
Abstract
Programmed cell death (PCD) plays a critical role in cancer biology, influencing tumor progression and treatment response. This study aims to investigate the role of PCD-related genes in hepatocellular carcinoma (HCC), identifying potential prognostic biomarkers and therapeutic targets to enhance patient outcomes. Data from the GEO, TCGA, and ICGC databases were analyzed to identify differentially expressed genes associated with PCD in HCC. A cell death signature (CDS) model was constructed based on seven key PCD genes using machine learning techniques, including Random Survival Forest and Cox regression models. The model was validated across multiple cohorts to evaluate its predictive accuracy for clinical outcomes, immune infiltration, and therapeutic response, and further validation of the relationship between NNMT overexpression and clinical prognosis using tumor tissue microarray data, and in vitro experiments to confirm the impact of NNMT overexpression on cell proliferation and apoptosis. A total of 183 differentially expressed genes were identified, leading to the construction of a CDS model that incorporates seven key PCD-related genes (PRGs). The CDS showed significant associations with overall survival, immune cell infiltration, and therapeutic response in HCC patients. High CDS scores were linked to poorer prognosis, increased tumor immune exclusion, and decreased efficacy of immunotherapy and conventional treatments. The model demonstrated strong predictive performance across independent validation cohorts, underscoring its potential as a valuable prognostic tool. Additionally, NNMT overexpression promotes HepG2 proliferation, inhibits apoptosis, and correlates with poor prognosis in HCC patients. This study established a prognostic model for HCC based on PCD, and the CDS holds promise as a powerful tool for personalized risk assessment and treatment planning in HCC. Moreover, the model gene NNMT may serve as a potential therapeutic target for HCC.
Similar content being viewed by others


Introduction
Liver cancer remains one of the most formidable challenges in oncology, with HCC being the most prevalent type1. Despite advances in medical research and clinical interventions, the prognosis for HCC patients remains dismal due to late-stage diagnosis, high recurrence rates, and resistance to conventional therapies2. This underscores the urgent need for innovative diagnostic markers and therapeutic targets to enhance early detection and improve treatment outcomes.
PCD plays a pivotal role in maintaining cellular homeostasis and eliminating damaged cells3. Dysregulation of PCD pathways is a hallmark of cancer, contributing to tumor initiation, progression, and resistance to therapy4. In recent years, the exploration of PRGs has gained traction as a promising avenue for understanding cancer biology and developing novel interventions5,6,7. PRGs encompass a diverse array of cellular demise mechanisms, such as apoptosis, autophagy, necroptosis, cuproptosis, pyroptosis, and necroptosis, each fulfilling unique yet interrelated functions in the dynamics of cancer8.
Utilizing bioinformatics methods to develop potential biomarkers for cancer has been proven feasible9,10. Our study aims to bridge the gap between molecular insights and clinical applications by identifying key PRGs in HCC and evaluating their potential as prognostic biomarkers and therapeutic targets. Utilizing comprehensive datasets from the gene expression omnibus (GEO), The cancer genome atlas (TCGA), and the international cancer genome consortium (ICGC), we performed an integrative analysis to pinpoint PRGs that are differentially expressed in HCC. We further assessed the predictive value of these PRGs using advanced machine learning models, aiming to develop a robust prognostic signature for HCC.
This study’s significance lies in its potential to enhance the diagnostic and therapeutic landscape of HCC. By leveraging cutting-edge bioinformatics and machine learning techniques, we aim to provide a deeper understanding of PCD mechanisms in HCC and identify actionable targets for intervention. Ultimately, based on clinical samples and in vitro experiments, we discovered that the expression of the PCD - related gene NNMT is closely linked to cancer cell proliferation and the prognosis of HCC patients, which has not been reported in previous studies. Our research aims to make contributions to the advancement of precision oncology strategies, thereby enhancing the survival rates and quality of life of HCC patients.
Results
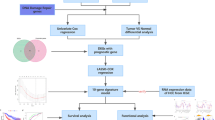
The workflow of this study
As illustrated in Fig. 1, we utilized advanced machine learning techniques to create an innovative seven-gene-based CDS for liver hepatocellular carcinoma (LIHC) data sourced from the TCGA cohort. To ensure the robustness of our model, we assessed its predictive capabilities across two independent testing cohorts: GSE14520 and ICGC. Our thorough analysis demonstrated a noteworthy association between the CDS and multiple clinical outcomes in LIHC, encompassing prognosis, immune cell infiltration, responses to immunotherapy, and essential cancer hallmarks, all of which were elucidated through the TCGA datasets.

The workflow of this study.
Insights into programmed cell death genes in HCC patients
In the present investigation, we performed an extensive examination of PCD genes in patients diagnosed with LIHC. Utilizing datasets from the TCGA-LIHC, GSE14520, and GSE25097 cohorts, we successfully identified 183 differentially expressed genes (DEGs) that exhibited statistical significance (adjusted p < 0.05 and log2FC > 1) (Fig. 2a–c and Supplementary Table 3). Among these DEGs, we pinpointed 31 genes that are characteristic of PCD, which are derived from a range of cell death pathways: specifically, 1 gene associated with parthanatos, 9 genes linked to necrosis, 2 genes related to necrosis-like morphology, 9 genes involved in necroptosis, 1 gene pertaining to lysosome-dependent cell death, 8 genes associated with intrinsic apoptosis, 12 genes related to immunogenic cell death, 6 genes involved in ferroptosis, 11 genes linked to extrinsic apoptosis, 1 gene associated with autophagy, and 2 genes related to apoptosis-like morphology. Additionally, gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses indicated that these DEGs participate in multiple pathways associated with carcinogenesis, including cell killing signaling and the Rap1 signaling pathway.

(a–c) Volcano plots showing differentially expressed genes in three datasets: (a) TCGA-LIHC, with 1,069 downregulated and 519 upregulated genes; (b) GSE14520, with 491 downregulated and 402 upregulated genes; and (c) GSE25097, with 751 downregulated and 869 upregulated genes. Blue and red dots represent significantly downregulated and upregulated genes, respectively, while gray dots represent genes with no significant changes. (d) Venn diagram depicting the overlap of differentially expressed genes among the TCGA-LIHC, GSE14520, GSE25097 datasets, and PCD-related genes. Bubble diagram of GO functional enrichment analysis (e) and KEGG pathway enrichment analysis37,38,39 (f) of intersect genes.
Construction of a CDS via machine learning for HCC patients
In this study, we employed a machine learning-based integrative procedure to develop a prognostic signature for LIHC patients using the expression profiles of 31 PCD signature genes. Utilizing the TCGA-LIHC dataset, we evaluated 101 predictive models through a leave-one-out cross-validation (LOOCV) framework and calculated the concordance index (C-index) for each model (Fig. 3a). The model combination with the highest C-index was found to be stepwise (backward) Cox selection combined with RSF. These two algorithms were applied for gene selection and model construction, identifying the top 10 genes with the highest variable importance by stepwise (backward) Cox (Fig. 3b). RSF analysis ranked the genes based on their importance, and we selected the top 7 genes (Fig. 3c). Subsequently, we constructed the prognostic signature model using the RSF method and calculated the prognostic cell death index (PCDI) for each patient. Patients from the TCGA-LIHC, GSE14520, and ICGC cohorts were stratified into high-risk and low-risk subgroups based on their median PCDI. Our findings indicated a significant association between a high PCDI and shorter overall survival (OS) (Fig. 3d–f). The model’s accuracy in predicting patient survival was evaluated using the ROC curve, yielding values of 0.88 and 0.92 for 3- and 5-year survival, respectively, in the TCGA cohort (Fig. 3h–j). For the GSE14520 cohort, the ROC values for 3- and 5-year survival were 0.8 and 0.72, respectively. Similarly, the ICGC cohort yielded ROC values of 0.77, 0.71, and 0.78 for 1-, 3-, and 5-year survival, respectively. These results collectively demonstrate the strong performance of our prognostic model across different cohorts. The visualized risk scores and corresponding groupings are displayed in (Fig. 4a–c).

A consensus PCDI was developed and validated via the machine learning-based integrative procedure. (a) A total of 10 kinds of prediction models via a ten-fold cross-validation framework further calculated the C index of each model. (b) The variable importance of the hub genes through stepcox COX regression. (c) The error rate of the RSF result and the variable relative importance of screened genes based on RSF. (d–f) Overall survival in the low- and high-PCDI group patients in the TCGA-LIHC, GSE14520, and ICGC cohorts. (h–j) the time ROC curve in the TCGA, GSE14520, and ICGC cohorts.

Risk factor association map for the TCGA-LIHC, GSE14520, and the ICGC cohort. Risk score, survival status, and the expression of the seven PCD-related genes are shown, respectively.
Single-cell RNA sequencing (scRNA-seq) reveals PCD-associated immune landscape in HCC
In this study, we conducted an in-depth analysis of 10 HCC samples to elucidate the immune landscape of LIHC. Using the t-SNE method, we classified all cells into 15 distinct clusters based on the expression levels of canonical marker genes. Further classification with SingleR identified these clusters as seven major cell types (Fig. 5a, b). These cell types included T cells, NK cells, B cells, monocytes, hepatocytes, tissue stem cells, and endothelial cells (ECs), identified by well-recognized gene markers. In addition, the specific number of each cell type was presented in (Fig. 5c). Moreover, the proportion of cell type was also investigated in each HCC sample (Fig. 5d). The violin plot revealed the most representative marker gene in each cell type (Fig. 5e). Using the AUCell algorithm, we calculated the active scores for each cell in the scRNA-seq data based on the expression of seven PCD model genes (Fig. 6a, b). Differential gene enrichment analysis was conducted between the active group and non-active group, revealing significant differences in gene expression profiles (Fig. 6d). Similarly, our results demonstrated that ECs exhibited markedly higher active scores compared to other subpopulations (Fig. 6c), suggesting that these cells play a critical role in the progression of HCC through mechanisms related to PCD. To further investigate this finding, we reclassified the ECs into distinct subtypes and conducted a detailed analysis of the evolutionary trajectories within the EC subpopulation to understand their differentiation processes (Fig. 7a, d). Our evolutionary trajectory analysis of ECs offers valuable insights into the dynamic processes and pathways involved in cancer development, emphasizing the significant influence of PCD pathways on EC function and tumor progression.

Prognostic analysis using Cox proportional hazards model and nomogram for survival prediction. (a) Forest plot of the Cox proportional hazards model showing the hazard ratios and 95% confidence intervals for various prognostic factors, including risk group, AFP levels, age, gender, microvascular invasion (MVI), Child-Pugh score, and tumor stage. (b) Nomogram for predicting 1-year, 3-year, and 5-year overall survival probabilities in patients. (c) Calibration curves for the nomogram-predicted survival probabilities at 1-year (yellow), 3-year (blue), and 5-year (red) intervals.

Analysis of somatic mutation landscape and TMB. (a) A waterfall plot illustrating the single nucleotide variant (SNV) frequencies of the top 20 genes among patients categorized into high- and low-risk groups. (b) KM survival curves comparing patients within high- and low-TMB groups, where yellow denotes the low-TMB cohort and blue signifies the high-TMB cohort. (c) A scatter plot depicting the relationship between RS and TMB, with red indicating the low-risk group and green representing the high-risk group.

Immune infiltration and immune-related gene expression in high and low-risk groups. (a–c) Violin plots illustrate the variations in exclusion scores and MSI scores between high-risk and low-risk groups across the TCGA-LIHC, GSE14520, and ICGC cohorts. (d) Heat map illustrates the abundance of immune cells within both high-risk and low-risk cohorts. (e) box plot that delineates the variations in the proportions of 22 infiltrating immune cell types between patients categorized into high-risk and low-risk groups. (f) The correlation heat map illustrates the expression levels of seven model genes alongside immune checkpoints, and (g) the expression of immune checkpoints in relation to high-risk and low-risk groups within the TCGA dataset.
Development and evaluation of the nomogram survival model
To assess the independent prognostic significance of PCDI in LIHC patients, we conducted multivariate Cox regression analyses using the TCGA-LIHC datasets. In the multivariate analysis, PCDI retained its independent prognostic value in LIHC patients even after adjusting for other confounding factors (HR 0.036, 95% CI 0.014–0.09, P < 0.001, Fig. 8a). Based on the results from the multivariable Cox and stepwise regression analyses, we constructed a prognostic nomogram model in the TCGA cohort to predict the 1-, 3-, and 5-year OS of LIHC patients (Fig. 8b). The nomogram demonstrated good calibration in predicting 1-, 3-, and 5-year OS, as shown by the calibration curves, where the predicted probabilities closely matched the observed probabilities (Fig. 8c).

Relationship between risk scores and response to immunotherapy, Sorafenib, and TACE. (a) Box plot comparing risk scores between responders (CR/PR) and non-responders (SD/PD) and (b) KM survival curve comparing overall survival between low-risk and high-risk groups in the IMvigor210 cohort. (c) Box plot comparing risk scores between responders (CR/PR) and non-responders (SD/PD) to Sorafenib. (d) Stacked bar chart showing the distribution of responders and non-responders to Sorafenib in low-risk and high-risk groups. (c) Box plot comparing risk scores between responders and non-responders to TACE. (d) Stacked bar chart showing the distribution of responders and non-responders to TACE in low-risk and high-risk groups.
Differences in genetic mutations
Genetic mutations are critical drivers in the occurrence and progression of cancer. To deepen our understanding of genetic mutations in HCC, we conducted an analysis of the top 20 genes with the highest mutation frequency in patients from both high- and low-risk groups. This analysis was visualized using a waterfall diagram, which highlighted distinct differences in gene mutation frequencies between the high- and low-risk groups (Fig. 9a). Additionally, we calculated the tumor mutation burden (TMB) and stratified patients into high-TMB and low-TMB groups based on the median TMB value. Kaplan-Meier (KM) survival curves were generated for these groups, revealing that patients in the high-TMB group had a better prognosis compared to those in the low-TMB group (Fig. 9c). A scatter plot further demonstrated a significant positive correlation between RS and TMB (Fig. 9b, R = 0.17, p = 0.0015).

Analysis of drug sensitivity. (a) The variation in the distribution of IC50 values for ten chemotherapeutic agents between the high-risk and low-risk cohorts is depicted. The high-risk group is indicated in red, while the low-risk group is represented in blue. (b) Scatter plots with correlation coefficients (R) and p-values showing the relationships between CD274 (PD-L1) expression and key genes: CDKN3, FOS, and NNMT. (c) Heatmap showing correlations between various drugs and key genes, including CDKN3, DNASE1L3, ESR1, FOS, NNMT, CXCL2, and LCN2.
Analysis of immune cell infiltration characteristics of the CDS
Our study utilized the TIDE algorithm to assess tumor immune exclusion and microsatellite instability (MSI) scores in HCC samples. Consistently, across both the training and validation sets, patients in the high-risk group exhibited significantly higher tumor immune exclusion scores and lower MSI scores compared to those in the low-risk group (Fig. 10a–c). These findings suggest that high-risk tumors are more proficient at evading immune surveillance, which may contribute to their aggressive behavior and poorer prognosis. Additionally, the lower MSI scores indicate that these tumors are less likely to be immunogenic, potentially reducing their responsiveness to certain immunotherapies. Immune cell infiltration in the tumor microenvironment (TME) plays a crucial role in the initiation and progression of cancers. To investigate the differences in the TME between high-risk and low-risk groups, we utilized the cell-type identification by estimating relative subsets of RNA transcript (CIBERSORT) algorithm. This algorithm estimated the proportion of 22 immune cell types infiltrating HCC samples (Supplementary Table 4). Our analysis revealed significant differences in the proportions of immune cells between high- and low-risk groups. Notably, substantial differences were observed in the proportions of M1 macrophages and monocytes (Fig. 10e). The distribution of the 22 immune cell types in high- and low-risk groups is illustrated in (Fig. 10d). Immune checkpoints are molecules expressed by immune cells that regulate the level of immune activation and play a significant role in the development of autoimmune diseases. Consequently, we analyzed checkpoint genes and assessed their correlation with the expression of the seven model genes (Fig. 10f). The results indicated that these seven model genes had a strong positive correlation with immune checkpoints. Additionally, 28 immune checkpoints exhibited significant differences between the high-risk and low-risk groups (Fig. 10g).

Single-Cell RNA sequencing analysis of HCC. (a) t-SNE plot showing the clustering of single cells from the dataset into 15 distinct clusters (0–14) based on their gene expression profiles. (b) t-SNE plot with cell types annotated, including T cells, B cells, NK cells, monocytes, hepatocytes, endothelial cells, and tissue stem cells. (c) Bar plot displaying the frequency of identified cell types in the dataset. (d) Stacked bar plot showing the proportion of different cell types across multiple samples. (e) Violin plots illustrate the distribution of marker genes specific to each distinct cell types.
Predictive effects of CDS in various therapies
To assess the prognostic utility of the CDS in predicting therapeutic responses, we evaluated its performance across multiple treatment cohorts. In the IMvigor210 immunotherapy cohort, we examined the ability of CDS to predict patient response to anti-PD-L1 treatment. Our analysis revealed that non-responders exhibited higher PCDI levels compared to responders (Fig. 11a). Moreover, patients with high PCDI displayed worse survival outcomes compared to those with low PCDI (Fig. 11b), underscoring the predictive value of CDS in immunotherapy contexts. In the GSE109211 sorafenib treatment cohort, we observed similar trends. Non-responders to sorafenib exhibited higher PCDI levels than responders (Fig. 11c). The percentage of patients responding to sorafenib in the high PCDI group was significantly lower than that in the low PCDI group (Fig. 11d), indicating that higher PCDI is associated with poorer response to sorafenib treatment. In the GSE104580 transarterial chemoembolization (TACE) treatment cohort, our findings were consistent. Non-responders to TACE also exhibited higher PCDI levels compared to responders (Fig. 11e). The percentage of patients responding to TACE in the high PCDI group was markedly lower than in the low PCDI group (Fig. 11f), further demonstrating the utility of CDS as a predictive marker for therapy response.

Analysis of PCD Signature in single-cell RNA sequencing data. (a) AUC histogram plot showing the distribution of area under the curve (AUC) scores for the PCD signature across the dataset. The vertical line marks the threshold for significant AUC values (AUC > 0.15). (b) t-SNE plot illustrating the expression levels of the PCD signature across different cell types in the dataset. Cells are colored based on their AUC scores, with higher scores indicating stronger expression of the PCD signature. (c) Violin plots showing the distribution of PCD scores across various cell types. (d) Bubble plot showing the KEGG pathway enrichment analysis37,38,39 of genes associated with the PCD signature. (e) GSEA plot depicting the enrichment of various hallmark pathways related to the PCD signature.
CDS correlates with the therapeutic response to medication in HCC patients
To further understand the therapeutic implications of our CDS, we investigated the correlation between gene expression profiles and drug sensitivity in HCC. Utilizing the expression data from TCGA-LIHC samples, we predicted the half-maximal inhibitory concentration (IC50) values for 138 drugs from the GDSC database (Supplementary Table 5). A box plot was generated to illustrate the 10 drugs with the most significant differences in IC50 values between high-risk and low-risk populations. The distribution of IC50 values for these ten chemotherapy drugs is shown in (Fig. 12a). Similarly, given the importance of CD274 as a target for immunotherapy, our analysis indicated that higher expression levels of FOS and NNMT are often associated with elevated CD274 expression in HCC, whereas lower expression levels of CDKN3 are also linked to increased CD274 expression (Fig. 12b). We further calculated the correlation between the expression of the model genes and the IC50 values of the chemotherapeutic drugs. The results demonstrated a strong correlation between model gene expression and the IC50 values of these drugs (Fig. 12c). These findings suggest that the CDC comprising the seven PRGs can be effectively used to guide therapy selection for HCC patients in the foreseeable future. The above data underscore the potential of CDS as a predictive marker for drug sensitivity, providing valuable insights for personalized treatment strategies. By leveraging these predictive capabilities, clinicians can optimize therapeutic approaches, ultimately improving outcomes for HCC patients.

Analysis of endothelial cell subtypes and pseudotime trajectory. (a) t-SNE plot visualizing the distribution of different endothelial cell subtypes, including arterial endothelial (blue), capillary endothelial (red), vascular endothelial (green), and sinusoidal endothelial (purple). (b) Pseudotime trajectory analysis showing the differentiation trajectory of endothelial cells. (c) Clustered pseudotime trajectory with cells colored by subtype. (d) Dendrogram plot showing the hierarchical structure of the endothelial cell subtypes based on pseudotime trajectory analysis.
NNMT overexpression promotes HepG2 proliferation, inhibits apoptosis, and correlates with poor prognosis in HCC patients
We compared the expression levels of NNMT protein between tumor tissues and adjacent non-tumor tissues in HCC patients using immunofluorescence (MIF). Immunohistochemical staining was performed on tissue microarrays from 29 HCC patient tumor samples, revealing that NNMT expression was significantly lower in tumor tissues compared to adjacent non-tumor tissues (Fig. 13a). Further analysis of the relationship between NNMT expression and clinical characteristics of the patients showed that high NNMT expression in cancer tissues was associated with PVTT, recurrence, and mortality in HCC patients (Fig. 13b–d). These findings suggest that NNMT is a gene associated with poor prognosis in HCC patients. Clinical evidence indicates that NNMT may play a potential role in the progression of HCC. To explore this further, the CCK − 8 assay was employed to measure cell viability, and PI single - staining flow cytometry was utilized to analyze the cell cycle, while Annexin V - APC/PI double - staining flow cytometry was applied to determine cell apoptosis. The results of the CCK − 8 assay and PI single - staining flow cytometry demonstrated that overexpression of NNMT significantly promoted the proliferation of HepG2 cells and reduced cell apoptosis (Fig. 13e–h).

NNMT expression in relation to clinical outcomes in patients. (a) Immunofluorescence staining of NNMT expression in tumor (T) and adjacent non-tumor (AT). Scale bars represent 50 μm. (b) Violin plot showing NNMT expression in HCC tissues with or without macrovascular invasion. (c) NNMT expression in HCC tissues with or without relapse. (d) NNMT expression in relation to OS. (e) mRNA expression of NNMT in HepG2 cells transfected with control (NC) or NNMT overexpression (OE-NNMT) plasmid. (f) Cell viability measured by CCK-8 assay at 24, 48, and 72 h. (g) Flow cytometry analysis of cytotoxicity in OE-NNMT and NC HepG2 cells. h: Flow cytometry analysis of cell cycle distribution in OE-NNMT and NC HepG2 cells.
Discussion
In light of our findings, it is imperative to consider the broader implications of the seven-gene CDS developed for HCC. The ability of this gene signature to correlate with survival outcomes and immune cell infiltration highlights its potential utility not only as a prognostic tool but also as a guide for personalized therapeutic strategies. Previous studies have similarly established prognostic gene signatures for HCC, emphasizing various biological aspects such as epithelial-mesenchymal transition and metabolic pathways, which play crucial roles in tumor progression and therapy resistance11,12,13.
The unique mutation profiles identified in high- and low-risk patients highlight the pivotal role of TMB as a prognostic indicator across various cancer types, including HCC. Recent literature affirms the idea that elevated TMB correlates with unfavorable outcomes, bolstering the results of our analysis. For example, a study by Liu et al.14 emphasized that mutations in LRP1B, a commonly altered gene in HCC, were linked to increased TMB and diminished survival rates, suggesting that TMB could function as a significant biomarker for risk stratification in HCC patients. Immunotherapy is currently one of the important treatment modalities for unresectable HCC. In different risk groups, we also found differences in immune escape and immune infiltration characteristics. The immune escape score in the high risk group was higher than that in the low risk group, and the proportion of M1 macrophages was significantly lower than that in the low risk group. All these findings suggest that the CDS can better guide whether liver cancer patients can benefit from immunotherapy. Moreover, the validation of CDS as a prognostic indicator for treatment response in HCC patients marks an important advancement in personalized medicine. The correlation between persistently high levels of the PCDI and diminished responses to sorafenib, TACE, and anti-PD-1 treatment emphasizes the potential role of CDS in guiding therapeutic decision making. This is especially relevant considering the heterogeneity of HCC and the varying efficacy of treatment modalities among different patient populations. This is in line with the existing literature that emphasizes the significance of molecular profiling in predicting treatment outcomes15.
In this study, based on the CDS, we revealed the immune landscape related to PCD in HCC and found that the PCD score of ECs was significantly higher than that of other cells. On the one hand, in the tumor microenvironment, ECs can regulate the recruitment and activation of immune cells16,17,18. ECs can express adhesion molecules and chemokines to attract immune cells such as T cells and natural killer cells to the tumor site. These immune cells can recognize and kill tumor cells, promoting tumor cell death. On the other hand, ECs may also be exploited by tumor cells to evade immune surveillance and killing. For example, the up-regulation of PD - L1 on the surface of ECs can bind to PD-1 on the surface of immune cells, inhibiting the activity of immune cells and enabling tumor cells to evade immune attack, thus reducing tumor cell death19,20,21. The complex relationship between them still needs further exploration.
Our study found that there is a strong correlation between the upregulation of NNMT and the prognosis of liver cancer as well as the development of cancer emboli. In vitro experiments also confirmed that high expression of NNMT promotes cell proliferation. NNMT has been associated with metabolic processes that affect tumor growth and metastasis. For example, research has demonstrated that NNMT plays a role in the methylation of nicotinamide, which subsequently influences cellular signaling pathways linked to cancer progression22. Previous studies23 have indicated that, compared to adjacent non-tumor tissues, the mRNA levels of NNMT are significantly reduced in HCC, and the expression of NNMT in tumors is significantly correlated with tumor staging. Patient stratification based on tumor NNMT mRNA levels reveals that individuals with higher NNMT mRNA levels tend to have shorter OS times (P = 0.053) and significantly reduced disease-free survival (DFS) durations (P = 0.016). Our research further corroborates this perspective at the protein level. The downregulation of NNMT in tumor tissues may signify a shift in metabolic requirements during tumorigenesis, resulting in a more aggressive phenotype24. This metabolic reprogramming could potentially provide insights into the tumor microenvironment and its interaction with systemic metabolism. Additionally, identifying NNMT as a biomarker could aid in the development of targeted therapies aimed at modulating its activity, thereby potentially enhancing treatment outcomes for HCC patients. Future investigations should concentrate on validating these results across larger populations and examining the therapeutic implications of targeting NNMT within the context of HCC. This could pave the way for more tailored treatment strategies, ultimately improving patient outcomes in this challenging cancer type.
Despite these promising findings, our study has several limitations. First, although our CDS was validated in multiple independent cohorts, all data were retrospective. Prospective validation in clinical trials is necessary to confirm the utility of CDS in real-world settings. Second, while we identified significant correlations between CDS and various clinical outcomes, the underlying biological mechanisms driving these associations remain unclear. Future studies should focus on elucidating the pathways linking CDS to immune evasion and therapeutic resistance. Additionally, our study primarily focused on a single cancer type, HCC. It would be beneficial to explore the applicability of this CDS across other cancer types to determine its broader relevance. Finally, the study’s reliance on publicly available datasets may introduce biases related to sample selection and data quality, which could impact the generalizability of our findings.
In summary, this research offers persuasive proof of the prognostic and therapeutic significance of a seven-gene CDS in HCC. By incorporating this model into clinical practices, we could markedly enhance the accuracy of patient stratification and treatment strategies, ultimately improving patient outcomes. Subsequent studies should concentrate on further validating this CDS in prospective clinical trials and investigating its potential to direct therapeutic interventions in practical clinical environments.
Methods
Data collection and processing
We enhanced the methodology for consolidating PRGs derived from 15 distinct cell death modalities, employing various resources including GSEA gene sets, KEGG pathways, relevant review articles25, and manual selection. This comprehensive approach resulted in a meticulously curated compilation of 2173 PRGs, as outlined in (Supplementary Table 1). The expression matrix and associated metadata for the single-cell RNA-sequencing dataset GSE235057 were acquired from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). To pinpoint significant PRGs, we also collected bulk RNA-seq data pertaining to HCC and normal liver specimens from the TCGA database (https://portal.gdc.cancer.gov/repository), as well as datasets GSE14520 and GSE25097. For the evaluation of the predictive capacity of the PRGs, we sourced data from the TCGA database (343 cases), GEO datasets GSE14520 (221 cases), and the ICGC database (dcc.icgc.org, release 28, 231 cases). Furthermore, datasets IMvigor210 (348 cases), GSE109211 (140 cases), and an additional instance of GSE104580 (148 cases) were utilized to assess the efficacy of PRGs in forecasting therapeutic outcomes. The data processing steps included: the elimination of samples lacking clinical follow-up information, the exclusion of samples with unspecified or non-positive survival durations, absent survival status, or survival units not based on days. Additionally, probe IDs were converted to gene symbols, probes mapping to multiple genes were discarded, median expression values for multiple gene symbols were calculated, and FPKM values were transformed using Log2 (FPKM + 1). Patient survival and mutation data were obtained and adjusted based on the findings of Liu et al.26.
Development and evaluation of a prognostic PRGs
The “limma” package27 identified DEGs in the LIHC, GSE14520, and GSE25097 datasets, using |Log fold change| ≥ 1.0 and P < 0.01 cutoffs. The “VennDiagram” package was used to identify PRGs by intersecting the DEGs from the HCC datasets with known PCD genes. Identified PCD biomarkers underwent an integrative machine learning analysis using the “Mime1” R package28, incorporating: Random survival forest (RSF), Elastic network (Enet), Lasso, Ridge, Stepwise Cox, CoxBoost, Partial least squares regression for Cox (plsRcox), Supervised principal components (SuperPC), Generalized boosted regression modeling (GBM), Survival support vector machine (survival-SVM), The procedure for generating significant signatures included: Applying 10 algorithm combinations to the prognostic signature, fitting prediction models based on LOOCV framework using the TCGA dataset. Evaluating models in the GSE14520 and ICGC cohorts. Calculating Harrell’s concordance index (C-index) for each model across the TCGA, GEO, and ICGC datasets. The model with the highest average C-index was deemed optimal. Using the median as the cutoff value, LIHC cases were divided into CDS high score and low score groups. A predictive nomogram was developed using the“nomogramEx”R package, and forest plots were constructed using the “ggforest” R package based on risk scores (RS) and clinical features.
Immune infiltration and gene mutation analyses
To investigate the correlation between CDS and immune infiltration, the TIDE algorithm was used to evaluate tumor immune exclusion and MSI scores of HCC samples29. Visualization was done using violin plots and boxplots with the“ggpubr”package. Relationships between 79 immune checkpoint genes, sourced from literature30, and model genes were examined. The CIBERSORT and the“ESTIMATE”algorithm, using the “IBOR” R package31, calculated proportions of infiltrating immune cells in the TCGA-LIHC datasets. The“maftools”R package generated waterfall plots showing gene mutation distributions in LIHC patients, with patients classified into high- and low-risk groups based on mutation frequency differences. CNV data from TCGA were visualized using the oncoplot function in the“maftools”R package, integrating clinical data with model groups. TMB was calculated for each sample to examine relationships between CDS and TMB, and survival differences among high-TMB and low-TMB groups were predicted using Kaplan-Meier survival curves and log-rank tests.
Single cell RNA-seq analysis
For the analysis of single-cell RNA sequencing data, we employed the Seurat R package (version 5.0)32, which facilitated various processes including the construction of objects, data normalization, dimensionality reduction, clustering, and the identification of marker genes. The CreateSeuratObject function was utilized to generate Seurat objects, ensuring a minimum inclusion of 5 cells and 200 features. We selected the top 2000 hypervariable genes for principal component analysis (PCA). The ElbowPlot function was instrumental in determining the first 15 significant principal components. To mitigate batch effects, we implemented the “harmony” R package33 for batch correction. Tumor cell subsets were delineated using the FindClusters algorithm at a resolution parameter set to 0.3, while t-distributed stochastic neighbor embedding (tSNE) was employed for the visualization of cell populations. The clustering outcomes were annotated through the “SingleR” package34. For low-dimensional data visualization, we utilized the DimPlot function. Additionally, cell proportion plots and violin plots were generated using the ggplot2 package. The “AUCell” R package was utilized to compute activity scores, and the “Monocle2” R package was employed to investigate the evolutionary trajectories of cells exhibiting high activity.
Enrichment analysis
GSEA was executed utilizing the “ClusterProfiler”35 and “hallmark” pathway gene set R packages to pinpoint genes that are enriched within active cellular subgroups, with a significance threshold set at p-values less than 0.01. Additionally, enrichment analyses for GO and KEGG pathways were performed.
Evaluation of therapeutic benefit
RS derived from the RSF analysis were used to rank patients in both the training and validation sets. The RS formula, based on the importance scores of the model genes identified from the RSF analysis in the training set, uses a weighted sum of the gene expression levels, where the weights are the importance scores. The RS can be calculated using the following formula:
The distribution of RS and the corresponding survival curves were evaluated to assess the prognostic value of the model. Immunotherapy effectiveness was evaluated using the IMvigor210 datasets, sorafenib efficacy using the GSE109211 datasets, and TACE efficacy using the GSE104580 datasets.
Drug sensitivity
Utilizing the “pRRophetic” R package36, we estimated the IC50 values for 138 pharmacological agents sourced from the genomics of drug sensitivity in cancer (GDSC) database. To assess the relationship between IC50 values and copy number alterations, we employed Spearman’s rank correlation analysis. Furthermore, a box plot was generated to illustrate the distribution of IC50 values across high-risk and low-risk cohorts, thereby examining the association between our predictive model and various chemotherapeutic agents.
MIF detection of CDS gene expression in tissue microarrays
In order to facilitate a standardized and concurrent evaluation of protein expression across various tissue specimens, we developed tissue microarray slides intended for subsequent multiplex MIF analysis. These tissue microarrays were constructed from 29 HCC tumor samples alongside adjacent non-tumor tissues, sourced from patients who underwent surgical resection at the First Affiliated Hospital of Fujian Medical University during the period from January 2014 to December 2019. All participating patients were treatment-naive, having received no prior chemotherapy or targeted therapies, and pertinent clinical data were collected. The study protocol received approval from the Ethics Committee (MTCA, ECFAH of FMU [2015]084 − 2). It is important to note that two tumor regions within the microarrays experienced tissue loss during the processing phase. The MIF methodology encompassed several steps, including deparaffinization, antigen retrieval, blocking of endogenous peroxidase activity, sequential incubation with primary antibodies followed by HRP-conjugated secondary antibodies, TSA amplification, nuclear staining utilizing DAPI, autofluorescence quenching (G1211, Servicebio, Wuhan, China), and the final image acquisition conducted using fluorescence microscopy (NIKON ECLIPSE C1, Nikon, Japan). The specifics of the primary antibodies utilized are provided in (Supplementary Table 2).
Experimental assays for NNMT overexpression in HepG2 cells
HepG2 cells were cultured in DMEM(PM150210, Procell, USA, China) supplemented with 10% fetal bovine serum at 37 °C in an atmosphere of 5% CO2. Once thawed, the cells were transferred into culture flasks. When the cell confluence reached 80–90%, the cells were passaged. The coding sequence of NNMT was synthesized and cloned into the pcDNA3.1 plasmid. Subsequently, the recombinant plasmid was transfected into HepG2 cells using Lipofectamine 3000 (L3000015, Invitrogen, USA). After 48 h, quantitative polymerase chain reaction (qPCR) was carried out to verify the overexpression of NNMT. For the CCK − 8 assay, the cells were seeded in 96 - well plates. The CCK − 8 reagent was added, and the absorbance at 450 nm was measured at 24, 48, and 72 h. Apoptosis was detected by staining the cells with Annexin V - allophycocyanin (APC)/propidium iodide (PI) (A6030M, UE, China) followed by flow cytometry analysis. Meanwhile, cell cycle analysis was performed by staining the cells with PI (GK3601-50T, GenView, China) and then analyzing them using flow cytometry. The experimental groups consisted of the wild-type group and the NNMT overexpression group.
Statistical analyses
The Wilcoxon rank-sum test and the Kruskal-Wallis test were employed to conduct comparisons among groups. The levels of significance were denoted as follows: NS (p > 0.05), * (p ≤ 0.05), ** (p ≤ 0.01), *** (p ≤ 0.001), and **** (p ≤ 0.0001).
Data availability
The datasets analyzed for this study can be found in the TCGA database (http://www.cancer.gov/tcga), the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo), the International Cancer Genome Consortium (https://dcc.icgc.org/releases/current/Projects) and IMvro210 (http://research-pub.Gene.com/imvigor210corebiologies).
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 74, 229–263. https://doi.org/10.3322/caac.21834 (2024).
Ren, Z. et al. Nanoparticle conjugation of ginsenoside Rg3 inhibits hepatocellular carcinoma development and metastasis. Small (Weinheim Der Bergstrasse Germany). 16, e1905233. https://doi.org/10.1002/smll.201905233 (2020).
Newton, K., Strasser, A., Kayagaki, N. & Dixit, V. M. Cell death. Cell 187, 235–256. https://doi.org/10.1016/j.cell.2023.11.044 (2024).
Bedoui, S., Herold, M. J. & Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 21, 678–695. https://doi.org/10.1038/s41580-020-0270-8 (2020).
Zhang, B. et al. Exploration of pyroptosis-associated prognostic gene signature and LncRNA regulatory network in ovarian cancer. Comput. Biol. Med. 164 https://doi.org/10.1016/j.compbiomed.2023.107343 (2023).
Deng, M. et al. The pyroptosis-related gene signature predicts prognosis and indicates immune activity in hepatocellular carcinoma. Mol. Med. (Cambridge Mass) 28, 16. https://doi.org/10.1186/s10020-022-00445-0 (2022).
Wang, S. et al. Machine learning reveals diverse cell death patterns in lung adenocarcinoma prognosis and therapy. NPJ Precis. Oncol. 8 https://doi.org/10.1038/s41698-024-00538-5 (2024).
Wu, X., Cao, J., Wan, X. & Du, S. Programmed cell death in hepatocellular carcinoma: mechanisms and therapeutic prospects. Cell. Death Discov. 10 https://doi.org/10.1038/s41420-024-02116-x (2024).
Ye, Y. et al. Identification of a cancer associated fibroblasts-related index to predict prognosis and immune landscape in ovarian cancer. Sci. Rep. 13, 21565. https://doi.org/10.1038/s41598-023-48653-w (2023).
Bian, G. et al. Identification and validation of a cancer-testis antigen-related signature to predict the prognosis in stomach adenocarcinoma. J. Cancer 15, 3596–3611. https://doi.org/10.7150/jca.91842 (2024).
Xiao, S. et al. Identification of a novel epithelial-to-mesenchymal-related gene signature in predicting survival of patients with hepatocellular carcinoma. Comb. Chem. High Throughput Screen. 25, 1254–1270. https://doi.org/10.2174/1386207324666210303093629 (2022).
Hu, W. et al. A panel of E2F target gene signature predicting the prognosis of hepatocellular carcinoma. Front. Genet. 13, 879299. https://doi.org/10.3389/fgene.2022.879299 (2022).
Tang, Y. et al. Identification of hypoxia-related prognostic signature and competing endogenous RNA regulatory axes in hepatocellular carcinoma. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms232113590 (2022).
Liu, L., Wu, L., Shan, D. & Han, B. Characterization and clinical relevance of PDGFRA pathway copy number variation gains across human cancers. Mol. Genet. Genom. MGG 297, 561–571. https://doi.org/10.1007/s00438-022-01860-y (2022).
Shao, J., Ma, J., Zhang, Q., Li, W. & Wang, C. Predicting gene mutation status via artificial intelligence technologies based on multimodal integration (MMI) to advance precision oncology. Sem. Cancer Biol. 91, 1–15. https://doi.org/10.1016/j.semcancer.2023.02.006 (2023).
Kim, S. W., Kim, C. W., Moon, Y. A. & Kim, H. S. Reprogramming of tumor-associated macrophages by metabolites generated from tumor microenvironment. Anim. Cells Syst. 28, 123–136. https://doi.org/10.1080/19768354.2024.2336249 (2024).
Geyer, M., Gaul, L. M., Corbo, S. L. D. A., Queiroz, K. & V. & The tumor stroma influences immune cell distribution and recruitment in a PDAC-on-a-chip model. Front. Immunol. 14, 1155085. https://doi.org/10.3389/fimmu.2023.1155085 (2023).
Zhu, Y. et al. Endothelial cell Notch signaling programs cancer-associated fibroblasts to promote tumor immune evasion. Res. Square https://doi.org/10.21203/rs.3.rs-4538031/v1 (2024).
Taguchi, K. et al. Tumor endothelial cell-mediated antigen-specific t-cell suppression via the PD-1/PD-L1 pathway. Mol. Cancer Res. MCR 18, 1427–1440. https://doi.org/10.1158/1541-7786.Mcr-19-0897 (2020).
Luo, J. et al. Vascular immune evasion of mesenchymal glioblastoma is mediated by interaction and regulation of VE-cadherin on PD-L1. Cancers 15 https://doi.org/10.3390/cancers15174257 (2023).
Palicelli, A. et al. What do we have to know about PD-L1 expression in prostate cancer? A systematic literature review. Part 3: PD-L1, intracellular signaling pathways and tumor microenvironment. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms222212330 (2021).
Wang, Z. et al. NNMT enriches for AQP5(+) cancer stem cells to drive malignant progression in early gastric cardia adenocarcinoma. Gut 73, 63–77. https://doi.org/10.1136/gutjnl-2022-328408 (2023).
Couto, J. P. et al. Nicotinamide N-methyltransferase sustains a core epigenetic program that promotes metastatic colonization in breast cancer. EMBO J. 42, e112559. https://doi.org/10.15252/embj.2022112559 (2023).
Eckert, M. A. et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728. https://doi.org/10.1038/s41586-019-1173-8 (2019).
Zhou, Y. et al. Implications of different cell death patterns for prognosis and immunity in lung adenocarcinoma. NPJ Precis. Oncol. 7, 121. https://doi.org/10.1038/s41698-023-00456-y (2023).
Liu, J. et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416e411. https://doi.org/10.1016/j.cell.2018.02.052 (2018).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 https://doi.org/10.1093/nar/gkv007 (2015).
Liu, H. et al. A flexible machine-learning framework to construct and visualize models for clinical characteristics prediction and feature selection. Comput. Struct. Biotechnol. J. 23, 2798–2810. https://doi.org/10.1016/j.csbj.2024.06.035 (2024).
Fu, J. et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 12 https://doi.org/10.1186/s13073-020-0721-z (2020).
Hu, F. F., Liu, C. J., Liu, L. L., Zhang, Q. & Guo, A. Y. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief. Bioinform. 22 https://doi.org/10.1093/bib/bbaa176 (2021).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587e3529. https://doi.org/10.1016/j.cell.2021.04.048 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods 16, 1289–1296. https://doi.org/10.1038/s41592-019-0619-0 (2019).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172. https://doi.org/10.1038/s41590-018-0276-y (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics: J. Integr. Biol. 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PloS One 9, e107468. https://doi.org/10.1371/journal.pone.0107468 (2014).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. Public. Protein Soc. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–d592. https://doi.org/10.1093/nar/gkac963 (2023).
Acknowledgements
We thank Dr. Jianming Zeng (University of Macau) and all the members of his bioinformatics team, Biotrainee, for generously sharing their experience and codes.
Funding
This study was funded by the Fujian Province Double High Project.
Author information
Authors and Affiliations
Contributions
Z.W.C. and Y.P.C. contributed to the study concept and design. F.Q.W. wrote the first draft of the manuscript. P.S.H., B.Z., R.G., Y.X. and Z.W.C. supervised and oversaw the study. P.S.H. contributed to the statistical analysis. Y.P.C. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, F., Huang, P., Zhang, B. et al. Machine learning analysis identified NNMT as a potential therapeutic target for hepatocellular carcinoma based on PCD-related genes. Sci Rep 15, 7494 (2025). https://doi.org/10.1038/s41598-025-91625-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91625-5
