
Abstract
Breast cancer (BC) is the second leading cause of cancer-related death in females, followed by lung cancer. Disadvantages exist in conventional diagnostic techniques of BC, such as radiation risk. The present study integrated bioinformatics analysis with machine learning to elucidate potential key candidate genes associated with the tumorigenesis of BC. Eleven datasets were downloaded from the Gene Expression Omnibus (GEO) database and were consolidated into two independent cohorts (training cohort and validation cohort) after batch-effect removal. We employed “limma” package to screen differentially expressed genes (DEGs) between BC and adjacent normal breast samples. Subsequently, the most reliable diagnostic indicators were identified utilizing LASSO-Logistic regression, SVM-RFE and multivariate stepwise Logistic regression analysis. Logistic model and nomogram were created based on these hub genes and applied in external validation cohort to verify the robustness of the model. As a result, a total of six hub genes connected with BC pathogenesis were identified, including CD300LG, IGSF10, FAM83D, MAMDC2, COMP and SEMA3G. Then, a diagnostic model of BC on the basis of these genes was established. ROC analysis of the diagnostic model illustrated that AUC of the training cohort was 0.978 (0.962, 0.995). In the validation cohort, AUC of training set and validation set were 0.936 (0.910, 0.961) and 0.921 (0.870, 0.972), respectively. This indicated that the model was reliable in separating BC patients from healthy individuals. The model may assist in early diagnosis of BC with implications for improving the prognosis of BC patients.
Similar content being viewed by others

Introduction
Breast cancer (BC) is the second leading cause of cancer-related death in females, followed by lung cancer1. In 2020, it has been estimated that there were 2.26 million new cases of BC in females globally, accounting for approximately 11.7% of all new diagnosed cancer cases2, and 680,000 deaths, accounting for 6.9% of all cancer-related deaths in females2. Most cases are diagnosed at advanced stage due to the insidious early symptoms of BC, leading to missing the best opportunity for treatment. Consequently, 5-year survival rate of advanced BC is relatively low. Therefore, it is essential to identify effective biomarkers that aid in early diagnosis and developing novel treatment for improving BC prognosis3.
It has been claimed that genetic mutations leading to changes in DNA sequence, activation of oncogenes and inactivation of tumor suppressor genes are the main causes of tumorigenesis4,5. BC is a complex heterogeneous disease with primary risk factors including age, sex, high-fat diet, alcohol intake and obesity6. In addition, its occurrence is also associated with two types of genetic mutations: (1) germline mutations (a combination of egg and sperm cell)7,8; and (2) somatic mutations, which occurs due to gene repair misalignment of DNA recombination9. The occurrence of BC can be described as a multi-step process in which each step is thought to be linked to one or more different mutations in the major regulatory genes.
Early diagnosis of cancer can improve the treatment effectiveness. To date, clinical detection of BC patients is usually carried out by imaging and histopathology10. Imaging diagnosis of BC incorporates Mammography, Ultrasound, Magnetic resonance imaging (MRI), Computed tomography (CT) and Positron emission tomography (PET). Of these, Mammography, CT and PET exist the risk of radiation10. Moreover, pathological diagnosis of BC is not applicable for rapid diagnosis because of insufficient pathologists, difficulties in sample labeling and heavy workload of manual diagnosis11. Increasing high-throughput data facilitate to identify tumor biomarkers that contribute to accurately reflecting the oncogenesis and progression of BC, which is of great significance for clinical monitoring of tumors12. Hence, it may be a better option for early detection of BC via utilizing collaborative analysis of multiple biomarkers at the molecular level.
Rapid development of bioinformatics promotes to exploit plenty of BC related potential diagnostic biomarkers at molecular level13. Jia et al. and Liu et al. constructed BC diagnostic models based on deep learning and bioinformatics analysis, in an attempt to find biomarkers for clinical diagnosis of BC, with AUC values of 0.99 and 0.93, respectively, but the robustness of these findings has yet to be validated in independent external cohorts14,15. Li et al. established a prediction model for BC based on protein–protein interaction (PPI) network and support vector machines (SVM)16. Although the prediction accuracy of the SVM model reached 95.394%, the results also have yet to be verified in independent external cohorts or experiments16. Moreover, Chen et al. identified hub genes related to the occurrence of BC based on PPI network, but defects exist in this research17: firstly, the research only analyzed the independent diagnostic performance of genes for BC instead of jointly constructing a classifier; secondly, the research was limited to only one dataset with insufficient sample size; thirdly, how robust these hub genes in BC diagnosis are open to evaluate.
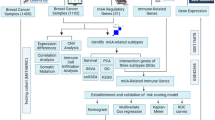
In the current study, we sought to explore novel genes connected with the pathogenesis of BC. We downloaded gene expression profiles from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database and identified differentially expressed genes (DEGs) between BC tissue and normal breast tissue samples via bioinformatics analysis. Subsequently, functional enrichment analysis was performed to further scout the biological processes of the DEGs. Finally, the most robust hub genes were extracted using LASSO-Logistic regression, support vector machine-recursive feature elimination (SVM-RFE) and Logistic analysis, and Logistic model and Nomogram prediction model were established based on the hub genes to further understand the pathogenesis of BC. The workflow of this study is shown in Fig. 1.

Flowchart for identifying breast cancer (BC) candidate diagnostic biomarkers.
Discussion
BC is one of the most common malignancies in females1. Most patients are diagnosed at advanced stage, which poses a serious threat to women’s health. BC, similar to other cancers, occurs due to environmental and genetic factors8. Imaging techniques and pathological histology are the major diagnostic tools for BC10. However, due to various limitations, neither is suitable for rapid diagnosis. The European Group on Tumor Markers (EGTM) guidelines stated that the application of various biomarkers is considered as one of the substantial aspects of BC diagnosis and monitoring, contributing to a better understanding of the cellular and molecular pathways involved in the pathogenesis of BC18. Accordingly, it is urgent to explore specific and sensitive biomarkers for early BC diagnosis.
In the present study, a total of 154 DEGs were identified between BC and adjacent normal samples via a combination of integrated bioinformatics analysis and machine learning. Subsequently, six genes were selected as diagnostic biomarkers of BC by Lasso-Logistic regression, SVM-RFE and Logistic regression analysis: CD300LG, IGSF10, FAM83D, MAMDC2, COMP and SEMA3G. It is well known that the mutation status of BC-related genes is closely associated with the occurrence and progression of BC, especially mutations in BRCA1 and BRCA2. In this study, 6-gene biomarker set was screened based on gene expression profiles. Gene expression is not equivalent to gene mutation, and the difference is whether the gene structure is altered or not. Therefore, it’s different status of genes under investigating that leads to the lack of overlap between the 6-gene biomarker set obtained in this study and the BC-related genes. These six genes were discussed in detail as follows.
CD300LG (CD300 molecule like family member G), encoding proteins belonging to the family of CD300 membrane-bound molecules, possesses a wide variety of immune effects, including the ability to recognize and interact with extracellular lipids19,20. Umemoto et al. declared that CD300LG is selectively expressed in microvessels and has a specific expression pattern in tissue, which may play a crucial role in immune function21. Meanwhile, its expression is rapidly down-regulated by acute inflammation or tumor-related signals21. Bloomstein and colleagues claimed that CD300LG may come into play in the pathophysiology of cervical cancer metastasis and may serve as a potential biomarker for cervical cancer lymph node metastasis22. It has been showed that CD300LG expression is down-regulated in BC tissue compared with normal tissue, which is in agreement with our findings23,24,25. Besides, latest research implied that there is a link between polymorphism Arg82Cys of CD300LG and high-density lipoprotein cholesterol (HDLc) metabolism26.
IGSF10 (Immunoglobulin superfamily member 10), the genetic basis of delayed puberty and neuronal development27, is connected with differentiation and developmental processes28. Up to now, the biological role of IGSF10 in most cancers is poorly investigated, especially in the pathogenesis of BC. Chang et al. identified a potential association between the potential passenger gene IGSF10 and endometrial carcinoma via whole-exome sequencing29. Ling and colleagues reported that IGSF10 expression is down-regulated in lung cancer patients; meanwhile, it is confirmed that IGSF10 knockdown significantly promotes lung cancer cell proliferation, enhances the adhesion between cells and stroma, and activates the integrin-β1/FAK pathway30. Additionally, IGSF10 mutations may increase the risk of rectal and gastric cancer28. As a gene that is down-regulated in BC, IGSF10 is a novel potential biomarker for BC diagnosis, treatment and prognosis31,32, which is in accordance with our findings. IGSF10 is positively correlated with BC prognosis, and it is hypothesized that the underlying pathogenesis of BC is the activation of the integrin-β1/FAK pathway and the reduction of IGSF10 expression32.
FAM83D (Family with sequence similarity 83 member D) belongs to FAM83 family, which can regulate cell proliferation, growth, migration and epithelial–mesenchymal transition (EMT)33,34. It has been indicated that FAM83D is not an oncogenic factor, but may be an indirect indicator of tumors that is more dependent on cell division and mitosis35. Overexpression of FAM83D can not only promote cell proliferation, migration and invasion as well as EMT via down-regulating the expression of tumor suppressor gene FBXW7 protein36, but also involve in the activation of MAPK signaling, which promotes BC cell growth and drives epithelial cell transition37. Snijders and colleagues pointed out that in BC, alterations in FAM83 family genes are significantly correlated with TP53 mutations38. FAM83D involved in the development and progression of multifarious cancers is up-regulated in BC39, endometrial cancer40, colorectal cancer41 and lung cancer42, which can be regarded as a potential candidate gene for various cancers diagnosis and prognosis. According to recent researches, FAM83D expression is significantly up-regulated in primary BC tissue, which is associated with poor prognosis and distant metastasis in BC patients35,36,39,43.
Beckmann et al. identified the present of MAM domain in the functionally diverse receptors meprin, A5 protein and protein tyrosine phosphatase μ, and proposed that it may widely present in various adhesive proteins44. MAMDC2 (MAM domain containing 2), a member of the MAM family, is recognized as a kind of secreted protein. Although MAMDC2 is differentially expressed in several types of cancer, including oral squamous cell carcinoma45, gastric cancer46 and BC25, its exact molecular function has not been determined. In line with our findings, Lee et al. argued that MAMDC2 expression is down-regulated in BC tissue and has good diagnostic performance47. Moreover, MAMDC2 may be an extracellular regulator of cell proliferation, exerting its growth inhibition function through attenuating MAPK signaling pathway47.
COMP (Cartilage oligomeric matrix protein), a metabolic marker of cartilage, is an extracellular matrix protein that can modulate cell phenotype in the course of histogenesis and remodeling48. Intriguingly, COMP can be expressed by cancer cells and aggravate malignant diseases33,49. It has been already established that COMP expressing in cancer cells renders them resistant to apoptosis, enhances their invasive potential and warburg metabolic effect33,49,50. The mechanisms of COMP in BC progression are still under investigation. Papadakos et al. confirmed that COMP affects the migration, invasion and metabolism of BC cells as well as the abundance of cancer stem cells51. High expression level of COMP in BC makes the disease more aggressive52. The present study uncovered that COMP expression level was significantly elevated in BC tissue compared with adjacent normal tissue.
It has been proposed that SEMA3G (Semaphorin 3G) is a regulator of angiogenesis that conduces to the patterning of vascular networks53. Simultaneously, as a potential transcription gene associated with cancer susceptibility candidate 9 (CASC9), SEMA3G is involved in the pathogenesis of lung squamous cell carcinoma (LUSC)54. Ji et al. noted that SEMA3G is considered as a predictor of poor prognosis in testicular germ cell tumor patients55. Additionally, SEMA3G expression is also connected with the prognosis of glioblastoma or lung cancer patients56,57. It has been revealed that SEMA3G is a protective factor for lung cancer and glial tumor patients, and its high expression forebodes good clinical outcomes of patients57,58. However, it seems that SEMA3G exerted the opposite effect on the pathogenesis of BC in the current study. Since there are a few studies on the relationship between SEMA3G and BC, its exact mechanism of action needs to be further investigated.
Next, in the training cohort, we applied the six hub genes to construct a diagnostic nomogram for individual risk quantification. It was demonstrated that the AUC value of ROC analysis in the model was 0.978 (0.962, 0.995), indicating that the model possessed considerable risk prediction power. The calibration curve illustrated that the nomogram predicted probability was in agreement with the actual results observed in the training cohort. Meanwhile, the decision curve and clinical impact curve uncovered that the model had good clinical benefit. Besides, the predictive performance of the model was satisfactory in training and validation sets of validation cohort: the AUC values were greater than 0.900. In conclusion, these results suggested that the six-gene diagnostic model was connected with BC pathogenesis, and may carry good net benefit in clinical practice, which was a potential diagnostic biomarker for BC.
Our study also found that the DEGs related to the pathogenesis of BC were mainly enriched in cell cycle pathways. Of note, conferring to the GSEA analysis, we discovered that the E2F targets and G2M checkpoint were strongly enriched in BC. E2F targets and G2M checkpoint are involved in regulating the cell cycle, and play crucial roles in tumor progression and metastasis. The proteins encoded by E2F target genes are associated with DNA replication, cyclin and the E2F transcription factor59. Dysregulated expressions of E2F target genes are common in a variety of malignancies, including BC59. The transcription factor E2F family acts as an indispensable downstream effector in pathways that control cell cycle progression, G1/S conversion and DNA replication-related gene expression60. Components that regulate the E2F pathway are found in almost all human malignancies, and many of these components, including the E2F transcription factor themselves, serve as imperative parts in BC progression, metastasis and therapeutic response61. Masanori with colleagues reported that the E2F transcription factor is a marker of BC aggressiveness and can predict the response to neoadjuvant chemotherapy in ER-positive/HER2-negative patients59. In addition, the G2M checkpoint is regarded as biomarkers of cancer progression, which is strongly correlated with the E2F targets62. Both E2F targets and G2M checkpoint are associated with TP53 mutations62. These results showed that these DEGs may be associated with BC development through cell cycle-related E2F transcription factor and G2M checkpoint pathways.
Limitations were presented in studies on biomarkers used for BC diagnosis14,15,16,17. The robustness of detection remains to be improved and validated in independent external cohorts or experiments. Small sample size and insufficient markers used compromised the diagnosis reliability, suggesting a promising classifier that includes more hub genes might convince accuracy. This study presented a novel diagnostic model that can effectively distinguish BC from healthy individuals through integrated bioinformatics analysis with machine learning.
There were some limitations that need to be cautious when interpret the data. First, the samples were obtained from publicly accessible databases and further verification of diagnostic performance is warranted. The characteristics that are risky for BC such as age, family history, and menstrual status, should be taken into consideration, and this prompted us to evaluate the potential influence of these factor on the classifier in future work. Second, the datasets in the study mainly come from Asian, European, and North American populations, and it remains unclear whether the conclusions apply to other populations. The diagnostic efficacy and therapeutic potential of these hub genes for BC patients with different stages, degrees of differentiation, and other characteristics still need to be verified in multiple clinical cohorts, which will help to achieve clinical translational utility. Finally, the model constructed in this paper needs to be further compared with other cutting-edge advanced algorithms, so that the advantages of the prediction model can be more comprehensively demonstrated.
Conclusions
In this study, we construct a six-gene based diagnostic model and verify it in an external independent cohort, which reliably distinguishes BC from healthy individuals. Our study revealed that these hub genes played complex roles in immune regulation, cell adhesion, cell proliferation and signaling pathway regulation of BC, and may serve as diagnostic markers and therapeutic targets for BC, helping to improve the prognosis of BC. Enrichment analysis illustrates that these DEGs related to BC are enriched in cell cycle, E2F targets and G2M checkpoint pathways, which may shed some new light on the underlying mechanism of tumor progression as well as aid in clinical diagnosis of BC.
Materials and methods
Gene expression datasets preparation
The microarray datasets used in the current study were downloaded from the GEO database63. Keywords “breast cancer” and “normal” were searched in GEO database, and appropriate datasets were selected according to the following criteria for subsequent analysis: (1) samples were collected from human BC and normal breast tissue; (2) gene expression profiles were complete; and (3) at least 40 tissue samples were included. Training cohort was comprised of a total of 287 BC and 76 adjacent normal samples from GSE29044, GSE42568 and GSE162228. Validation cohort contained a total of 360 BC and 218 adjacent normal samples from GSE37751, GSE29431, GSE109169, GSE115144, GSE120129, GSE103512, GSE134359, and GSE38959. After batch-effect was removed using the combined association test (ComBat) empirical Bayes method in the surrogate variable analysis (“SVA”) package64, these datasets were further integrated as the training cohort and the validation cohort, respectively. Additionally, the validation cohort was split into a new set of training and validation sets at a ratio of 7:3, respectively. The data in the datasets were normalized and log2 transformed. Supplementary Fig. 1 showed PCA and box plotsbefore/after batch-effect removal. Details of these datasets in this study are exhibited in Table 1.
Identification of differentially expressed genes and functional enrichment analysis
In the training cohort, the DEGs between BC and adjacent normal samples were identified via linear models for microarray data (“limma”) package in R65. Genes satisfied the threshold of absolute log2FoldChange (log2FC) > 2 and adjusted p-value < 0.05 were considered as significantly differentially expressed and selected for subsequent analysis.
In order to elucidate the potential biological processes and molecular functions of the DEGs, “org.Hs.eg.db” package in R was utilized to convert gene symbols into corresponding EntrezIDs. Subsequently, “clusterProfiler” R package was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis at the cutoff criteria of p-value < 0.05 and false discovery rate (FDR) < 0.0566. Furthermore, to explore biomarker performance of the DEGs in the training cohort, Gene Set Enrichment Analysis (GSEA) was performed utilizing gene set variation analysis (“GSVA”) R package, with annotated h.all.v7.0.entrez.gmt gene set retrieved from the molecular signatures database (MsigDB, https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) as the reference gene set67,68. Absolute value of normalized enrichment score (|NES|) > 1, FDR q-value < 0.25 and p-value < 0.05 were regarded as critical criteria.
Candidate diagnostic biomarkers evaluation and signature establishment
LASSO-Logistic regression was performed with ten-fold cross-validation, as implemented in the R package “glmnet”69, and λ value with the minimum cross-validation error was selected as the criterion for screening the best diagnostic features in the training cohort. Subsequently, SVM-RFE algorithm was executed to further dimensionality reduction based on the results of LASSO analysis70: the classification model with the highest accuracy of tenfold cross-validation was selected as the optimal model71,72. Ultimately, multivariate stepwise Logistic regression was used to extract genes significantly associated with BC as hub genes (p < 0.05), and then the optimal diagnostic model of BC was obtained based on these genes. Moreover, a diagnostic nomogram was created to visualize the predicted rate for each individual. The Hosmer–Lemeshow test was applied to verify the calibration curve that was established to improve and assess the accuracy and reliability of the nomogram prediction. Decision curve and clinical impact curve analysis were conducted via R package “rmda” so as to evaluate the clinical effectiveness of the prediction model, and the results were visualized by “DecisionCurve” R package73. The performance and discriminative powers of the final diagnostic model were assessed using the area under the curve (AUC) approach.
Validation of the diagnostic signature
In order to further verify the stability of the diagnostic model, we applied the model to the training and validation sets of validation cohort, respectively. The expression profiles of the hub genes in the validation cohort were extracted for subsequent analysis. Split violin plot was drawn to evaluate the differential expression of the hub genes between BC and adjacent normal samples. Additionally, the predictive performance of the model was evaluated by receiver operating characteristic (ROC) curve and confusion matrix analysis, and the sensitivity and specificity of the model in BC diagnosis were assessed utilizing the AUC values.
Statistical analysis
All statistical analysis and graphs were performed in R software (version 4.1.2, https://www.r-project.org/). The “glmnet” package in R was conducted for LASSO-Logistic regression analysis, and the “caret” and “glm” R packages were executed for SVM-RFE analysis and construction of Logistic diagnostic model, respectively. Nomogram was established by the root mean squares (“RMS”) package, and “pROC” and “InformationValue” packages were applied to ROC curve and confusion matrix analysis, respectively. Odds ratio (OR) and the corresponding 95% confidence interval (CI) were evaluated by the Logistic regression model. Independent sample t-test or Mann–Whitney U-test was executed to compare continuous variables. The p-value was adjusted by the Benjamini–Hochberg method to control the FDR74. Statistical significance was defined as p < 0.05.
Results
Identification of the DEGs in the training cohort
We utilized the “limma” R package to recognize the DEGs between BC tissue and adjacent normal breast tissue. Out of 20,862 genes profiles in the training cohort after Log transformation and normalization, 154 DEGs were identified at the threshold criteria of adjusted p-value < 0.05 and |log2FC|> 2, with 51 DEGs being up-expression and 103 DEGs being down-expression in BC group (Fig. 2A, B, Supplementary Table 1).

Identification of DEGs in training cohort at the cutoff criteria of adjusted p-value < 0.05 and |log2FoldChange|> 2. (A) Volcano plot of gene expression profiles between samples from BC to adjacent normal tissue; (B) Heatmap represented the expression of 154 DEGs between samples from BC to adjacent normal tissue.
Functional enrichment analysis of the DEGs
To further reveal the potential biological mechanisms of the DEGs, we performed functional enrichment analysis on 154 DEGs in GO terms including biological process (BP), cellular component (CC) and molecular function (MF), and KEGG pathways75,76,77. GSEA analysis was also conducted based on hallmark gene sets in MSigDB. Of the three categories in GO terms, the most abundant groups were nuclear division, collagen-containing extracellular matrix and extracellular matrix structural constituent, respectively (Fig. 3A–C). We discovered that PPAR, Cell cycle, p53, Oocyte meiosis, Tyrosine metabolism and AMPK were the most enriched KEGG pathways (Fig. 3D). Moreover, the GSEA analysis demonstrated that HALLMARK_E2F_TARGETS (NES = 4.47, FDR q-value < 0.0001), HALLMARK_G2M_CHECKPOINT (NES = 4.36, FDR q-value < 0.0001) and HALLMARK_MITOTIC_SPINDLE (NES = 3.88, FDR q-value < 0.0001) gene sets were the most enriched gene sets in BC (p-value < 0.0001) (Fig. 3E–H).

Enrichment analysis of the DEGs. (A) Biological process. (B) Cellular component. (C) Molecular function. (D) KEGG pathway analysis. (E-H) GSEA analysis using hallmark gene sets from MSigDB.
Identification of diagnostic biomarkers and establishment of diagnostic signature in the training cohort
To obtain the most robust diagnostic indicators for BC, we performed the LASSO-Logistic regression on 154 DEGs in the training cohort for dimensionality reduction. We utilized tenfold cross-validation to adjust parameters so as to overcome over-fitting. The LASSO regression results showed that the λ value corresponding to the minimum cross-validation error was 0.057, at which point the model with 26 genes was the optimal predictor (Fig. 4A). In this model, all 26 candidate genes associated with diagnosis had non-zero LASSO coefficients (Fig. 4B, C). Subsequently, we carried out the SVM-RFE algorithm to further detect diagnostic candidates that can optimize the performance of the classifier. Grid search and tenfold cross-validation were utilized to train and estimate SVM hyperparameters. The radial basis function (RBF) kernel function was employed in this study, with a cost parameter of 0.25 and sigma of 0.15. We chose the optimal classification model with the highest accuracy, which contained 10 candidate genes (Fig. 4D–F). The accuracy of the SVM-RFE model was 93.44%, with a kappa of 0.791, a positive predictive value (PPV) of 87.50% and a F1 score of 0.851 (Supplementary Table 2). Finally, multivariate stepwise Logistic regression was applied to check the relation of candidate gene expression levels on the disease status, and six biomarkers with the most robust diagnostic performance were obtained to construct a diagnostic model for BC. In terms of the Logistic analysis results, the elevated expression of FAM83D, COMP and SEMA3G were risk factors for BC. In contrast, CD300LG, IGSF10 and MAMDC2 expression were protective factors (Fig. 5A, Table 2). Moreover, cluster heatmap of the six biomarkers revealed that the expression of these genes in the training cohort differed significantly between BC and adjacent normal breast tissue. All these data indicated that our classifier was robust in distinguishing BC from normal tissue (Fig. 5B).

LASSO-Logistic regression and SVM-RFE results of the DEGs. (A–C) 26 genes considered the more correlated with BC were identified by LASSO-Logistic regression. (D–F) Accuracy, Kappa values and confusion matrix heatmap of the SVM-RFE results.

Construction and evaluation of the diagnostic model. (A) Multivariate Stepwise Logistic regression model. (B) Cluster heatmap of six diagnostic biomarkers in the training cohort. (C) Diagnostic nomogram for quantifying risk assessment of individual. (D) Calibration curve illustrated the calibration of the nomogram in terms of the agreement between the predicted risk of BC and the observed outcomes. (E, F) The decision curve and clinical impact curve evaluated the clinical benefit of the model. (G) ROC analysis of the model for predicting BC risk. (H) Confusion matrix heatmap of the model in the training cohort.
In order to quantify the risk assessment for each individual, we constructed a nomogram involving six candidate genes. Subsequently, we randomly calculated the values of each covariate of an individual in the training cohort and mapped them to the corresponding scores to predict the probability of developing BC by calculating the total score. The probability value of No.25 individual (GSM719696) being attacked by BC was 0.247 (Fig. 5C). We carried out the calibration curve analysis for the sake of evaluating prediction performance of the diagnostic model, and the results revealed that the nomogram predicted rate was in agreement with the actual outcomes observed in the training cohort (Fig. 5D), indicating that the nomogram had good accuracy and reliability. Moreover, the decision curve drawn with net benefit (NB) as vertical coordinate and high risk threshold as horizontal coordinate illustrated that the NB of the model was greater than 0, which was of clinical significance. When the high risk threshold was 0.6, the NB was 0.52, meaning that 52 out of every 100 BC patients would benefit from diagnosis and treatment of the model without harming the benefit of others (Fig. 5E, Supplementary Table 3). The clinical impact curve of the model exhibited that the number of people classified as high risk by the diagnostic model was basically consistent with the number of true positive people at the threshold probability of 0.6 (Fig. 5F). Both the decision curve and the clinical impact curve disclosed that the diagnostic model had good net benefit and predictive performance. Additionally, the ROC curve analysis indicated that the AUC of the model was 0.978 (0.962, 0.995) (Fig. 5G), with the resulting confusion matrix heatmap of the classifying performance shown in Fig. 5H. This indicated that the diagnostic model had a high sensitivity and specificity of 0.829 and 0.976, respectively, in distinguishing BC patients from healthy individuals in the training cohort (Table 3). Besides, accuracy, recall and precision of the model were 0.945 (0.916, 0.966), 0.829 and 0.900, respectively, indicating that the classifier performed well (Table 3).
Validation of the diagnostic signature in the independent validation cohort
We validated the diagnostic model in validation cohort to confirm its robustness. The expression levels of six genes in the model were significantly different between samples from BC tissue to normal tissue (Fig. 6A). Notably, the ROC analysis uncovered that the AUC values of the model in training set and validation set of validation cohort were 0.936 (0.910, 0.961) and 0.921 (0.870, 0.972), respectively (Fig. 6B, D, Table 3). The confusion matrices of classification performance were shown in Figs. 6C and E. These results revealed that the prediction performance of the diagnostic model was satisfactory, and it performed well in distinguishing BC patients from healthy individuals in training set and validation set.

Evaluating the diagnostic model in the validation cohort. (A) Expression profiles of six diagnostic biomarkers between samples from BC to adjacent breast normal tissue. (B, D) ROC analysis of the model for predicting BC risk. (C, E) Confusion matrix heatmap of the model.
Contribution to the field statement
Breast cancer (BC) is the second leading cause of cancer-related death in females, followed by lung cancer. Disadvantages exist in conventional diagnostic techniques of BC, such as radiation risk. Therefore, it is essential to identify effective biomarkers that aid in early diagnosis and developing novel treatments for improving BC prognosis. The present study integrated bioinformatics analysis with machine learning to elucidate potential key candidate genes associated with the tumorigenesis of BC. As a result, a total of six hub genes connected with BC pathogenesis were identified. Then, a diagnostic model of BC on the basis of these genes was established. ROC analysis of the diagnostic model illustrated that AUC of training set was 0.978 (0.962, 0.995). In the validation cohort, AUC of training set and validation set were 0.936 (0.910, 0.961) and 0.921 (0.870, 0.972), respectively. The results indicated that the model was reliable in separating BC patients from healthy individuals. Our findings may assist in early diagnosis of BC with implications for improving the prognosis of BC patients.
Data availability
All data are available and are based upon public data extracted from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- ANN:
-
Artificial neural network
- AUC:
-
Area under the curve
- BC:
-
Breast cancer
- BP:
-
Biological processes
- CASC9:
-
Cancer susceptibility candidate 9
- CC:
-
Cellular component
- CD300LG:
-
CMRF35-like molecule 9
- COMBAT:
-
Combined association test
- COMP:
-
Cartilage oligomeric matrix protein
- CT:
-
Computed tomography
- DEGs:
-
Differentially expressed genes
- EGTM:
-
European group on tumor markers
- EMT:
-
Epithelial–mesenchymal transition
- FAM83D:
-
Protein FAM83D
- GEO:
-
Gene expression omnibus
- GO:
-
Genome ontology
- GSEA:
-
Gene set enrichment analysis
- GSVA:
-
Gene set variation analysis
- IGSF10:
-
Immunoglobulin superfamily member 10
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LASSO:
-
Least absolute shrinkage and selection operator
- LUSC:
-
Lung squamous cell carcinoma
- MAMDC2:
-
MAM domain-containing protein 2
- MF:
-
Molecular function
- MRI:
-
Magnetic resonance imaging
- NB:
-
Net benefit
- NES:
-
Normalized enrichment score
- OR:
-
Odds ratio
- SEMA3G:
-
Semaphorin-3G
- PET:
-
Positron emission tomography
- PPI:
-
Protein-protein interaction
- PPV:
-
Positive predictive value
- RBF:
-
Radial basis function
- ROC:
-
Receiver operating characteristic
- SVA:
-
Surrogate variable analysis
- SVM:
-
Support vector machine
- SVM-RFE:
-
Support vector machine-recursive feature elimination
- TP53:
-
Tumor protein p53
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Sung, H. et al. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Subramanian, S. & Keating, N. L. Delays in breast cancer diagnosis after a state policy limiting medicaid enrollment. 123, 3219–3221.
Hahn, W. C. & Weinberg, R. A. Rules for making human tumor cells. N. Engl. J. Med. 347, 1593–1603 (2002).
Poillet-Perez, L. et al. Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573 (2018).
Mohandass, J. et al. BCDB—A database for breast cancer research and information. Bioinformation 5, 1–3 (2010).
Cobain, E. F., Milliron, K. J. & Merajver, S. D. Updates on breast cancer genetics: clinical implications of detecting syndromes of inherited increased susceptibility to breast cancer. Semin. Oncol. 43, 528–535 (2016).
Walsh, T. & King, M.-C. Ten genes for inherited breast cancer.
Walker-Smith, T. L. & Peck, J. Genetic and genomic advances in breast cancer diagnosis and treatment. Nurs. Women’s Health 23, 518–525 (2019).
McDonald, E. S., Clark, A. S., Tchou, J., Zhang, P. & Freedman, G. M. Clinical diagnosis and management of breast cancer. J. Nucl. Med. 57(Suppl 1), 9S-16S (2016).
Chongyang, C., Shangchun, F., Han, L., Xiaolei, Q., & Dezhi, Z. Deep learning-based research on the influence of training data size for breast cancer pathology detection. J. Eng. 2019 (2019).
Li, G., Hu, J. & Hu, G. Biomarker studies in early detection and prognosis of breast cancer. Adv. Exp. Med. Biol. 1026, 27–39 (2017).
Ayyildiz, D. & Piazza, S. Introduction to bioinformatics. Methods Mol. Biol. 1986, 1–15 (2019).
Jia, D. et al. Breast cancer case identification based on deep learning and bioinformatics analysis. Front. Genet. 12, 628136 (2021).
Liu, S. et al. Identification of candidate biomarkers correlated with the pathogenesis and prognosis of breast cancer via integrated bioinformatics analysis. Medicine 99, e23153 (2020).
Li, M., Guo, Y., Feng, Y. M. & Zhang, N. Identification of triple-negative breast cancer genes and a novel high-risk breast cancer prediction model development based on PPI data and support vector machines. Front. Genet. 10, 180 (2019).
Chen, D. & Yang, H. Integrated analysis of differentially expressed genes in breast cancer pathogenesis. Oncol. Lett. 9, 2560–2566 (2015).
Duffy, M. J. et al. Clinical use of biomarkers in breast cancer: Updated guidelines from the European group on tumor markers (EGTM). Eur. J. Cancer 75, 284–298 (2017).
Borrego, F. The cd300 molecules: An emerging family of regulators of the immune system. Blood 121, 1951–1960 (2013).
Cannon, J. P., O’Driscoll, M. & Litman, G. W. Specific lipid recognition is a general feature of cd300 and TREM molecules. Immunogenetics 64, 39–47 (2012).
Umemoto, E. et al. Dynamic changes in endothelial cell adhesion molecule nepmucin/cd300lg expression under physiological and pathological conditions. PLoS One 8, e83681 (2013).
Bloomstein, J. D. et al. Validated limited gene predictor for cervical cancer lymph node metastases. Oncotarget 11, 2302–2309 (2020).
Bao, Y. et al. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell. Mol. Biol. Lett. 24, 38 (2019).
Ren, C., Tang, X. & Lan, H. Comprehensive analysis based on DNA methylation and RNA-seq reveals hypermethylation of the up-regulated wt1 gene with potential mechanisms in pam50 subtypes of breast cancer. PeerJ 9, e11377 (2021).
Sultan, G., Zubair, S., Tayubi, I. A., Dahms, H.-U. & Madar, I. H. Towards the early detection of ductal carcinoma (a common type of breast cancer) using biomarkers linked to the PPAR(γ) signaling pathway. Bioinformation 15, 799–805 (2019).
Metz, S. et al. The arg82cys polymorphism of the protein nepmucin implies a role in HDL metabolism. J. Endocr. Soc. 6, bvac034 (2022).
Howard, S. R. et al. IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol. Med. 8, 626–642 (2016).
Thutkawkorapin, J. et al. Exome sequencing in one family with gastric- and rectal cancer. BMC Genet. 17, 41 (2016).
Chang, Y.-S., Huang, H.-D., Yeh, K.-T. & Chang, J.-G. Identification of novel mutations in endometrial cancer patients by whole-exome sequencing. Int. J. Oncol. 50, 1778–1784 (2017).
Ling, B. et al. Identification of prognostic markers of lung cancer through bioinformatics analysis and in vitro experiments. Int. J. Oncol. 56, 193–205 (2020).
Wang, M. et al. Immunoglobulin superfamily member 10 is a novel prognostic biomarker for breast cancer. PeerJ 8, e10128 (2020).
Wu, M., Li, Q. & Wang, H. Identification of novel biomarkers associated with the prognosis and potential pathogenesis of breast cancer via integrated bioinformatics analysis. Technol. Cancer Res. Treat. 20, 1533033821992081 (2021).
Li, M. X. et al. Identification of potential core genes in triple negative breast cancer using bioinformatics analysis. Onco Targets Ther. 11, 4105–4112 (2018).
Santamaria, A., Nagel, S., Sillje, H. H. W. & Nigg, E. A. The spindle protein chica mediates localization of the chromokinesin kid to the mitotic spindle. Curr. Biol. 18, 723–729 (2008).
Pérez-Peña, J. et al. Mitotic read-out genes confer poor outcome in luminal a breast cancer tumors. Oncotarget 8, 21733–21740 (2017).
Wang, Z. et al. FAM83D promotes cell proliferation and motility by downregulating tumor suppressor gene fbxw7. Oncotarget 4, 2476–2486 (2013).
Cipriano, R. et al. Conserved oncogenic behavior of the fam83 family regulates MAPK signaling in human cancer. Mol. Cancer Res. 12, 1156–1165 (2014).
Snijders, A. M. et al. FAM83 family oncogenes are broadly involved in human cancers: An integrative multi-omics approach. Mol. Oncol. 11, 167–179 (2017).
Zhai, X., Yang, Z., Liu, X., Dong, Z. & Zhou, D. Identification of nuf2 and fam83d as potential biomarkers in triple-negative breast cancer. PeerJ 8, e9975 (2020).
Uyar, D. S., Huang, Y.-W., Chesnik, M. A., Doan, N. B. & Mirza, S. P. Comprehensive serum proteomic analysis in early endometrial cancer. J. Proteomics 234, 104099 (2021).
Mu, Y., Zou, H., Chen, B., Fan, Y. & Luo, S. FAM83D knockdown regulates proliferation, migration and invasion of colorectal cancer through inhibiting fbxw7/notch-1 signalling pathway. Biomed. Pharmacother. Biomed. Pharmacother. 90, 548–554 (2017).
Yin, C. et al. FAM83D promotes epithelial-mesenchymal transition, invasion and cisplatin resistance through regulating the akt/mtor pathway in non-small-cell lung cancer. Cell. Oncol. 43, 395–407 (2020).
Fu, Y., Zhou, Q.-Z., Zhang, X.-L., Wang, Z.-Z. & Wang, P. Identification of hub genes using co-expression network analysis in breast cancer as a tool to predict different stages. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 25, 8873–8890 (2019).
Beckmann, G. & Bork, P. An adhesive domain detected in functionally diverse receptors. Trends Biochem. Sci. 18, 40–41 (1993).
Fox, S. A., Vacher, M. & Farah, C. S. Transcriptomic biomarker signatures for discrimination of oral cancer surgical margins. Biomolecules 12 (2022).
Wu, K.-Z., Xu, X.-H., Zhan, C.-P., Li, J. & Jiang, J.-L. Identification of a nine-gene prognostic signature for gastric carcinoma using integrated bioinformatics analyses. World J. Gastrointest. Oncol. 12, 975–991 (2020).
Lee, H. et al. MAM domain containing 2 is a potential breast cancer biomarker that exhibits tumour-suppressive activity. Cell Prolif. 53, e12883 (2020).
Saxne, T. & Heinegård, D. Cartilage oligomeric matrix protein: A novel marker of cartilage turnover detectable in synovial fluid and blood.
Englund, E. et al. Cartilage oligomeric matrix protein promotes prostate cancer progression by enhancing invasion and disrupting intracellular calcium homeostasis. Oncotarget 8, 98298–98311 (2017).
Englund, E. et al. Cartilage oligomeric matrix protein contributes to the development and metastasis of breast cancer. Oncogene 35, 5585–5596 (2016).
Papadakos, K. S. et al. Cartilage oligomeric matrix protein initiates cancer stem cells through activation of jagged1-notch3 signaling. Matrix Biol. 81, 107–121 (2019).
Papadakos, K. S., Hagerling, C., Rydén, L., Larsson, A.-M. & Blom, A. M. High levels of expression of cartilage oligomeric matrix protein in lymph node metastases in breast cancer are associated with reduced survival. Cancers 13 (2021).
Hyun, J., Lee, M., Rehman, J., Pajcini, K. V. & Malik, A. B. Notch1 promotes ordered revascularization through semaphorin 3g modulation of downstream vascular patterning signalling factors. J. Physiol. 600, 509–530 (2022).
Gao, L. et al. The expression, significance and function of cancer susceptibility candidate 9 in lung squamous cell carcinoma: A bioinformatics and in vitro investigation. Int. J. Oncol. 54, 1651–1664 (2019).
Ji, C. et al. Immune-related genes play an important role in the prognosis of patients with testicular germ cell tumor. Ann. Transl. Med. 8, 866 (2020).
Hu, B. et al. Identification of three glioblastoma subtypes and a six-gene prognostic risk index based on the expression of growth factors and cytokines. Am. J. Transl. Res. 12, 4669–4682 (2020).
Liu, L., Zeng, P., Yang, S. & Yuan, Z. Leveraging methylation to identify the potential causal genes associated with survival in lung adenocarcinoma and lung squamous cell carcinoma. Oncol. Lett. 20, 193–200 (2020).
Karayan-Tapon, L. et al. Semaphorin, neuropilin and VEGF expression in glial tumours: sema3g, a prognostic marker?. Br. J. Cancer 99, 1153–1160 (2008).
Oshi, M. et al. The E2F pathway score as a predictive biomarker of response to neoadjuvant therapy in ER+/HER2- breast cancer. Cells 9 (2020).
Ivanova, I. A., D’Souza, S. J. A. & Dagnino, L. Signalling in the epidermis: The E2F cell cycle regulatory pathway in epidermal morphogenesis, regeneration and transformation. In. J. Biol. Sci. 1, 87–95 (2005).
Lin, X. et al. Integrative analyses of gene expression and DNA methylation profiles in breast cancer cell line models of tamoxifen-resistance indicate a potential role of cells with stem-like properties. Breast Cancer Res. 15, R119 (2013).
Oshi, M. et al. G2M checkpoint pathway alone is associated with drug response and survival among cell proliferation-related pathways in pancreatic cancer. Am. J. Cancer Res. 11, 3070–3084 (2021).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 16, 284–287 (2012).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 (2005).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Duan, K.-B., Rajapakse, J. C., Wang, H. & Azuaje, F. Multiple SVM-RFE for gene selection in cancer classification with expression data.
Guo, P. et al. Gene expression profile based classification models of psoriasis. Genomics 103, 48–55 (2014).
Huang, M.-L., Hung, Y.-H., Lee, W. M., Li, R. K. & Jiang, B.-R. SVM-RFE based feature selection and Taguchi parameters optimization for multiclass SVM classifier. TheScientificWorldJournal 2014, 795624 (2014).
Vickers, A. J. & Elkin, E. B. Decision curve analysis: A novel method for evaluating prediction models. Med. Decis. Mak. 26, 565–574 (2006).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 57 (1995).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Colak, D. et al. Age-specific gene expression signatures for breast tumors and cross-species conserved potential cancer progression markers in young women. PLoS One 8, e63204 (2013).
Clarke, C. et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis 34, 2300–2308 (2013).
Chen, Y.-J. et al. Molecular subtyping of breast cancer intrinsic taxonomy with oligonucleotide microarray and nanostring ncounter. Biosci Rep 41, BSR20211428 (2021).
Tang, W. et al. Integrated proteotranscriptomics of breast cancer reveals globally increased protein-mrna concordance associated with subtypes and survival. Genome Med 10, 94 (2018).
Cuadros, M., Cano, C., López, F. J., López-Castro, R., & Concha, A. Expression profiling of breast tumors based on human epidermal growth factor receptor 2 status defines migration-related genes. Pathobiology 80, 32–40 (2013).
Chang, J.-W. et al. Wild-type p53 upregulates an early onset breast cancer-associated gene gas7 to suppress metastasis via gas7-cyfip1-mediated signaling pathway. Oncogene 37, 4137–4150 (2018).
Sun, W. et al. Transcriptome analysis of luminal breast cancer reveals a role for lol in tumor progression and tamoxifen resistance. Int J Cancer 145, 842–856 (2019).
Dhage, S. et al. A genomic ruler to assess oncogenic transition between breast tumor and stroma. PLoS One 13, e0205602 (2018).
Brouwer-Visser, J. et al. Regulatory t-cell genes drive altered immune microenvironment in adult solid cancers and allow for immune contextual patient subtyping. Cancer Epidemiol Biomarkers Prev 27, 103–112 (2018).
Cedro-Tanda, A. et al. A lncrna landscape in breast cancer reveals a potential role for ac009283.1 in proliferation and apoptosis in her2-enriched subtype. Sci Rep 10, 13146 (2020).
Komatsu, M. et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int J Oncol 42, 478–506 (2013).
Acknowledgements
The authors wish to acknowledge Denggang Fu from Indiana University in editing the manuscript.
Funding
This work was supported by the Jiangxi Provincial Natural Science Foundation (20224ACB206038).
Author information
Authors and Affiliations
Contributions
SQY, CMW and XW conceived and designed the present study. TL, FFZ and YZ conducted the literature search and drafted the initial manuscript. JOU and LYG analyzed the data. SXH confirmed the authenticity of all the raw data. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, S., Wang, C., Ouyang, J. et al. Identification of candidate biomarkers correlated with the pathogenesis of breast cancer patients. Sci Rep 15, 8770 (2025). https://doi.org/10.1038/s41598-025-93208-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93208-w
