leptospiral chromosome18,19.
In several bacteria, DnaK is essential for cell growth not only at elevated temperatures but also under optimal conditions55. When CRISPRi with constitutive expression of dCas9 was used, no L. biflexa colonies could be recovered when the dnaK gene was targeted for silencing17. IPTG-inducible CRISPRi was used to re-validate these findings, since this novel tool can discern between a failed conjugation or a lethal phenotype by comparison between colony recovery with and without IPTG. Accordingly, agar plates after L. biflexa conjugation revealed that the presence of IPTG, and therefore dnaK gene silencing, completely annulled colony formation, whereas plates without IPTG displayed similar colony growth rates in comparison to the control group with no sgRNA. Of note, the ability to plate the same conjugation reaction into plates with and without IPTG provides a valuable and more definitive assessment of essential genes, ruling out unsuccessful conjugation experiments.
Evaluation of conditional dnak mutants in liquid media revealed that knockdown mutants for the DnaK chaperone are not viable at higher temperatures (37 °C for Leptospira), and in agreement with previous observations for E. coli at 42 °C56 also, and consistent with the slower growth of null dnaK mutants in E. coli at 30 and 37 °C, L. biflexa mutants reached lower cell densities (~ 100-fold) and presented with an abnormal morphology, compared to cells without IPTG. The mutant growth in liquid medium at 29 °C could also be due to some residual DnaK concentrations in cells that were gradually “titrated” after silencing as cells divided, until it reached critical levels interrupting cell growth. Of note, E. coli mutants that could grow in liquid but not form colonies in solid media were identified in a library of temperature-sensitive mutants57.
Deep RNA sequencing has been used for transcriptome analysis and regulatory RNA discovery in several pathogenic bacteria41,58,59,60. So far, little is known about ncRNA function in Leptospira spp. and whether they have an impact upon virulence. To verify the feasibility of the application of CRISPRi to ncRNA silencing in Leptospira, the non-coding RNase P target was selected, based on its high expression profile41 and its ubiquitous and essential role, since RNase P is involved in the processing of multiple RNA substrates, including tRNAs and small non-coding RNAs61. Inactivation of RNase P leads to accumulation of 5′- extended tRNAs and nonviability in E. coli62,63.
Using a constitutive CRISPRi strategy, it was shown that the complete absence of RNase P is not tolerated by Leptospira cells, even though colony formation was observed when partial silencing was performed. To confirm that indeed the absence of colonies was due to silencing lethality rather than a failed conjugation, the IPTG-induced dCas9 system was used, in conjunction with the sgRNA for complete silencing. Accordingly, while colonies could be recovered in the absence of IPTG, the presence of the inducer completely abolished colony formation, confirming the essential role of RNase P to Leptospira and further demonstrating the feasibility of CRISPRi for silencing ncRNAs in these bacteria.
Because of the previously published inability to recover colonies after silencing of OmpL1, this porin was proposed as essential for leptospiral biology35. Now, taking advantage of the IPTG-inducible silencing, we revisited the role of OmpL1. When both L. biflexa and L. interrogans cells harbored plasmids for IPTG-inducible dCas9 and sgRNA targeting their respective ompL1 genes, colonies could be equally recovered in plates with or without IPTG, and conditional mutants displayed similar growth rates in liquid media. Target protein absence and dCas9 conditional expression was demonstrated by immunoblotting in pathogenic L. interrogans, corroborating that the recovered cells in liquid media in the presence of IPTG were truly knockdown mutants, expressing dCas9, instead of spontaneous spectinomycin-resistant mutants. In this sense, the lack of transconjugant recovery in our previous publication35 could have been due to failed conjugations, consolidating the importance of using the IPTG-inducible CRISPRi.
The concurrent expression of RNA-guided Cas9 and M. smegmatis proteins LigD and Ku, which are responsible for the ligation of DSBs, has allowed the recovery of knockout mutants in Leptospira spp., with the possibility of plasmid elimination after establishment of mutations24. Since both conjugation and DNA repair efficiencies are interdependent events, mutant recovery is somewhat low in this system. To address this gap, a repressed Cas9 expression would allow transconjugant recovery in a first step that is limited only by conjugation efficiencies. This is followed by propagation of cells and Cas9 expression by IPTG and now making mutant recovery dependent on DNA repair frequency. But because of basal leakage of Cas9, DSB and therefore knockout mutagenesis occurred even in the absence of IPTG, but with an apparent better efficiency compared to when conjugation reactions were plated in the presence of the inducer. In addition, strain LAD-1 displayed overall higher conjugation efficiencies. Since a lipL21 mutant colony for LAD-1 displayed a dual population, it suggests that, since Cas9 is expressed in minor basal concentrations in the cytoplasm of these cells, DSB could be happening after at least one cell replication cycle, also giving time for LigD and Ku proteins to accumulate and more readily and efficiently deal with DNA repair.
In conclusion, we show that the IPTG-controlled expression of heterologous proteins in Leptospira spp. and its application for dCas9 and Cas9 expression, further advances the potential for genetic manipulation in these bacteria. The regulated expression of dCas9 enables conditional silencing of target genes and validation of essential genes and non-coding RNAs, providing valuable tools for deeper insights into leptospiral biology and virulence mechanisms. Additionally, the improved recovery of knockout mutants through the controlled Cas9 system, combined with constitutive NHEJ expression, was achieved even in the absence of IPTG due to basal Cas9 expression. This approach facilitates the generation of mutant strains that hold promise for studying virulence phenotypes and enhancing bacterin vaccines for veterinary applications.
Data availability
The original contributions presented in the study are included in the article/Supplementary material. Further inquiries can be directed to the corresponding author.
References
Bharti, A. R. et al. Leptospirosis: A zoonotic disease of global importance. Lancet Infect. Dis. 3(12), 757–771 (2003).
Haake, D. A. & Levett, P. N. Leptospirosis in humans. Curr. Top. Microbiol. Immunol. 387, 65–97 (2015).
Levett, P. N. Leptospirosis. Clin. Microbiol. Rev. 14(2), 296–326 (2001).
Costa, F. et al. Global morbidity and mortality of Leptospirosis: A systematic review. PLoS Negl. Trop. Dis. 9(9), e0003898 (2015).
Fouts, D. E. et al. What makes a bacterial species pathogenic?: Comparative genomic analysis of the genus Leptospira. PLoS Negl. Trop. Dis. 10(2), e0004403 (2016).
Nally, J. E., Artiushin, S. & Timoney, J. F. Molecular characterization of thermoinduced immunogenic proteins Q1p42 and Hsp15 of Leptospira interrogans. Infect. Immun. 69(12), 7616–7624 (2001).
Nally, J. E., Timoney, J. F. & Stevenson, B. Temperature-regulated protein synthesis by Leptospira interrogans. Infect. Immun. 69(1), 400–404 (2001).
Eshghi, A., Cullen, P. A., Cowen, L., Zuerner, R. L. & Cameron, C. E. Global proteome analysis of Leptospira interrogans. J. Proteom. Res. 8(10), 4564–4578 (2009).
Matsunaga, J., Sanchez, Y., Xu, X. & Haake, D. A. Osmolarity, a key environmental signal controlling expression of leptospiral proteins LigA and LigB and the extracellular release of LigA. Infect. Immun. 73(1), 70–78 (2005).
Nally, J. E., Chow, E., Fishbein, M. C., Blanco, D. R. & Lovett, M. A. Changes in lipopolysaccharide O antigen distinguish acute versus chronic Leptospira interrogans infections. Infect. Immun. 73(6), 3251–3260 (2005).
Nally, J. E., Whitelegge, J. P., Bassilian, S., Blanco, D. R. & Lovett, M. A. Characterization of the outer membrane proteome of Leptospira interrogans expressed during acute lethal infection. Infect. Immun. 75(2), 766–773 (2007).
Evangelista, K. V. & Coburn, J. Leptospira as an emerging pathogen: a review of its biology, pathogenesis and host immune responses. Futur. Microbiol. 5(9), 1413–1425 (2010).
Daroz, B. B. et al. A review on Host-. Front. Cell Infect. Microbiol. 11, 777709 (2021).
Kochi, L. T., Fernandes, L. G. V. & Nascimento, A. L. T. O. Heterologous expression of the pathogen-specific LIC11711 gene in the Saprophyte. Pathogens 9(8), 599 (2020).
Picardeau, M. Virulence of the zoonotic agent of leptospirosis: still terra incognita?. Nat. Rev. Microbiol. 15(5), 297–307 (2017).
Picardeau, M. Conjugative transfer between Escherichia coli and Leptospira spp. as a new genetic tool. Appl. Environ. Microbiol. 74(1), 319–322 (2008).
Fernandes, L. G. V. et al. Gene silencing based on RNA-guided catalytically inactive Cas9 (dCas9): A new tool for genetic engineering in Leptospira. Sci. Rep. 9(1), 1839 (2019).
Fernandes, L. G. V., Hornsby, R. L., Nascimento, A. L. T. O. & Nally, J. E. Application of CRISPR interference (CRISPRi) for gene silencing in pathogenic species of Leptospira. J. Vis. Exp. https://doi.org/10.3791/62631-v (2021).
Fernandes, L. G. V., Hornsby, R. L., Nascimento, A. L. T. O. & Nally, J. E. Genetic manipulation of pathogenic Leptospira: CRISPR interference (CRISPRi)-mediated gene silencing and rapid mutant recovery at 37 °C. Sci. Rep. 11(1), 1768 (2021).
Croda, J. et al. Targeted mutagenesis in pathogenic Leptospira species: Disruption of the LigB gene does not affect virulence in animal models of leptospirosis. Infect. Immun. 76(12), 5826–5833 (2008).
Fernandes, L. G. V. et al. Evaluation of LipL32 and LigA/LigB Knockdown Mutants in. Front. Microbiol. 12, 799012 (2021).
Murray, G. L. et al. Major surface protein LipL32 is not required for either acute or chronic infection with Leptospira interrogans. Infect. Immun. 77(3), 952–958 (2009).
Wunder, E. A. et al. A novel flagellar sheath protein, FcpA, determines filament coiling, translational motility and virulence for the Leptospira spirochete. Mol. Microbiol. 101(3), 457–470 (2016).
Fernandes, L. G. V. & Nascimento, A. L. T. O. A novel breakthrough in. Front. Microbiol. 13, 915382 (2022).
Fernandes, L. G. V. et al. CRISPR-prime editing, a versatile genetic tool to create specific mutations with a single nucleotide resolution in. mBio 15(9), e0151624 (2024).
Giraud-Gatineau, A. et al. Evolutionary insights into the emergence of virulent Leptospira Spirochetes. PLoS Pathog. 20(7), e1012161 (2024).
Jiang, W., Bikard, D., Cox, D., Zhang, F. & Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31(3), 233–239 (2013).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096), 816–821 (2012).
Bowater, R. & Doherty, A. J. Making ends meet: repairing breaks in bacterial DNA by non-homologous end-joining. PLoS Genet. 2(2), e8 (2006).
Shuman, S. & Glickman, M. S. Bacterial DNA repair by non-homologous end joining. Nat. Rev. Microbiol. 5(11), 852–861 (2007).
Mao, Z., Bozzella, M., Seluanov, A. & Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7(18), 2902–2906 (2008).
Aniukwu, J., Glickman, M. S. & Shuman, S. The pathways and outcomes of mycobacterial NHEJ depend on the structure of the broken DNA ends. Genes Dev. 22(4), 512–527 (2008).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5), 1173–1183 (2013).
Fernandes, L. G. V., Foltran, B. B., Teixeira, A. F. & Nascimento, A. L. T. O. LipL41 and LigA/LigB gene silencing on a LipL32 knockout. Pathogens 12(10), 1191 (2023).
Fernandes, L. G. V., Teixeira, A. F. & Nascimento, A. L. T. O. Evaluation of. Front. Microbiol. 14, 1199660 (2023).
Nascimento, A. L. et al. Genome features of Leptospira interrogans serovar Copenhageni. Braz. J. Med. Biol. Res. 37(4), 459–477 (2004).
Hornsby, R. L., Alt, D. P. & Nally, J. E. Isolation and propagation of leptospires at 37 °C directly from the mammalian host. Sci. Rep. 10(1), 9620 (2020).
Demarre, G. et al. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res. Microbiol. 156(2), 245–255 (2005).
Malmström, J. et al. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 460(7256), 762–765 (2009).
Jewett, M. W., Jain, S., Linowski, A. K., Sarkar, A. & Rosa, P. A. Molecular characterization of the Borrelia burgdorferi in vivo-essential protein PncA. Microbiology 157(Pt 10), 2831–2840 (2011).
Zhukova, A. et al. Genome-wide transcriptional start site mapping and sRNA identification in the pathogen. Front. Cell Infect. Microbiol. 7, 10 (2017).
Pappas, C. J., Benaroudj, N. & Picardeau, M. A replicative plasmid vector allows efficient complementation of pathogenic Leptospira strains. Appl. Environ. Microbiol. 81(9), 3176–3181 (2015).
Zhao, Y. et al. CRISPR/dCas9-mediated multiplex gene repression in Streptomyces. Biotechnol. J. 13, e1800121 (2018).
Lewis, M. The lac repressor. C R Biol. 328(6), 521–548 (2005).
Matthews, K. S. & Nichols, J. C. Lactose repressor protein: Functional properties and structure. Prog. Nucl. Acid. Res. Mol. Biol. 58, 127–164 (1998).
Wilson, C. J., Zhan, H., Swint-Kruse, L. & Matthews, K. S. The lactose repressor system: paradigms for regulation, allosteric behavior and protein folding. Cell Mol. Life Sci. 64(1), 3–16 (2007).
Takacs, C. N. et al. A CRISPR interference platform for selective downregulation of gene expression in. Appl. Environ. Microbiol. https://doi.org/10.1128/AEM.02519-20 (2021).
Murphy, B. T. et al. Inducible CRISPRi-Based Operon Silencing and Selective in. J. Bacteriol. 205(2), e0046822 (2023).
Aviat, F., Slamti, L., Cerqueira, G. M., Lourdault, K. & Picardeau, M. Expanding the genetic toolbox for Leptospira species by generation of fluorescent bacteria. Appl. Environ. Microbiol. 76(24), 8135–8142 (2010).
Simas, R. G., Pessoa Junior, A. & Long, P. F. Mechanistic aspects of IPTG (isopropylthio-β-galactoside) transport across the cytoplasmic membrane of Escherichia coli-a rate limiting step in the induction of recombinant protein expression. J. Ind. Microbiol. Biotechnol. https://doi.org/10.1093/jimb/kuad034 (2023).
Nascimento, A. L. et al. Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. J. Bacteriol. 186(7), 2164–2172 (2004).
King, A. M. et al. Leptospiral outer membrane protein LipL41 is not essential for acute leptospirosis but requires a small chaperone protein, lep, for stable expression. Infect. Immun. 81(8), 2768–2776 (2013).
Nielsen, B. L., Willis, V. C. & Lin, C. Y. Western blot analysis to illustrate relative control levels of the lac and ara promoters in Escherichia coli. Biochem. Mol. Biol. Educ. 35(2), 133–137 (2007).
Caimano, M. J. et al. A model system for studying the transcriptomic and physiological changes associated with mammalian host-adaptation by Leptospira interrogans serovar Copenhageni. PLoS Pathog. 10(3), e1004004 (2014).
Schramm, F. D., Heinrich, K., Thüring, M., Bernhardt, J. & Jonas, K. An essential regulatory function of the DnaK chaperone dictates the decision between proliferation and maintenance in Caulobacter crescentus. PLoS Genet 13(12), e1007148 (2017).
Paek, K. H. & Walker, G. C. Escherichia coli dnaK null mutants are inviable at high temperature. J. Bacteriol. 169(1), 283–290 (1987).
Nosho, K. et al. Isolation of colonization-defective Escherichia coli mutants reveals critical requirement for fatty acids in bacterial colony formation. Microbiology (Reading) 164(9), 1122–1132 (2018).
Putz, E. J. et al. Distinct transcriptional profiles of Leptospira borgpetersenii serovar Hardjo strains JB197 and HB203 cultured at different temperatures. PLoS Negl. Trop. Dis. 15(4), e0009320 (2021).
Sahr, T. et al. Deep sequencing defines the transcriptional map of L. pneumophila and identifies growth phase-dependent regulated ncRNAs implicated in virulence. RNA Biol. 9(4), 503–519 (2012).
Wurtzel, O. et al. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog. 8(9), e1002945 (2012).
Loveland, J. L., Rice, J., Turrini, P. C., Lizotte-Waniewski, M. & Dorit, R. L. Essential is not irreplaceable: fitness dynamics of experimental E. coli RNase P RNA heterologous replacement. J. Mol. Evol. 79(3–4), 143–152 (2014).
Mohanty, B. K. & Kushner, S. R. Ribonuclease P processes polycistronic tRNA transcripts in Escherichia coli independent of ribonuclease E. Nucl. Acid. Res. 35(22), 7614–7625 (2007).
Mohanty, B. K. & Kushner, S. R. New Insights into the Relationship between tRNA processing and Polyadenylation in Escherichia coli. Trend. Genet. 35(6), 434–445 (2019).
Acknowledgements
This research was supported by USDA and in part by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and the U.S. Department of Agriculture (USDA). ORISE is managed by ORAU under DOE contract number 60-5030-3-003. USDA is an equal opportunity provider and employer. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. We thank Dr. David Haake and Dr. James Matsunaga for the anti-OmpL1 antibodies.
Funding
U.S. Department of Agriculture, Oak Ridge Associated Universities, Fundação Butantan
Author information
Authors and Affiliations
Contributions
Writing – original draft: LGVF, JEN Writing – review & editing: LGVF, JEN, ALTON Data curation: LGVF, JEN, ALTON Formal analysis: LGVF, JEN Investigation: LGVF, JEN Methodology: LGVF Validation: LGVF, JEN, ALTON Supervision: JEN Funding acquisition: JEN Project administration: JEN.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernandes, L.G.V., Nascimento, A.L.T.O. & Nally, J.E. Induced protein expression in Leptospira spp. and its application to CRISPR/Cas9 mutant generation. Sci Rep 15, 4334 (2025). https://doi.org/10.1038/s41598-025-88633-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88633-w
