
Abstract
Identifying viral replication within cells demands labor-intensive isolation methods, requiring specialized personnel and additional confirmatory tests. To facilitate this process, we developed an AI-powered automated system called AI Recognition of Viral CPE (AIRVIC), specifically designed to detect and classify label-free cytopathic effects (CPEs) induced by SARS-CoV-2, BAdV-1, BPIV3, BoAHV-1, and two strains of BoGHV-4 in Vero and MDBK cell lines. AIRVIC utilizes convolutional neural networks, with ResNet50 as the primary architecture, trained on 40,369 microscopy images at various magnifications. AIRVIC demonstrated strong CPE detection, achieving 100% accuracy for the BoGHV-4 DN-599 strain in MDBK cells, the highest among tested strains. In contrast, the BoGHV-4 MOVAR 33/63 strain in Vero cells showed a lower accuracy of 87.99%, the lowest among all models tested. For virus classification, a multi-class accuracy of 87.61% was achieved for bovine viruses in MDBK cells; however, it dropped to 63.44% when the virus was identified without specifying the cell line. To the best of our knowledge, this is the first research article published in English to utilize AI for distinguishing animal virus infections in cell culture. AIRVIC’s hierarchical structure highlights its adaptability to virological diagnostics, providing unbiased infectivity scoring and facilitating viral isolation and antiviral efficacy testing. Additionally, AIRVIC is accessible as a web-based platform, allowing global researchers to leverage its capabilities in viral diagnostics and beyond.
Similar content being viewed by others
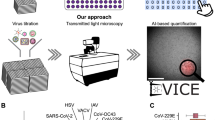


Introduction
Worldwide, virology laboratories use cell cultures to produce viruses, which are obligate intracellular parasites. During virus isolation, some viruses replicate with cytopathic effects (CPEs), while others do not. The underlying virus can trigger various structural and functional alterations in host cells. The underlying virus can induce various structural and functional changes in host cells, including rounding, cell enlargement, granulation, syncytia formation, cell lysis, and detachment, among others1,2,3. Over time, observing these CPE changes allows researchers to track viral infection progression and gain insights into viral mechanisms, disease development, and antiviral treatment efficacy. However, some viruses have subtle or delayed effects, making them difficult to identify based solely on CPE observation. Additionally, identifying and interpreting CPEs can be subjective and requires trained personnel. Furthermore, cellular stress or other factors can mimic some CPEs, requiring additional tests for confirmation. Therefore, cell cultures are laborious and expensive due to the materials used, the time required, and the need for additional testing to detect the virus after production, which can be further complicated by genetic diversity and co-infections4. To avoid these challenges, artificial intelligence systems can be used to recognize and differentiate viral CPE. Deep learning neural network models provide various benefits, such as enhanced performance due to their capacity to manage extensive datasets and intricate architectures5,6. Creating a dataset of virus-induced CPEs and developing models to recognize the virus will contribute to the isolation process and prevention and treatment of many viral diseases. For similar purposes, various models have been tested for recognition of CPE by artificial intelligence using different human viruses such as respiratory syncytial virus (RSV), influenza, Corona 229E, herpes simplex virus type 1 (HSV-1), enterovirus, SARS-CoV-2, polio, parainfluenza, adenovirus, modified vaccinia ankara (MVA), human immunodeficiency virus (HIV), parainfluenza virus, and herpes simplex virus type 2 (HSV-2)7,8,9,10. In this study, virus-induced datasets and convolutional neural network (CNN) models were developed using a local strain of SARS-CoV-2 and various viruses associated with the Bovine Respiratory Disease Complex (BRDC), without the need for labeling.
The respiratory virus responsible for COVID-19, SARS-CoV-2 belongs to the Coronaviridae family’s Betacoronavirus genus. It is highly contagious and has caused significant disruptions to world health and economies because of its rapid transmission and devastating disease outcomes11,12. Numerous cell lines have been identified to be infected by SARS-CoV-2, with differing degrees of cytopathogenicity and susceptibility. Among these, Vero cells—derived from the kidney epithelial cells of the African green monkey—are widely used in virology laboratories for isolating viruses and producing viral stocks because of their ability to support the replication of various viruses and their distinct cytopathic effects (CPE), such as cell rounding, detachment, and lysis. Notably, SARS-CoV-2-induced CPE in Vero/Vero E6 cells can be observed as early as 48 h post-infection13,14.
BRDC, also known as shipping fever pneumonia, imposes significant financial burdens on the cattle industry, including costs related to lost productivity, treatments, mortality, and diagnostics15. These factors threaten the long-term sustainability of farms and the industry as a whole16. This study focuses on several pathogens that contribute to BRDC, including bovine herpesvirus type 1 (BoAHV-1), bovine herpesvirus type 4 (BoGHV-4), bovine parainfluenza virus type 3 (BPIV3), and bovine adenovirus-1 (BAdV-1). Within this complex, BoAHV-1 is belonging to the genus Varicellovirus within the subfamily Alphaherpesvirinae and the Herpesviridae family. Another one; BoGHV-4, also belonging to the same family located in the Rhadinovirus genus, Gammaherpesvirinae subfamily. Both BoAHV-1 and BoGHV-4 are enveloped, double-stranded linear DNA viruses that can cause respiratory disease and genital system infections while also exhibiting the latency characteristic of herpesviruses17. The third one; BPIV3, which is alternatively referred to as Bovine Respirovirus 3, is a negative strand, enveloped RNA virus in Paramyxoviridae family, Respirovirus genus and frequently causes moderate or undetectable symptoms in cattle and sheep, but it can cause more serious infections if co-infected with other respiratory viruses and bacteria. Lastly, BAdV-1 is a non-enveloped, double-stranded DNA virus belonging to the genus Mastadenovirus within the family Adenoviridae, known to cause enteritis or pneumoenteritis in affected cattle18,19. These viruses can manifest numerous cytopathologies, such as cytoplasmic inclusions, shrinkage, cell enlargement, vacuolation, nuclear alterations, image of grape clusters, plaques and syncytia development19,20,21,22,23,24,25.
CPEs caused by different viruses can appear visually similar, complicating their identification and requiring additional methods post-replication. The aim of this study was to utilize existing architectures to establish deep learning models for AI recognition of viral CPEs (AIRVIC) under a range of conditions. Specifically, the objective was to create models capable of distinguishing viral CPEs across different cell lines from morphological changes due to non-viral factors, such as cellular aging. AIRVIC was further enhanced to differentiate among multiple viruses, reducing the need for supplementary post-isolation procedures. The results aligned well with laboratory findings, supporting the AIRVIC’s efficiency. Although deep learning studies have begun to be conducted for human viruses7,8,9,10,26 to our knowledge, this study is the initial investigation of utilizing CNN models for the identification and discrimination of animal virus infections.
Results
Deep learning models accurately and efficiently detect viral CPE across infections, but achieving high performance requires a large and diverse dataset
Virus-induced datasets were generated, and convolutional neural network (CNN) models were developed to enhance viral detection and discrimination, focusing on bovine viruses and SARS-CoV-2. Bovine viruses were successfully propagated in MDBK cells, whereas Vero cells were used to produce SARS-CoV-2. Following propagation, TCID50 assays were conducted to calculate the multiplicities of infection (MOIs) for inoculating bovine viruses in MDBK and Vero cells, and SARS-CoV-2 in Vero cells at various doses. Viral genes were amplified, which yielded fragments of the expected size, and PCR products were confirmed via Sanger sequencing by a commercial firm. Data collection across various MOIs and time points allowed for a detailed analysis of viral progression and CPE across multiple cell lines and viral strains. This comprehensive dataset, highlighting specific CPE examples, is illustrated in Fig. 1. For SARS-CoV-2 in Vero cells, MOIs ranged from 0.0001 to 0.5, with post-infection imaging conducted at time intervals from 8 to 73 h, yielding 969 images. The BoGHV-4 (MOVAR 33/63) was inoculated in MDBK cells, with MOIs ranging from 0.0001 to 1.0, and imaging occurred between 8- and 150-hours post-infection, resulting in 5,053 images. In Vero cells, BoGHV-4 (MOVAR 33/63) produced 5,009 images across 7 time points between 8- and 150-hours post-infection. Similarly, the BoGHV-4 (DN-599) strain in MDBK cells produced 2,545 images between 8- and 72-hours post-infection, whereas its growth in Vero cells resulted in 4,208 images collected from 6 time points spanning 8 to 126 h. BPIV3, in MDBK cells, formed 4,718 images over a range of 8 to 128 h, while in Vero cells, 3,259 images were captured at 7 time points from 8 to 96 h. The propagation of BoAHV-1 in MDBK cells generated a total of 2,823 images over a time frame of 8 to 72 h, while its propagation in Vero cells produced 4,678 images within 8 to 128 h. Lastly, BAdV-1 generated 2,393 images in MDBK cells at MOIs between 0.0001 and 0.5, with time points from 8 to 76 h, while in Vero cells, 4,714 images were captured at 8 time points ranging from 8 to 120 h. In total, 40,369 images were collected across various conditions, highlighting the range and diversity of the dataset (Supplementary Table S1, Fig. 2).

Dataset composition. Distribution of control and virus-infected cell images among various virus and cell line combinations collected in this study.

Effects of magnification, MOI, and post-infection time on the visualization of viral-induced CPE in various cell lines. (a) Vero cells infected with SARS-CoV-2 at an MOI of 0.01, captured 46 h post-infection. Images display the cells at varying magnifications. (b) MDBK cells infected with BoAHV-1 at 20x magnification, 24 h post-infection, with varying MOIs. (c) MDBK cells infected with BoGHV-4 (DN-599) at 20x magnification and an MOI of 0.001 at various hours post-infection.
In selecting an architecture, we focused on optimizing model performance to address the complexities and unique characteristics of the dataset. The goal was to identify architectures that minimize overfitting while enhancing generalization across different augmentations and dataset variations. To balance model complexity with predictive capability, we compared architectures such as ResNet18, ResNet50, MobileNetV2, and YGNet.
In the initial training phase, we only used ResNet50 with 20X magnification images on the BoAHV-1/MDBK dataset. This setup yielded an accuracy of 0.8439 but an F1 score of just 0.4576, emphasizing the effect of dataset imbalance. The problem originated from the fact that for every control cell image, there were approximately five to six infected cell images, which posed a significant challenge for the model’s learning process. The F1 score, which revealed the presence of this imbalance, is the harmonic mean of precision (the ratio of true positives to all predicted positives) and recall (the ratio of true positives to the sum of true positives and false negatives (Supplementary Note C). Data augmentation was employed to balance the control and infected cell images, resulting in notable improvement. After augmentation, the F1 score increased to 0.9523, and accuracy reached 0.9514 (Supplementary Table S2). To avoid overfitting, we enriched the dataset by incorporating 10X magnification images. This enhancement significantly improved the model’s evaluation metrics in fewer epochs while maintaining consistent hyperparameters (Supplementary Table S3). Subsequently, we added 5X and 40X magnification images to the dataset when evaluating the ResNet18 model using the same hyperparameters (Supplementary Table S4). The analysis indicated that the model struggled to accurately detect CPE with 5x magnification images. However, with images at 10X-20X and 40X magnifications, the model’s detection capability improved markedly across all architectures (Supplementary Table S5). To broaden our analysis, the architectures were trained on a different dataset, SARS-CoV-2/Vero, to compare their adaptability across varied data. Although MobileNetV2 achieved the highest accuracy on the SARS-CoV-2/Vero dataset (Supplementary Table S6), the stability demonstrated by the ResNet50 model on this dataset, along with its better performance on the BoAHV-1/MDBK dataset makes it the preferred choice for handling more complex tasks in later stages of the research. The consistent efficiency of ResNet50 across various datasets highlights its reliability and supports its adoption as a baseline model. Optimized use of data augmentation, hyperparameter tuning, and model selection further enhances virus-specific CPE detection accuracy of the models.
AIRVIC introduces a structured approach to virus classification via CPE detection
To optimize viral detection and classification through CPE analysis, AIRVIC employs a hierarchical system consisting of three distinct deep learning models, each tailored to a specific aspect of detection. The Broad-Spectrum CPE Detection Model functions as an initial screening tool, identifying any CPE and assessing likely viral activity. When a CPE is detected, the system transitions to the Virus-Specific CPE Detection Model, which provides a probabilistic assessment to identify the viruses most likely responsible for the observed cytopathologies. Finally, the Targeted CPE Detection Model refines this analysis, confirming findings with a more focused, virus-specific approach. Together, these three-tiered models streamline the diagnostic workflow, making it both more efficient and more accurate. Figure 3 illustrates this workflow and demonstrates how AIRVIC combines the capabilities of these models for enhanced CPE detection.

AIRVIC workflow. The process begins with a broad-spectrum CPE detection model for initial screening. Cases with positive detections proceed to analysis by virus-specific CPE detection models to identify probable viral agents. High-probability cases are subsequently validated using targeted CPE detection models to ensure accurate confirmation.
In the Targeted CPE Detection (Fig. 4), the performance in detecting the cytopathic effects of various viruses on specific cell lines was thoroughly evaluated. Each virus-cell line combination was tested under controlled conditions at magnifications of 10X, 20X, and 40X. To strengthen the dataset, systematic data augmentation was applied, followed by precise filtering based on MOI values. This filtering process was carefully tailored for each combination by considering specific post-infection hours and MOI values (Supplementary Table 7). Consequently, images where the cytopathic effect had not yet appeared were excluded, significantly enhancing the model’s learning accuracy. This approach allowed for the exclusion of early infection hours, when virus-induced CPE was not yet evident, and enabled the selection of optimal MOI ranges to capture the critical stages of virus-induced CPE on the cells.

Overview of CPE detection models. Targeted CPE Detection for specific virus-cell line combinations, Broad-Spectrum CPE Detection across multiple viruses and cell lines, and Virus-Specific CPE Detection to pinpoint the exact virus causing CPE. In the input stage, the term ‘new image’ refers to images that were not seen by the model during training or validation and are exclusively used for testing purposes.
Model parameters, unless otherwise specified in the Supplementary Table S7, were standardized with a batch size of 8, dropout of 0.5, learning rate of 5e-4, weight decay of 5e-2, step size of 2, and gamma of 0.8 across 100 epochs of training. Most models showed strong performance. In particular, the BoGHV-4(DN-599)/MDBK model achieved perfect scores, with 100% accuracy. A BPIV3/Vero model with 98.74% accuracy and 0.9820 F1-score also performed well. The BoAHV-1/MDBK model followed closely, with 97.94% accuracy and 0.9797 F1-score (Fig. 5). Detailed results of Targeted CPE Detection models are presented in Table 1.

Model performance metrics across virus-cell line combinations. F1 score, precision, recall, and accuracy of the model across various virus-cell line combinations, highlighting the model’s effectiveness in detecting specific viruses in different cell environments. BoGHV-4 (M) represents the MOVAR 33/63 strain of Bovine herpesvirus 4, while BoGHV-4 (D) refers to the DN-599 strain.
Broad-spectrum CPE detection model’s performance, comparable to targeted models, offers an advantage in identifying virus-induced CPE in field samples, including new viruses
The Broad-Spectrum CPE Detection approach (Fig. 4) was trained using the entire dataset to detect CPE across a wide range of viruses and cell lines, utilizing the ResNet50 architecture. The utilized virus-cell pairs are: BAdV-1, BPIV3, BoAHV-1, BoGHV-4 in both Vero and MDBK cells, and SARS-CoV-2 in Vero cells. This model was trained with the same hyperparameters as those used in targeted detection scenarios and with filtering criteria applied per virus and cell line, ensuring consistency in data preprocessing across different detection tasks. During the testing phase, images featuring all virus-cell pairs that were not used in training or validation were employed. Testing was conducted on 10% of the complete dataset which corresponds to approximately 4,000 images across all virus-cell pairs. After completing 100 epochs, the model achieved an F1 score of 0.9495 and an accuracy of 0.9528. This highlights the model’s effectiveness in the broad-spectrum context, showcasing its capacity to adeptly manage the diverse and complex viral and cellular interactions within the dataset. However, it is important to note that no viruses outside the training dataset were used during testing, and the model’s performance on such viruses remains untested. While the Broad-Spectrum CPE Detection model offers the potential to detect CPE from new viruses, this capability has not yet been experimentally validated. This accuracy is specific to the viruses included in the study. Although the broad-spectrum model’s performance metrics are slightly lower than the average of targeted models (Fig. 6), using a universal model to cover all scenarios provides computational efficiency advantages for identifying virus-induced CPE in field samples, not only for this virus dataset but also potentially for new viruses.

Comparative performance of targeted vs. broad-spectrum CPE detection models. F1 score, precision, recall, and accuracy of the average targeted CPE detection models compared to the broad-spectrum CPE detection model, illustrating the adaptability and efficiency of the universal model in diverse cellular environments.
Table 1 provides a detailed analysis of the performance metrics for both detection strategies. It aggregates the results for various virus-cell line combinations and includes evaluations conducted in the broad-spectrum detection context.
AIRVIC achieved success at the level of viral gene amplification, and further: virus and strain differentiation with notable accuracy
In Virus-Specific CPE Detection (Fig. 4), the goal was not only to identify the presence of CPE but also to determine the specific virus and cell line combinations responsible for it. Models were trained using varying magnification levels and data augmentation, with specific filtering to exclude early post-infection hours where cellular responses were uniform across different viruses. Unless otherwise specified in Supplementary Table 6, the hyperparameters used for these models were a batch size of 16, dropout rate of 0.5, learning rate of 5e-4, weight decay of 5e-2, step size of 2, and gamma of 0.8. The test set for each model comprised approximately 10% of the dataset specific to each virus-cell line combination, with the exact number of images depending on the dataset size (e.g., approximately 4,000 test images for the 11-class mixed cell line model and approximately 1,750 test images for the MDBK-specific 5-class model). These images were entirely excluded from training and validation. The models exhibited varying performance across different configurations. In the 11-class mixed cell line model, aimed at detecting both the cell line and virus causing CPE, the model achieved an F1 score of 0.7032 and an accuracy of 0.6960 after 100 training epochs (Fig. 7a). A 6-class generalized model, which was developed to detect the virus causing CPE without differentiating by cell line, yielded an F1 score of 0.6260 and an accuracy of 0.6344 over 100 epochs. For MDBK cells, a more specific 5-class model was developed, achieving an accuracy of 0.8539 and an F1 score of 0.8489 after 400 epochs of training. Incorporating data from the BoGHV-4 (MOVAR 33/63) and BoGHV-4 (DN-599) strains further enhanced the model’s performance, with the accuracy improving to 0.8761 and the F1 score to 0.8729. Figure 7b illustrates the steady progression of the MDBK-specific model, showcasing improvements in multi-class accuracy, F1 score, and loss reduction across the epochs. Similarly, for Vero cells, the initial 6-class model showed suboptimal performance, with an F1 score of 0.6550. This led to the development of a more targeted 4-class model, which excluded two poorly performing viruses. The updated model achieved an accuracy of 0.7540 and an F1 score of 0.7387 after 400 epochs of training, as shown in Fig. 7c. This version demonstrated clear improvements in accuracy and loss reduction, indicating effective optimization over the course of training.

Training performance of virus-specific CPE detection models. The figure shows the evolution of accuracy, F1 score, and loss over training epochs: (a) mixed cell lines, (b) MDBK cell lines, and (c) Vero cell lines. In each graph, the metrics highlight the model’s ability to distinguish between various CPE presentations induced by different viruses. The multi-class accuracy graphs for the MDBK and Vero models (b, c) display individual virus performance, with the black dashed line indicating the average accuracy across classes.
In contrast to the more focused targeted CPE detection models, the observed decrease in performance for the virus-specific CPE detection models underscores the heightened complexity and challenge involved in concurrently identifying not only the type of virus but, in some cases, the specific cell line as well. While the targeted models are designed to detect the presence of CPE, the virus-specific CPE detection models aim for a more detailed classification, determining both the presence of CPE and the exact virus or virus-cell line combination responsible. Despite these challenges, the virus-specific CPE detection models provide a detailed and holistic understanding of the viral landscape in a single analytical step, which is essential for the rapid and effective execution of diagnostic processes. In addition to that, strain differentiation is not achievable in many PCR reactions that rely on traditional primer binding, and the PCR assay used in this study was no exception. However, AIRVIC was able to distinguish between two viral strains in MDBK cells with an 81.29% success rate. Results and detailed metrics are compiled in Table 2 for a complete performance analysis.
Discussion
Artificial intelligence (AI) is transforming health sciences, with significant advancements in drug design, medical imaging, ophthalmology, dermatology, and radiology27,28,29,30,31,32,33,34. Recently, AI applications have expanded into virology35,36. Over the past five years, studies using deep learning to detect virus-induced morphological defects have primarily focused on human viruses in cell lines7,8,9,10,26,37. To the best of our knowledge, no English-language research articles have been published on the detection of animal viruses in cell culture. Studies detecting morphological defects caused by viruses using deep learning, particularly CNNs, are also relatively scarce8,10,26. Even fewer studies have focused on virus discrimination, such as Petkidis et al.9 using EfficientNet B0 and Chen, et al.7 employing ResNet 50 for this task.
In these studies, when considering the F1 score as the primary performance metric, Chen, et al.7 achieved an accuracy of 72.90% with a single-task model and 85.4% with a multi-task model on a total of 9,739 microscopic images. In comparison, Petkidis et al.9 demonstrated 80.2% accuracy in virus differentiation with their multi-task model on a total of 58,619 images comprising different viruses. In this study, utilizing light microscopy images, we developed AIRVIC, a system that automatically identifies CPE across a diverse range of unlabeled viruses and cell lines. On a dataset of 40,369 images, the single-task model demonstrated an average performance of 95.31% across all tested viruses, with F1 scores ranging from a maximum of 100% to a minimum of 87.28% for individual viruses. The multi-task model achieved 87.29% accuracy in distinguishing between four different viruses produced in MDBK cells. Additionally, it successfully classified two strains of BoGHV-4 with 84.89% accuracy. To our knowledge, no prior data exists on classifying CPEs caused by different strains of the same virus, which highlights AIRVIC’s refined detection capability. By classifying CPEs that appear identical to the human eye, AIRVIC underscores its potential for precise viral strain identification. The model’s performance in VERO cells was somewhat lower, demonstrating a 74.50% accuracy in virus differentiation, likely due to dataset imbalance, even though augmentation and filtering techniques were applied. Notably, even without information on cell line type, AIRVIC demonstrated a 69.60% accuracy in virus differentiation. It is essential to note that all comparisons involved different viruses and cell line datasets for each study mentioned. A more accurate evaluation could be achieved using a single, consistent dataset, underscoring the need for a benchmark dataset, similar to those established in many other research fields38,39,40.
Chen, et al.7 did not consider time-based differentiation. In contrast, our study attempted to detect and differentiate CPE even in images with minimal CPE presence. This approach aligns with the work of Petkidis et al.9 which performed simultaneous virus detection and viral titer calculation from images using a mathematical formula. Wang et al.10 used 1,190 images in their CNN model, developed to detect CPE caused by influenza virus in MDCK cells. They conducted additional experiments at different time intervals with the model achieving recognition rates of 71.80% at 16 h, 98.25% at 28 h, and 87.46% at 40 h. This supports our findings, indicating that CPE quantity impacts success rates, with higher CPE ratios improving performance. Poor performance on early infection data (8 h) in our study, due to insufficient viral replication, was resolved by applying replication-time and MOI-based filters. Similarly, Petkidis et al.9 reported challenges in detecting adenovirus in early stages and encountered similar difficulties with their DVICE model using GFP labeling Time-based imaging that considers viral load offers a more realistic approach, mirroring real-world conditions. These studies emphasize the limitations of current methods in detecting viruses before visible CPEs appear. Future research could explore imaging techniques that detect intracellular changes before visible CPEs, further improving early detection.
To prevent the machine learning model from memorizing background differences, we converted images to grayscale before training and eliminated non-CPE-related factors, enhancing AIRVIC’s performance. This preprocessing step avoided background memorization and improved the detection accuracy of CPE patterns caused by the virus. It should be considered essential for future studies, and comparisons between studies should be viewed through this lens, as many studies have prevented data noise memorization by employing similar techniques41,42,43.
Our analysis revealed that viral strain and cell line interactions significantly impacted model performance. This aligns with Chen et al.7 and Wang et al.10, who observed cell line-dependent variations in CPE detection. In our study, the models exhibited higher precision in MDBK cells compared to Vero cells, highlighting the significance of cell-specific factors. Models trained on smaller datasets underperformed, while accuracy improved as dataset size increased. Unexpectedly, combining images from different magnifications (10X, 20X, 40X) improved performance, while adding 5X images reduced success, suggesting that the model struggled to learn CPE features at lower magnifications.
AIRVIC performed well on simpler tasks using smaller models like ResNet18 and MobileNetV2, while ResNet50 was more effective for complex tasks, which is consistent with the literature44,45. In scenarios where resources are limited, such as future applications where models might run directly on microscopes or other edge devices, lightweight models provide effective results. As demonstrated in our study on SARS-CoV-2-induced CPE detection and supported by other studies in the literature46,47,48, MobileNetV2 is particularly well-suited for real-time inference and resource-constrained environments.
This study focused on SARS-CoV-2 and BRDC pathogens affecting cattle, both of which have significant economic and health impacts. Vaccination, early detection, and local isolate acquisition are crucial in controlling these diseases. For SARS-CoV-2, rapid diagnosis and isolation, aided by AI, play a pivotal role in accelerating research, vaccine development, and treatment efforts. To detect an unknown virus from the field, multiplex or monoplex PCR reactions with primers for various viruses are necessary after CPE formation. However, the CNN models developed here can differentiate viruses from the trained virus dataset in seconds using a single image.
AIRVIC employs a hierarchical model sequence to optimize CPE detection, starting with broad-spectrum CPE identification, then narrowing to specific viruses through targeted models. This structured approach streamlines the diagnostic process and emphasizes the importance of hyperparameter tuning. The flexibility of deep learning models enables AIRVIC to handle complex biological data, making it a valuable tool for virological diagnostics.
AIRVIC is primarily designed for detection, diagnosis, and classification, and is not intended for quantification. Therefore, it is presently unsuitable to calculate viral titers based on CPE. Viral titer calculation involves determining the number of infectious viral particles in a sample and is mostly performed using cell culture methods or PCR. Although the determination of viral titers using AI-based CPE detection is beyond the scope of this study, AIRVIC’s flexibility and potential adaptability underscore the necessity for further research in this area. Enhancing the ability to quantify infectious viral units, especially using label-free methods, will substantially improve AIRVIC’s functionalities and serve as a crucial instrument for forthcoming virological studies. Another limitation of AIRVIC is related to the type of microscope, as the models developed in this study were tested using images obtained exclusively with a ZEISS Axio Observer 5 microscope. These images were captured by three different individuals with the aim of creating variability in image acquisition, simulating real-world circumstances, and ensuring generalizability across various user techniques. The accuracy of the models depends on these specific imaging settings, and their efficacy with alternative microscopes has not been determined. Subsequent research will aim to assess the models’ performance across various microscopy equipment and at different levels of user proficiency.
Now accessible as a web-based application (https://airvic.turkai.com), AIRVIC will support researchers worldwide in detecting and classifying viral infections. AIRVIC is designed to support the development of additional algorithms, with the understanding that the virus database will expand through future studies, thereby increasing its utility in viral diagnostics and research. This research contributes to the field of CPE detection, demonstrating how AI can streamline diagnostic processes, reduce labor, and enhance viral studies.
Finally, this study demonstrates AI’s potential to streamline laboratory work by enabling rapid viral detection and strain differentiation. The use of AI tools like AIRVIC in detecting viral infections marks a critical step in improving both human and animal health, contributing to the One Health perspective by offering faster and more effective responses to global health threats.
Materials and methods
Cell lines and viruses
SARS-COV-2-Ank1 local isolate with the Genbank accession no: MT478018 were obtained from the cell culture collection of the Department of Virology, Ankara University Faculty of Veterinary Medicine and were propagated in culture flasks containing African green monkey kidney Vero E6 (ATCC CRL-1586) cell lines in the certified high containment animal facility (Animal Biosafety Level 3 plus - ABSL3+) of the department under appropriate conditions. The local Turkish BPIV-3c isolate recorded under Genbank accession no: MH357343 were obtained from Department of Virology, Faculty of Veterinary Medicine, Ondokuz Mayis University, Samsun49 and propagated in culture flasks containing Madin Darby Bovine Kidney (MDBK) (90050801; European Collection of Authenticated Cell Cultures, Salisbury, UK) cells. The BAdV type 1 (strain 11/66) that was previously obtained from Vienna Veterinary Institute isolate was used from the collection of our department50. BoAHV-1 Cooper with accession no: JX898220, BoGHV-4 MOVAR 33/63 European strain with accession no: AB035516 and American prototype strain DN-599 strain were obtained from the cell culture collection of the Department of Virology, Ankara University Faculty of Veterinary Medicine51,52 and were propagated in culture flasks containing MDBK cells.
Virus isolation and measuring viral titer
The stock viruses of all bovine viruses and SARS-COV-2 was produced in MDBK and Vero cells respectively; maintained with Dulbecco’s Modified Eagle Medium (DMEM) (Gibco™, ThermoFisher Scientific) including 4.5 g/L D-Glucose, L-glutamine and pyruvate, supplemented with 10% heat-inactivated fetal calf serum (FBS) and 100 IU/mL Penicillin 100U/mL penicillin and 100ug/mL streptomycin (Gibco™, ThermoFisher Scientific) under 5% CO2 at 37 °C. Daily observations of the cells were carried out to observe the appearance of CPEs. To retain the viruses, infected cell cultures were frozen at -80 °C once the CPE reached a substantial 80% coverage of the flask. The replication of viruses was assessed using the Tissue Culture Infectious Dose 50% (TCID₅₀) assay, which determines the quantity of virus required to infect 50% of cultured cells. In this procedure, cells seeded in 96-well plates were exposed to 100 µL of tenfold serial dilutions of each virus. The median infectious dose was then calculated using the Reed–Muench method53.
Polymerase chain reaction (PCR) for BoAHV-1, BoGHV-4 and BAdV-1
The DNA of BoAHV-1, BoGHV-4, and BAdV-1 viruses was extracted from cell culture supernatants using the guanidine isothiocyanate method, as described by Russel and Sambrook54. All PCR reactions were performed using DreamTaq™ DNA polymerase (Thermo Fisher Scientific, Waltham, MA). BoAHV-1 DNA amplification was carried out using the gC-specific forward primer 5′-CGGCCACGACGCTGACGA-3′ and reverse primer 5′-CGCCGCCGAGTACTACCC-3′, as reported by Esteves et al.55. The reaction conditions included an initial denaturation at 95°C for 10 minutes, followed by 30 cycles of denaturation at 95°C for 30 seconds, annealing at 56°C for 45 seconds, extension at 72°C for 1 minute, and a final extension at 72°C for 10 minutes. The expected product size was 575 bp. BoGHV-4 DNA amplification was conducted using primers 5’-CCCTTCTTTACCACCACCTACA-3’ and 5’-TGCCATAGCAGAGAAACAATGA-3′, which target the gB region of the virus56, following the protocol described by Bilge Dağalp et al.57. The expected product size was 615 bp. BAdV-1 DNA was amplified using a touchdown PCR protocol with primers MaAdF1 (CAGTGGTCHTACATGCACATC), MaAdR (GCATAAGACCCGTAGCAWGG), and MaAdF2 (CATGCACATCGCSGGNCAGGA), as developed by Sibley et al.58. PCR conditions adhered to the Sibley et al.58 protocol, with a total primer concentration of 1,600 nM. The expected product size ranged from 588 to 725 bp. PCR products for all three viruses were visualized under UV light following agarose gel electrophoresis and stained with Visafe Red gel stain (Vivantis, Malaysia).
Real-time reverse transcription-polymerase chain reaction (rRT-PCR) for SARS-COV-2 and reverse transcription-polymerase chain reaction (RT-PCR) BPIV3
Thermo Fisher Scientific’s TRIzol reagent was used to extract SARS-COV-2-Ank1 RNA from the cell line in accordance with the manufacturer’s instructions. Following purification, the RNA was used for real time rRT-PCR by dissolving it in 100 µL of sterile water. Using SF-1a 5’-GATCTCTGCTTTACTAATGTCTATGC-3’ and SR-1b 5’-CGCAGCCTGTAAAATCATCTG-3’ primers and SPrp FAM-TTGCCCTGGAGCATTTGTCTGA-BHQ1 probe partial S gene of the SARS-CoV-2-Ank1 was amplified according to Hanifehnezhad et al.59.
Following the methodology by Chomczynski and Sacchi60, RNA was extracted using the phenol: chloroform: isoamyl alcohol (25:24:1) method. Following purification, a one-step RT-qPCR reaction was carried out using the SCRIPT RT-qPCR ProbesMaster kit (Jena, Germany) to screen the partial region of the BPIV Fusion (F) protein. The forward primer 5’-CATTGAATTCATACTCAGCAC-3’ and the reverse primer 5’-AGATTGTCGCATTTAGCCTC-3’ were the specific primers employed in this reaction61. Reverse transcription was performed for 30 min at 50 °C, followed by denaturation at 95 °C for 15 min. Amplification involved 30 cycles of 95 °C for 45 s, 51 °C for 45 s, and 72 °C for 1 min, with a final extension at 72 °C for 10 min49. The PCR products were separated on a 1% agarose gel with Visafe Red (Vivantis, Malaysia) and visualized under UV light. A 400 bp fragment for the BPIV Fusion (F) protein was expected.
Infecting cells with different MOI
Multiplicity of Infection (MOI) refers to the number of infectious virus particles per cell in a culture. For SARS-CoV-2, Vero cells were exposed to MOIs ranging from 0.0001 to 0.5. BoGHV-4 (MOVAR 33/63) was used to infect both MDBK and Vero cells with MOIs from 0.0001 to 1.0, while the BoGHV-4 (DN-599) strain was applied to the same cell lines. BoAHV-1, BPIV3, and BAdV-1 infections were also performed on MDBK and Vero cells with similar MOI ranges. Each MOI of each virus was inoculated into a separate T25 flask, and throughout the imaging period, they were maintained alongside control cells in a separate T25 flask, prepared on the same day.
Imaging and data collection
After cells were infected with varying MOIs for various viruses, images were collected for both infected and control cells. Each image was classified based on virus type, cell line, MOI, magnification level, and post-infection time. Image collection occurred at various post-infection times to capture dynamic changes in cellular morphology as the infection progresses, and continued until complete infection of the cells was observed. As a result, the timing for post-infection imaging varied according to the virus and cell line involved. Images were acquired using a ZEISS Axio Observer 5 widefield microscope equipped with the Zeiss LD A-Plan 5×/0.15 Ph1, Zeiss LD A-Plan 10×/0.25 Ph1, Zeiss LD A-Plan 20×/0.4 Ph2, and Zeiss LD A-Plan 40×/0.6 Ph2 objectives. All objectives were non-oil, non-water immersion lenses, and no correction collar was used. A ZEISS Axiocam 506 color camera (CMOS sensor) was used to capture images. A white LED integrated into the ZEISS Axio Observer 5 system served as the light source. Phase contrast imaging was employed to enhance morphological visualization, with phase contrast rings set according to manufacturer specifications (Ph1 for low magnifications and Ph2 for high magnifications). Images were captured using the ZEISS ZEN lite 2.3 software and saved in JPEG format at 2752 × 2208 pixels, 96 dpi resolution and 24-bit color depth. Different scales of magnification setting including 5X, 10X, 20X and 40X were used to effectively capture the morphological changes caused by viral infections.
Deep learning architectures
Compared architectures
The models used in this study need to be able to learn efficiently from heterogeneous data, including various magnification levels, natural imbalances between time points, and uneven distributions. For this reason, the deep learning architectures ResNet1862, ResNet5062, MobilenetV263 and YGNet64—a lightweight, specialized architecture—were chosen for their ability to adapt to the dataset’s diverse complexities and characteristics.
ResNet18 and ResNet50 are members of the Residual Network (ResNet) family, which is recognized for its deep architectures that allow for training over many layers without losing performance due to the vanishing gradient problem. ResNet models accomplish this by skipping some layers of the neural network and enabling gradients to pass directly through it. ResNet50 has 50 layers, increasing its complexity and allowing it to learn more detailed information. The greater complexity of ResNet50 necessitates more computer resources and a larger dataset for optimal training, and it may be more prone to overfitting than the simpler ResNet18 model, which has only 18 layers.
MobilenetV2 architecture is specifically intended for mobile and resource-constrained devices. This method employs deeply separable convolutions to reduce the number of parameters and computational overhead, resulting in greater efficiency for real-time applications and devices with limited processing capacity. MobilenetV2 is smaller and faster than ResNet models, however it may be unable to capture complex patterns due to its lower complexity.
YGNet, which is based on the All-CNN architecture65, extends this framework by introducing maximum pooling layers, straying from the original architecture’s only convolutional nature. These YGNet architectural improvements are meant to shed light on the purpose and significance of different layers inside a convolutional neural network, especially when dealing with complex and diverse datasets.
Addressing dataset imbalance
The dataset was unbalanced due to a higher number of virus-infected cell images compared to control cell images. To avoid this, data augmentation strategies recommended by Shorten and Khoshgoftaar66 were employed, including specific techniques like rotation and scaling (Supplementary Note A). Importantly, these techniques were applied exclusively to the training dataset, ensuring that the validation and test datasets remained untouched. By generating additional control cell images, the proportion of virus-infected to control samples was stabilized, achieving a more uniform distribution. This approach enhanced the model’s learning efficiency.
Architecture and hyperparameter selection
The selection of architecture and hyperparameters was a deliberate approach targeted to prevent overfitting, essential for managing the dataset’s complexity. Consequently, models and their hyperparameters were selected to enhance generalization abilities, preventing overfitting to particular dataset characteristics. This aligned with the ideas outlined by Bergstra and Bengio67, who emphasized the significance of hyperparameter selection for improving model performance and relieving issues such as dataset imbalance and overfitting. A systematic method to hyperparameter tuning was employed to enhance the predicted accuracy and efficiency of the models, with each model carefully fine-tuned using hyperparameters appropriate to its architecture and dataset features (Supplementary Note B).
Training and evaluation
CPE detection tasks
Targeted CPE Detection detects virus-induced CPE in a known cell line. This method allows the model to learn specific patterns of virus’s effects on cells for accurate identification. Each virus-cell combination had its own model. Broad-Spectrum CPE Detection recognizes CPE across a wide range of viruses and cell lines. Training the model to generalize over diverse features makes it versatile in detecting CPE, regardless of the specific virus or cell line. Virus-Specific CPE Detection identifies the specific virus causing CPE. Distinguishing the virus responsible for CPE is a complex task, requiring a deep understanding of each virus’s effects on cells. For this task, models were trained both with and without cell line dependency.
Dataset partitioning
The dataset was divided into training (70%), validation (20%), and testing (10%) subsets to assess model performance and ensure generalizability. Data augmentation was performed only on the training set after this partitioning to prevent data leakage. The model was trained on the training set, with hyperparameters adjusted using the validation set to prevent overfitting. The test set provided an unbiased evaluation of the model’s effectiveness.
Training
The training process began with the selection of architecture. For this, Targeted CPE Detection models were evaluated using specific datasets, such as BoAHV-1 in MDBK cell lines and SARS-CoV-2 in Vero cell lines. In this phase, the optimal hyperparameters for CPE detection were determined. Following this, Targeted CPE Detection models for other viruses and cell lines were trained using the preferred architecture. Data filtering was applied to each virus-cell line dataset, excluding samples without significant viral activity in the early post-infection phase or with insufficient MOI to generate CPE. This ensured that the models were trained on data that most accurately reflected viral impact. Subsequently, the Broad-Spectrum CPE Detection model, designed to detect CPE regardless of virus type, was trained using the entire dataset. It was trained in two configurations: independent of and dependent on cell lines. Finally, Virus-Specific CPE Detection models were trained. As these models needed to precisely distinguish the virus causing CPE in each cell line, this training phase was the most complex, involving virus classification.
Model validation
Data augmentation and hyperparameter optimization were applied to prevent overfitting and improve generalizability. Data augmentation expanded training scenarios (Supplementary Note A). Hyperparameter tuning controlled model complexity, further improving performance (Supplementary Note B). Real-time monitoring with TensorBoard enabled quick identification and correction of overfitting or underfitting.
Performance evaluation
Key metrics assessed model accuracy and efficiency. Cross-Entropy Loss measured prediction error, while accuracy and multi-class accuracy evaluated overall and class-specific performance. Precision and recall highlighted the model’s ability to identify true CPE instances, while the F1 score provided a balanced view of both metrics (Supplementary Note C).
AIRVIC web application
The models developed in the study were made available through the AIRVIC web application (accessible at https://airvic.turkai.com), designed to provide ease of use and accessibility for researchers and laboratory staff. AIRVIC allows users to upload microscopy images for automated analysis and provides detailed results on CPE detection and potential virus identification through a user-friendly interface.
The interface includes an option for users to specify known details about the cell line and virus, increasing the accuracy of the model by adapting the analysis to the relevant parameters. Users can choose cell lines (e.g. Vero or MDBK) and viruses from a predefined list or leave the fields as “Unknown” if no information is available. This optional step helps to optimize detection sensitivity.
Uploaded images pass through the AIRVIC Workflow (Fig. 3) which provides results that include probabilities for detected CPE and potential virus matches. For accurate virus identification, results include likelihood scores and recommendations for interpretation. Users can access the Analysis History section to review results from previously analyzed images and explore the Example Images section to test the system with predefined samples.
Experimental setup and model development environment
Image acquisition
Images were acquired using the ZEISS Axio Observer 5 microscope and saved with ZEISS ZEN lite software (v 2.3).
Model development
The experiments were conducted using Python 3.8 and PyTorch machine learning framework. The server setup included an Intel Core i9-10920X CPU @ 3.50 GHz (12 cores and 24 threads), 128 GB DDR4 memory, and dual Nvidia A6000 graphics card (48GB each, Ampere architecture) with CUDA 11.3 drivers. Several Python packages were utilized for model training and validation, included PyTorch (v 1.11.0) for deep learning, torchvision (v 0.12.0) for computer vision tasks, pytorch-lightning (v 1.2.5) for training simplification, imageio (v 2.9.0) for image data handling, pandas (v 1.3.0) for data analysis, OpenCV (v 4.7.0.72) for image pre-processing and TensorBoard (v 2.16.2) for the training log monitoring.
Data availability
The datasets generated and analysed during the current study are not publicly available due to ongoing research, potential commercial applications by our partners, and a pending patent application, but are available from the corresponding author on reasonable request.
Code availability
The code is available under restricted access on Zenodo https://doi.org/10.5281/zenodo.14036204 under the CC BY-NC-ND license. Access will be provided from the corresponding author upon reasonable request.
References
Flint, J., Racaniello, V. R., Rall, G. F., Hatziioannou, T. & Skalka, A. M. Principles of Virology, Molecular Biology, Vol. 1 (Wiley, 2020).
Howley, P. M., Knipe, D. M. & Enquist, L. W. Fields Virology: Fundamentals (Lippincott Williams & Wilkins, 2023).
Vijayakrishnan, S., Jiu, Y. & Harris, J. R. Virus Infected Cells (Springer, 2023).
Reynolds, K. A. Integrated cell culture/PCR for detection of enteric viruses in environmental samples. In Public. Health Microbiology: Methods Protocols 69–78 (2004).
Vlimant, J. R. & Yin, J. Artificial Intelligence for High Energy Physics 243–264 (World Scientific, 2022).
Liu, W. et al. A survey of deep neural network architectures and their applications. Neurocomputing 234, 11–26 (2017).
Chen, J. J. et al. Detection of cytopathic effects induced by influenza, parainfluenza, and enterovirus using deep convolution neural network. Biomedicines 10, 70 (2021).
Dodkins, R. et al. A rapid, high-throughput, viral infectivity assay using automated brightfield microscopy with machine learning. SLAS Technol. 28, 324–333 (2023).
Petkidis, A., Andriasyan, V., Murer, L., Volle, R. & Greber, U. F. A versatile automated pipeline for quantifying virus infectivity by label-free light microscopy and artificial intelligence. Nat. Commun. 15, 5112 (2024).
Wang, T. E. et al. Differentiation of cytopathic effects (CPE) induced by influenza virus infection using deep convolutional neural networks (CNN). PLoS Comput. Biol. 16, e1007883 (2020).
Wu, S. C. Progress and concept for COVID-19 vaccine development. Biotechnol. J. 15, 1 (2020).
Liu, H. et al. The basis of a more contagious 501Y. V1 variant of SARS-CoV-2. Cell Res. 31, 720–722 (2021).
De Pires, G. A. et al. Choosing a cellular model to study SARS-CoV-2. Front. Cell. Infect. Microbiol. 12, 1003608 (2022).
14 Saccon, E. et al. Tropism of SARS-CoV-2 in commonly used laboratory cell lines and their proteomic landscape during infection. Biorxiv 1, 271684 (2020).
Zhou, Y. et al. Pathogenic infection characteristics and risk factors for bovine respiratory disease complex based on the detection of lung pathogens in dead cattle in Northeast China. J. Dairy Sci. 106, 589–606 (2023).
Sáfár, J. et al. The impact of environmental factors on bovine respiratory disease complex in dairy calves-a review. Acta Vet. Brno. 92, 213–231 (2023).
Gatherer, D. et al. ICTV virus taxonomy profile: Herpesviridae 2021. J. Gen. Virol. 102, 001673 (2021).
Gomez, D. E. & Weese, J. S. Viral enteritis in calves. Can. Veterinary J. 58, 1267 (2017).
Saxegaard, F. & Bratberg, B. Isolation of bovine adenovirus type 1 from a calf with pneumo-enteritis. Acta Vet. Scand. 12, 464–466 (1971).
Kumagai, A. et al. Phylogenetic and antigenic analysis of bovine parainfluenza virus type 3 isolated in Japan between 2002 and 2019. Vet. Microbiol. 247, 108774 (2020).
Lin, T. M. et al. Susceptibility of endothelial cells to bovine herpesvirus type 4 (BHV-4). J. Virol. Methods. 63, 219–225 (1997).
McLean, A. M. & Doane, F. W. The morphogenesis and cytopathology of bovine parainfluenza type 3 virus. J. Gen. Virol. 12, 271–279 (1971).
Nandi, S., Kumar, M., Manohar, M. & Chauhan, R. Bovine herpes virus infections in cattle. Anim. Health Res. Reviews. 10, 85–98 (2009).
Rosales, J. J., Verna, A., Marin, M. & Pérez, S. Bovine alphaherpesvirus type 5 replicates more efficiently than bovine alphaherpesvirus type 1 in undifferentiated human neural cells. Virus Res. 286, 198037 (2020).
Zhu, L., Yu, Y., Jiang, X., Yuan, W. & Zhu, G. First report of bovine herpesvirus 1 isolation from bull semen samples in China. Acta Virol. 61, 483–486 (2017).
Petkidis, A., Andriasyan, V. & Greber, U. F. Machine learning for cross-scale microscopy of viruses. Cell. Rep. Methods. 1, 1 (2023).
Floresta, G., Zagni, C., Gentile, D., Patamia, V. & Rescifina, A. Artificial intelligence technologies for COVID-19 de novo drug design. Int. J. Mol. Sci. 23, 3261 (2022).
Hogarty, D. T., Mackey, D. A. & Hewitt, A. W. Current state and future prospects of artificial intelligence in ophthalmology: a review. Clin. Exp. Ophthalmol. 47, 128–139 (2019).
Huang, S., Yang, J., Fong, S. & Zhao, Q. Artificial intelligence in the diagnosis of COVID-19: challenges and perspectives. Int. J. Biol. Sci. 17, 1581 (2021).
Johnson, K. W. et al. Artificial intelligence in cardiology. J. Am. Coll. Cardiol. 71, 2668–2679 (2018).
Lopes, B. et al. The use of Artificial Intelligence in predicting respiratory syncytial virus-inhibiting flavonoids. Brazilian J. Biology. 83, e270776 (2023).
Patel, S., Wang, J. V., Motaparthi, K. & Lee, J. B. Artificial intelligence in dermatology for the clinician. Clin. Dermatol. 39, 667–672 (2021).
Hegde, P. R. & Shenoy, M. M. Artificial intelligence in medicine and health sciences. Archives Med. Health Sci. 9, 145–150 (2021).
Young, A. T., Xiong, M., Pfau, J., Keiser, M. J. & Wei, M. L. Artificial intelligence in dermatology: a primer. J. Invest. Dermatology. 140, 1504–1512 (2020).
Ramachandran, A., Lumetta, S. S., Chen, D. & PandoGen Generating complete instances of future SARS-CoV-2 sequences using deep learning. PLoS Comput. Biol. 20, e1011790 (2024).
Rancati, S. et al. Forecasting dominance of SARS-CoV-2 lineages by anomaly detection using deep AutoEncoders. Brief. Bioinform. 25, 535 (2024).
Werner, J. et al. Deep transfer learning approach for automatic recognition of drug toxicity and inhibition of SARS-CoV-2. Viruses 13, 610 (2021).
Qazi, Z., Shiao, W. & Papalexakis, E. E. Companion Proceedings of the ACM on Web Conference 2024 842–846.
Sattarzadeh, S., Sudhakar, M. & Plataniotis, K. N. Proceedings of the IEEE/CVF International Conference on Computer Vision 4158–4167.
Welsh, R. M., Misas, E., Forsberg, K. & Lyman, M. Chow, N. A. Candida Auris whole-genome sequence benchmark dataset for phylogenomic pipelines. J. Fungi. 7, 214 (2021).
Wang, J. & Lee, S. Data augmentation methods applying grayscale images for convolutional neural networks in machine vision. Appl. Sci. 11, 6721 (2021).
Mikołajczyk, A. & Grochowski, M. International Interdisciplinary PhD Workshop (IIPhDW) 117–122 (IEEE, 2018).
Rusche, D. et al. Unraveling a histopathological needle-in-haystack problem: exploring the challenges of detecting tumor budding in colorectal carcinoma histology. Appl. Sci. 14, 949 (2024).
Młodzianowski, P. Conference on Multimedia, Interaction, Design and Innovation 3–11 (Springer).
Padmaja, D. L., Nikhil, B. & Akshaya, B. S. & Surya Deepak, G. International Conference on Data Science, Machine Learning and Applications 22–30 (Springer).
Yuan, H., Cheng, J., Wu, Y. & Zeng, Z. Low-res MobileNet: an efficient lightweight network for low-resolution image classification in resource-constrained scenarios. Multimedia Tools Appl. 81, 38513–38530 (2022).
Albanese, A., Nardello, M. & Brunelli, D. Low-power deep learning edge computing platform for resource constrained lightweight compact UAVs. Sustainable Computing: Inf. Syst. 34, 100725 (2022).
Sang, X. et al. A real-time and high-performance MobileNet accelerator based on adaptive dataflow scheduling for image classification. J. Real-Time Image Proc. 21, 4 (2024).
Albayrak, H. et al. Characterisation of the first bovine parainfluenza virus 3 isolate detected in cattle in Turkey. Veterinary Sci. 6, 56 (2019).
Akca, Y., Burgu, I., Gur, S. & Bilge Dagalp A study on investigation of occurrence of some virus infection in buffaloes in Turkey. Revue De Médecine Vétérinaire. 155, 268–271 (2004).
Bilge-Dagalp, S. et al. Development of a BoHV-4 viral vector expressing tgD of BoHV-1 and evaluation of its immunogenicity in mouse model. Brazilian J. Microbiol. 52, 1119–1133 (2021).
Dağalp, S. B., Dogan, F., Babaoglu, A. R., Farzani, T. A. & Alkan, F. Genetic variability of bovine herpesvirus type 4 (BoHV-4) field strains from Turkish cattle herds. Vet. Ital. 57, 49–59 (2021).
Reed, L. J. & Muench, H. A Simple Method of Estimating Fifty Per Cent Endpoints (1938).
Russel, D. & Sambrook, J. Molecular cloning: a laboratory manual. New (2001).
Esteves, P. et al. Phylogenetic comparison of the carboxy-terminal region of glycoprotein C (gC) of bovine herpesviruses (BoHV) 1.1, 1.2 and 5 from South America (SA). Virus Res. 131, 16–22 (2008).
Goltz, M. et al. Glycoprotein B of bovine herpesvirus type 4: its phylogenetic relationship to gB equivalents of the herpesviruses. Virus Genes. 9, 53–59 (1994).
Bilge Dağalp, S. et al. The Investigation of the Presence of Bovine Herpesvirus Type 4 (BoHV-4) in Cows with Metritis in a Dairy Herd (2010).
Sibley, S. D., Goldberg, T. L. & Pedersen, J. A. Detection of known and novel adenoviruses in cattle wastes via broad-spectrum primers. Appl. Environ. Microbiol. 77, 5001–5008 (2011).
Hanifehnezhad, A. et al. Characterization of local SARS-CoV-2 isolates and pathogenicity in IFNAR-/-mice. Heliyon 6, 1 (2020).
Chomczynski, P. & Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 (1987).
Lyon, M. et al. Presence of a unique parainfluenza virus 3 strain identified by RT-PCR in visna-maedi virus infected sheep. Vet. Microbiol. 57, 95–104 (1997).
He, K., Zhang, X., Ren, S. & Sun, J. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition 770–778.
Sandler, M., Howard, A., Zhu, M., Zhmoginov, A. & Chen, L. C. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition 4510–4520.
Gajjar, Y. CNN-CIFAR-10 Image Classification. https://github.com/yogeshgajjar/CNN-CIFAR-10-image-classification (2021).
Springenberg, J. T., Dosovitskiy, A., Brox, T. & Riedmiller, M. Striving for simplicity: The all convolutional net. Preprint at http://arXiv.org/1412.6806 (2014).
Shorten, C. & Khoshgoftaar, T. M. A survey on image data augmentation for deep learning. J. Big Data. 6, 1–48 (2019).
Bergstra, J. & Bengio, Y. Random search for hyper-parameter optimization. J. Mach. Learn. Res. 13, 1 (2012).
Acknowledgements
The authors gratefully acknowledge Prof. Dr. Seval Bilge Dağalp for providing the BoAHV-1, BoGHV-4, and BAdV-1 isolates, as well as Prof. Dr. Harun Albayrak and Assoc. Prof. Dr. Emre Özan for sharing BPIV3. We also thank Assistant Prof. Dr. Sıdıka Öztop for technical support with preliminary trials involving BRSV and BVDV, which were excluded from the study. All figures included in this manuscript were originally created by the author specifically for this publication. This study was conducted as part of the Technology Development Zone project (code STB/79708) between May 16, 2022, and November 30, 2023, and was approved by the Republic of Türkiye Ministry of Industry and Technology.
Author information
Authors and Affiliations
Contributions
Z.A.-Y., M.T.Y., and B.Ü. conceptualized and designed the experiments. Z.A.-Y., S.Ş., and F.G. created the dataset, while Z.A.-Y., Y.B.A., S.Ş., and F.G. conducted the experiments. Z.A.-Y., Y.B.A., and O.T. curated the data. Z.A.-Y., M.T.Y., Y.B.A., and O.T. analyzed, validated, and interpreted the results. Z.A.-Y. and M.T.Y. wrote the original manuscript, and Z.A.-Y., M.T.Y., A.Ö., and B.Ü. revised the paper. M.T.Y., O.C.Y. and B.E. developed and designed the interface. All contributors provided feedback on the text and discussed the findings.
Corresponding author
Ethics declarations
Competing interests
Z.A.-Y., M.T.Y., and B.Ü. have filed a patent application (TR/2025/000078). The remaining authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Akkutay-Yoldar, Z., Yoldar, M.T., Akkaş, Y.B. et al. A web-based artificial intelligence system for label-free virus classification and detection of cytopathic effects. Sci Rep 15, 5904 (2025). https://doi.org/10.1038/s41598-025-89639-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-89639-0
