
Abstract
This study aims to explore the effects and potential mechanisms of PKM2-mediated neuroinflammation leading to mitochondrial damage and its role in the progression of chronic fatigue syndrome (CFS). Bioinformatics methods were applied to predict and analyze PKM2 and downstream signaling factors. In vivo experiments were conducted with mice divided into four groups after different treatments: control group, model group, Model + PKM2-OE group, and Model + PKM2-KD group. Morris water maze and field tests were used to assess cognitive function, grip strength, and rotation tests to evaluate physical strength. HE and Nissl staining were used to observe cellular conditions in the CA1 region of the hippocampus. Immunohistochemistry was used to detect PKM2 levels in the CA1 region. Western blot was performed to assess protein expression, lactate assay kits measured serum and brain tissue lactate levels, and ELISA detected inflammatory factors in brain tissue. Bioinformatics analysis showed that PKM2 could promote the expression of glycolytic factors, leading to H4K12la histone lactylation modification, which enhances the expression of inflammatory factors such as NF-κB, resulting in mitochondrial damage. Compared to the control group, the cognitive function of the model group significantly declined, while the cognitive function of the Model + PKM2-OE group improved. However, cognitive function worsened in the Model + PKM2-KD group compared to the model group. The physical strength of the control group was normal, and no significant differences were observed in the model, Model + PKM2-OE, and Model + PKM2-KD groups. Cell quantity and arrangement in the control group were normal, while the model group showed fewer and disorganized cells. The Model + PKM2-OE group showed further deterioration compared to the model group, whereas the Model + PKM2-KD group showed improvement. Compared to the control group, the model group had increased expression of PKM2, H4K12la, H4, IL-1β, and TNFα. Compared to the model group, these markers were even higher in the Model + PKM2-OE group, but significantly reduced in the Model + PKM2-KD group. Serum lactate levels increased in the model group compared to the control group, but there was no significant difference between the Model + PKM2-OE and Model + PKM2-KD groups. Brain tissue lactate levels increased in the model group, further elevated in the Model + PKM2-OE group, but decreased in the Model + PKM2-KD group. PKM2 in hippocampal cells enhances glycolysis, lactate accumulation, and H4K12la/NF-κB-mediated neuroinflammation, leading to mitochondrial damage and accelerating the progression of chronic fatigue syndrome.
Similar content being viewed by others


Introduction
Chronic Fatigue Syndrome (CFS), also known as Myalgic Encephalomyelitis (ME), is a complex disorder primarily characterized by persistent fatigue that cannot be alleviated by rest. CFS affects millions of people worldwide. However, due to its unknown etiology and diverse symptom presentation, its diagnosis and treatment pose significant challenges. Patients often exhibit other symptoms, such as cognitive impairment, muscle pain, sleep disturbances, and autonomic dysfunction, which severely impact their daily lives and work capacity1.
The pathogenesis of CFS is still not fully understood, but recent research suggests that metabolic abnormalities, immune system dysregulation, and mitochondrial dysfunction may be central to its pathology2. Mitochondrial damage, in particular, is thought to be a key factor leading to fatigue and abnormal energy metabolism. Research on CFS has gradually shifted from symptom management to investigating its molecular mechanisms, with the role of PKM2 (pyruvate kinase M2) increasingly becoming a focus. In human cells, energy production primarily depends on glycolysis and mitochondrial oxidative phosphorylation. Under normal physiological conditions, cells break down glucose into pyruvate through glycolysis, which then enters the mitochondria for oxidative phosphorylation to produce ATP. However, the metabolic patterns in CFS patients have significantly changed. Studies have found increased glycolytic activity and reduced efficiency of mitochondrial oxidative phosphorylation in CFS patients, leading to energy deficiency and the accumulation of metabolic waste, such as lactate3.
PKM2 plays a crucial role in this process. PKM2 is a key enzyme in the glycolytic pathway, catalyzing the conversion of phosphoenolpyruvate to pyruvate, generating ATP. However, PKM2 is not solely involved in energy metabolism; it also regulates various functions such as cell proliferation, signal transduction, and gene expression4. Particularly under pathological conditions, PKM2 can influence cellular metabolism and signaling by altering its oligomerization state. When PKM2 exists in a dimeric form, its glycolytic activity is low, but when it switches to a tetrameric form, glycolytic efficiency increases significantly5.
Research suggests that abnormal activation of PKM2 in hippocampal cells may enhance glycolysis and lactate accumulation, leading to mitochondrial dysfunction and accelerating the progression of CFS6. Furthermore, the activation of PKM2 not only promotes glycolysis but also regulates lactate accumulation, which further alters the acidic environment within cells. This metabolic disorder is considered a crucial mechanism underlying energy production deficits in CFS patients.
Mitochondria play a critical role in cellular energy metabolism by producing ATP, the primary energy form required for cellular activities through oxidative phosphorylation. However, in CFS patients, the significant decline in mitochondrial function severely inhibits energy production, leading to fatigue, muscle weakness, and cognitive dysfunction7. The relationship between oxidative stress and mitochondrial damage has also received widespread attention. Elevated oxidative stress levels are common in CFS patients’ cells, contributing to continuous mitochondrial damage. Studies have shown that oxidative stress not only directly reduces mitochondrial membrane potential and ATP production but may also induce mitochondrial DNA damage, further exacerbating metabolic dysfunction8.
At the same time, excessive activation of PKM2 is closely related to mitochondrial dysfunction. PKM2 enhances glycolysis, increases lactate production, and leads to the accumulation of acidic metabolic byproducts, weakening the mitochondria’s ability to generate energy9. Additionally, lactate accumulation may further induce neuroinflammation through the regulation of epigenetic modifications (such as H4K12la) and the activation of the NF-κB signaling pathway, aggravating the pathological progression of CFS10. Neuroinflammation is considered a major contributor to cognitive dysfunction and emotional problems in CFS. Inflammatory responses not only impair the normal function of the central nervous system but may also cause neuronal damage and reduced synaptic plasticity through long-term neuroinflammation11. In CFS patients, elevated levels of various pro-inflammatory cytokines, particularly tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), have been found, which are closely associated with neuroinflammation12.
PKM2 activation plays an essential role in both metabolic regulation and the exacerbation of inflammation. Studies have shown that PKM2 can activate a series of pro-inflammatory signaling pathways, such as the NF-κB pathway, through its interaction with proteins13. NF-κB is the central regulator of cellular inflammatory responses; once activated, it induces the release of various inflammatory factors and maintains a sustained inflammatory response through a self-activation feedback mechanism14.
Specifically, the abnormal activation of PKM2 in hippocampal cells may promote lactate accumulation by enhancing glycolysis, further inducing neuroinflammatory responses mediated by H4K12la/NF-κB, and aggravating mitochondrial dysfunction15,16,17. This mechanism is particularly prominent in CFS, with studies showing that persistent neuroinflammation in CFS patients is closely related to their cognitive impairment and emotional disorders16,18. Long-term neuroinflammation not only damages the normal function of the central nervous system but may also exacerbate fatigue and metabolic disorders by affecting mitochondrial function19. Given the central role of PKM2 in glycolytic regulation, neuroinflammation, and mitochondrial dysfunction, targeting PKM2 and its associated signaling pathways may provide a new strategy for CFS treatment. Previous studies have shown that inhibiting PKM2 activity can reduce neuroinflammation and improve mitochondrial function20. Moreover, interventions targeting the NF-κB signaling pathway may alleviate the symptoms of CFS by reducing inflammatory responses21.
This study explored the expression levels of PKM2 and its downstream molecular mechanisms in the development of Chronic Fatigue Syndrome from both in vitro and in vivo perspectives. However, applying these findings to clinical practice is still quite complex and requires further research for validation and a deeper understanding.
Materials and methods
Experimental animals
Twenty-four SPF-grade C57BL/6J mice, 6 weeks old, half male and half female, weighing 16–18 g, were housed in groups and purchased from Henan Sikebeisi Biotechnology Co., Ltd. [SCXK (Yu) 2020-0015]. The animals were housed in the barrier environment animal laboratory at Hebei University [SYXK (Ji) 2020-0035]. During the experiment, the animal breeding room was maintained at a relative temperature of (23.0 ± 1.0)°C, with a relative humidity of 55–65%, under a 12-hour light/dark cycle. The experimental animals had free access to food and water. The experiment was approved by the Animal Ethics Committee of Hebei University (Approval No.: HBDX202310030005). All methods used in this experiment adhered to the 3R principles and the ARRIVE guidelines.
Experimental materials
The human hippocampal neuronal cell line ReNcell VM was purchased from PROCELL. PKM2-NC, PKM2-OE, and PKM2-KD were obtained from Genescript (Nanjing, China). The INTERFERin® kit was purchased from Selleck. The glycolysis inhibitor 3-bromopyruvate (3-BP), NF-κB inhibitor SN-50, and NF-κB activator activator-1 were purchased from MCE. ELISA kits were obtained from Shanghai Hengyuan Biotechnology Co., Ltd., and the Western blot electrophoresis apparatus was purchased from Visual Sonics, Canada.
Bioinformatics analysis
Single-cell RNA transcriptomic data for Chronic Fatigue Syndrome (CFS) were obtained from the publicly available gene expression database GEO. This process involved careful selection and analysis of the data. The acquired dataset included 7 control group patient samples and 8 CFS patient samples, forming a representative basis for comparison. To extract the relevant dataset GSE14577, we used the GEOquery package to efficiently retrieve the required data from the GEO database. After standardization, low-quality cells were removed to ensure the accuracy and reliability of subsequent analyses.
During the data processing stage, we applied the Seurat package for in-depth analysis of all single-cell RNA-seq data, aiming to integrate the significant differences between the control group and CFS patient samples. The selection criteria were set as |log2 Fold Change| > 0.5 and p-value < 0.05, ensuring that the gene expression changes we focused on were statistically significant. To further enhance the credibility of the results, we adopted the Benjamini-Hochberg method for multiple hypothesis testing correction of the p-values, reducing the possibility of false positives.
Subsequently, we used the Metascape analysis platform for functional enrichment analysis of the selected differentially expressed genes, covering GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) analyses, aiming to identify pathways related to signal transduction and to explore the homogeneity and heterogeneity of signaling responses in different subpopulations of chondrocytes. This analysis not only revealed potential mechanisms at the molecular level in CFS patients but also provided important clues and directions for future research.
Finally, we used the ggplot2 package to visualize the differential analysis results, allowing a more intuitive presentation of the research findings and enhancing the comprehensibility and dissemination of the results.
Animal model construction
Before starting the experiment, prepare lentiviruses carrying the PKM2 gene (PKM2-NC, PKM2-OE, PKM2-KD). The virus should have a high titer (1 × 10^9 TU/mL) to ensure effective infection of target cells after injection. Store at low temperatures (−80 °C) to maintain activity, and thaw thoroughly before use. Anesthetize the mice with 3% isoflurane to ensure a painless surgical process. After anesthesia, use eye ointment to protect the mice’s eyes from drying. Secure the mice on a stereotaxic apparatus, ensuring the head is stable and level. Use measuring tools to determine the coordinates of the lateral ventricle. The coordinates for the mouse lateral ventricle relative to the bregma are: Anteroposterior (AP): −0.5 mm (relative to bregma), Mediolateral (ML): ±1.0 mm (relative to midline), and Dorsoventral (DV): −2.0 mm (relative to the skull surface).
Use a scalpel to cut the scalp and expose the skull. Gently drill a small hole, approximately 0.5 mm in diameter, at the marked coordinates for virus injection. Connect a Hamilton syringe to the lentivirus mixture and fix it onto a microinjection pump. Insert the syringe to a depth of about 2.0 mm, and begin injecting the virus at a slow rate (0.2–0.5 µL per minute) with a total volume of 2 µL. After the injection is completed, leave the syringe in place for at least 5 min to ensure the virus diffuses into the target area. Then, slowly withdraw the syringe. After the injection is complete, suture the cut skin with a needle. Allow the mouse to recover from anesthesia in a warm environment and provide appropriate postoperative care. Carefully observe the mouse’s behavior changes, weight fluctuations, and other signs to ensure normal recovery.
Seven days after the injection, induce CFS modeling in the Model group, Model + PKM2-OE group, and Model + PKM2-KD group, excluding the control group. We followed the previous literature for CFS mouse modeling22,23. In the morning, use an exhaustive swimming method, and in the afternoon, perform chronic restraint, with a total modeling period of 21 days. The exhaustive swimming method: Place the mice in a custom-made swimming tank (60 cm long, 50 cm wide, 40 cm high, with a water depth of 30 cm) for forced swimming. Use methods to ensure that the mice maintain continuous swimming on the water’s surface, with no more than 5 s of inactivity. Exhaustion is defined when the mice show significant discoordination in swimming movements or when the head sinks into the water for 10 s without resurfacing.
The chronic restraint method: Place the mice in a custom-made restraint tube (20 cm long, with an inner diameter of 5.3 cm and an outer diameter of 6.0 cm), keeping them at a level of tension that does not induce strong resistance, with the head facing the ventilation hole. Observe and record the mice’s restraint condition, restraining each mouse for 3 h at a time. During the modeling period, observe the general condition of the mice daily, including body shape, fur color, activity, mental state, and daily water consumption. Measure the daily food intake of the mice and record any changes.
Morris water maze test
Following the completion of modeling, the Morris water maze test is performed, consisting of a spatial navigation test and a probe test. The experiment includes four daily training sessions over 4 days. The inner wall of the water maze is coated black, and the pool is surrounded by black opaque PVD reflective curtains. A thermometer is placed in the pool to monitor the water temperature, and hot water is prepared to maintain the water temperature at (25 ± 2)°C. The platform is placed in the center of the third quadrant and submerged 2 cm below the water’s surface. The spatial navigation test begins 24 h after the end of the modeling and treatment and lasts for 4 days. It trains the mice to explore and memorize the platform location within the water maze. At the start of each trial, the mice are placed at a randomly selected entry point with their heads facing the pool wall. The time it takes for each mouse to reach the platform (escape latency) and the movement trajectory are recorded, with the data from day 4 being used to assess the learning and memory abilities of the mice. Twenty-four hours after the last spatial navigation trial, the probe test is performed. The platform is removed, and the first quadrant is chosen as the entry point. The mice are placed in the water facing the pool wall, and the number of times the mice cross the platform’s previous location and the time spent in the target quadrant within 2 min are measured.
Open field test
The open field test is primarily used to evaluate the animals’ autonomous exploratory behavior and emotional responses, such as anxiety, in a novel environment. It is an important behavioral indicator used to assess the success of the CFS model in terms of psychological fatigue. Each mouse is placed in the center grid of the open field test box, and its activity is observed for a set period. The total distance traveled and the time spent in the center area are recorded.
Grip strength and Rotarod tests
Mouse grip strength test: Gently grasp the mouse by the tail, ensuring its limbs are suspended. Allow the mouse’s forelimbs to gently contact the metal rod or grid of the grip strength meter. Once the mouse naturally grasps the rod or grid, slowly pull the mouse backward until it releases the rod or grid. The instrument records the maximum grip strength at the point of release. The hind limb grip test is similar to the forelimb test but involves the hind limbs contacting the metal rod, and the maximum grip strength is recorded upon release. Each mouse is tested three times, and the average value is used as the final result.
Rotarod test: Prior to the formal experiment, the mice undergo a few adaptive training sessions to ensure familiarity with the apparatus and the experimental environment, usually consisting of 1–2 sessions per mouse, each lasting 1–2 min at a low speed (5 rpm). The mice are placed on the rotating rod, and the rotation begins. The speed is either fixed (20 rpm) or set to accelerate (e.g., starting at 5 rpm and increasing by 1–2 rpm per minute). The time each mouse maintains balance on the rotating rod is recorded, typically in seconds. Each mouse is tested three times, and the average time is calculated.
HE staining and Nissl staining
Mice are euthanized by CO₂ inhalation, followed by cardiac perfusion with 4% paraformaldehyde (PFA) buffer to fix the brain tissue thoroughly. After fixation, the brain is extracted and placed in 4% PFA for further fixation, typically for 24–48 h. The hippocampal region, especially the CA1 region, which is associated with learning and memory, is selected for sectioning. The fixed tissue is embedded in paraffin, sectioned into slices 5–10 microns thick, and mounted on slides. The slides are then deparaffinized, rehydrated, and stained using hematoxylin and eosin (HE). After staining, the slides are observed under a Motic microscope to assess tissue damage. Neuronal morphology, arrangement, and pathological changes such as nuclear shrinkage, dissolution, and inflammation can be observed in the hippocampal CA1 region. In normal hippocampal CA1 sections, neurons appear orderly, with round or oval nuclei and evenly distributed staining. In pathological models, neuronal nuclear shrinkage, dissolution, nuclear displacement, cytoplasmic degeneration, and tissue inflammation or glial proliferation may be observed.
Nissl staining: Paraffin sections are deparaffinized, rehydrated, and stained with toluidine blue for 5 min. After rinsing with water, 1% acetic acid is used for slight differentiation, followed by tap water washing to terminate the reaction. After washing, the slides are dried in an oven and cleared with xylene for 5 min, then mounted with neutral resin. The stained sections are observed under a microscope, focusing on the pathological morphology of neurons in the hippocampal CA1 region. CaseViewer software is used to view the images, and neuronal numbers are counted in random fields at a 50 μm scale. ImageJ software is used to count Nissl-positive cells in the ischemic side of the hippocampal CA1 region, and the degree of neuronal damage is compared between groups.
Immunohistochemistry
Frozen brain tissue stored at −80 °C is sectioned, then deparaffinized, rehydrated, and washed. The sections are incubated in sodium citrate buffer and blocked with BSA. Primary antibodies are added, and the sections are incubated overnight at 4 °C. The following day, secondary antibodies are applied at room temperature. After washing with PBS, DAB working solution is added for color development. Positive results appear as brown-yellow under a fluorescence microscope. ImagePro Plus software is used for quantitative analysis of PKM2 protein in the images, calculating the average optical density.
Western blot
After homogenizing brain tissues from each group, the samples are mixed with mammalian tissue protein lysis buffer and centrifuged at 1000 rpm for 20 min at 4 °C to collect the supernatant. Protein concentrations are determined using the BCA protein assay, followed by normalization. The samples are heated to denature the proteins and subjected to SDS-PAGE, membrane transfer, blocking, and washing. The membranes are incubated with diluted primary antibodies overnight at 4 °C. The next day, the membranes are washed three times with phosphate-buffered saline (PBST), each time for 15 min. Subsequently, the membranes are incubated with diluted rabbit anti-mouse secondary antibodies (1:1000), washed again, and developed using enhanced chemiluminescence (ECL) for imaging. ImageJ software is used for data analysis, with GAPDH as the internal reference, to calculate relative protein expression levels.
Elisa
After blood collection from the mice, the serum is separated using a centrifuge (typically at 3000 rpm for 10 min). The serum can be used directly for ELISA or stored at −80 °C for future use. After brain tissue collection, the tissue is homogenized using physiological saline (PBS) or an appropriate lysis buffer. After homogenization, the supernatant is collected by high-speed centrifugation (12000 rpm for 10–15 min) and used as the sample for ELISA, or stored at −80 °C for later use.
For lactate detection, a Lactate Assay Kit is used, based on the reaction of lactate oxidase. Prepare the working solution according to the kit instructions. Serum or brain tissue homogenates are diluted to the appropriate concentration, added to a 96-well plate, and lactate assay reagents are added. After the reaction, a colorimetric reaction is generated, with the intensity of the color proportional to the lactate concentration. Absorbance is read using a microplate reader at the specified wavelength (usually 450 nm). For IL-1β and TNFα detection, ELISA is the most commonly used method: Purchase corresponding IL-1β and TNFα ELISA kits. Follow the instructions provided with the kit to prepare all reagents, standards, and samples. Dilute the prepared brain tissue homogenates to an appropriate concentration. Add 100µL of the sample to each well of the 96-well ELISA plate, including standards and negative controls. After incubating with the capture antibody for 1–2 h and washing, add HRP-conjugated secondary antibody, followed by substrate (TMB) to produce a color reaction. The intensity of the reaction is proportional to the target protein’s concentration. After stopping the reaction with sulfuric acid, absorbance is measured using a microplate reader at 450 nm.
In vitro cell experiment
Human hippocampal neuron cell line ReNcell VM is divided into six groups: NC group (①); lactate group (②), with 15 mM lactate stimulation for 24 h; PKM2-OE group (③), with lactate stimulation and PKM2-OE lentiviral transfection; PKM2-OE + 3-BP group (④), treated with PKM2-OE and glycolysis inhibitor 3-BP at a concentration of 100 µM for 24 h; PKM2-OE + 3-BP + SN-50 group (⑤), treated with PKM2-OE + 3-BP and NF-κB inhibitor SN-50 at a concentration of 50 µM for 12 h; and PKM2-OE + 3-BP + activator-1 group (⑥), treated with PKM2-OE + 3-BP and NF-κB activator activator-1 at a concentration of 50 µM for 12 h. After collecting the supernatants, ELISA is used to detect IL-1β and TNFα levels. Glycolysis levels in the six groups are measured using a glycolysis assay kit (Shanghai QiYuan Biotechnology Co., Ltd.), and lactate levels are measured using a lactate assay kit (Shanghai XinFan Biotechnology Co., Ltd.).
JC-1 Immunofluorescence experiment
Samples from the six groups are fixed on slides. The cells are fixed with 4% paraformaldehyde (PFA) at room temperature for 10–20 min, then washed three times with PBS for 5 min each. The cells are stained with a 10 µg/mL JC-1 solution. After staining, the cells are gently washed 2–3 times with preheated PBS to remove unbound JC-1 dye. Positive control: Cells treated with FCCP (a mitochondrial uncoupler) to eliminate mitochondrial membrane potential, showing green fluorescence (monomer form) under a microscope. Negative control: Cells with normal culture conditions, showing high mitochondrial membrane potential with predominantly red fluorescence (aggregate form). Observations are made under a fluorescence microscope. Healthy cells show high mitochondrial membrane potential with predominantly red fluorescence, while damaged or apoptotic cells show reduced mitochondrial membrane potential with green fluorescence.
Statistical analysis
Statistical analysis is performed using SPSS 23.0 software. Quantitative data conforming to a normal distribution are expressed as mean ± standard deviation (SD). Independent samples t-test is used for inter-group comparisons. Differences are considered statistically significant at *P < 0.05, highly significant at **P < 0.01, and not significant at ns.
Results
Exploring the biological significance of these differentially expressed genes (degs)
Through in-depth analysis and screening of the data, a total of 1373 significantly differentially expressed genes were identified, including 638 upregulated and 735 downregulated genes. To further explore the biological significance of these DEGs, we used the ClusterProfiler tool to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses24. The results showed that these DEGs were significantly enriched in multiple biological pathways.
In the GO analysis, we found that these differentially expressed genes were significantly enriched in important biological processes such as neuron projection guidance, axon guidance, and muscle contraction. In the KEGG pathway analysis, the results revealed that these DEGs were significantly enriched in key pathways, including the transforming growth factor-beta (TGF-beta) signaling pathway, protein digestion and absorption, and neuroactive ligand-receptor interaction (Fig. 1). These findings provide important clues for understanding the role of these genes in biological processes.

A. Volcano plot of differential genes; B. Heatmap of differential genes; C. Bar plot of GO enrichment analysis; D. Bubble plot of GO enrichment analysis; E. Chord diagram of KEGG enrichment analysis; F. Circle plot of KEGG enrichment analysis.
PKM2 worsens cognitive impairment in chronic fatigue syndrome mice
Compared with the control group, the cognitive function of mice in the Model group significantly declined. Compared with the Model group, the cognitive function of mice in the Model + PKM2-OE group improved significantly, while cognitive function in the Model + PKM2-KD group worsened (Fig. 2A). The control group mice had normal physical abilities, and there were no significant differences in physical abilities between the Model, Model + PKM2-OE, and Model + PKM2-KD groups (Fig. 2B).

PKM2 Worsens Cognitive Function in Chronic Fatigue Syndrome Mice. A. Morris water maze test for control, Model, Model+PKM2-OE, and Model+PKM2-KD groups. B. Open field test for the four groups of mice. C. Grip strength test for the four groups. D. Rotarod test for the four groups. (*P<0.05, **P<0.01, ns indicates no difference).
PKM2 increases lactic acid and inflammatory factor levels in brain tissue
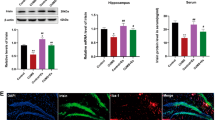
In HE and Nissl staining (Fig. 3A and B), the control group showed a normal hippocampal CA1 region with neatly arranged neurons, round or oval nuclei, even staining, and light pink cytoplasm. In the Model group, neuron karyopyknosis, karyolysis, or karyokinesis, along with cytoplasmic degeneration, inflammation, and astrocyte proliferation were observed. In the Model + PKM2-OE group, neuronal arrangement was more disordered compared to the Model group, while in the Model + PKM2-KD group, the condition improved. Compared to the control group, PKM2 levels increased significantly in the Model group; in the Model + PKM2-OE group, PKM2 levels were even higher; and in the Model + PKM2-KD group, PKM2 levels were lower compared to the Model group (Fig. 3C). In Western blot (Fig. 3D and F), PKM2, H4K12la, H4, IL-1β and TNFα, DRP1 expression was increased and PGC1α, MFN1 expression levels were decreased in the model group compared to the control group. Compared with the model group, PKM2, H4K12la, H4, IL-1β and TNFα, DRP1 protein levels were higher and PGC1α, MFN1 expression levels were decreased in the model + PKM2-OE group. While PKM2, H4K12la, H4, IL-1β and TNFα, DRP1 expression levels were decreased and PGC1α, MFN1 expression levels were increased in the model + PKM2-KD group. Compared to the Model group, the Model + PKM2-OE group had even higher levels of these proteins, while the Model + PKM2-KD group had decreased expression levels. Serum lactic acid levels were elevated in the Model group compared to the control group, with no significant differences between the Model + PKM2-OE and Model + PKM2-KD groups. Brain tissue homogenate lactic acid levels were higher in the Model group compared to the control group; the Model + PKM2-OE group showed further increases in brain tissue lactic acid levels, while the Model + PKM2-KD group showed a decrease (Fig. 3E). In the ELISA assay (Fig. 3G), brain tissue homogenates of the Model group showed elevated levels of IL-1β and TNFα compared to the control group; these levels were even higher in the Model + PKM2-OE group and lower in the Model + PKM2-KD group compared to the Model group.

PKM2 Increases Lactate and Inflammatory Cytokine Levels in Brain Tissue. A. HE staining of brain tissues in the four groups of mice. B. Nissl staining of brain tissues in the four groups. C. Immunohistochemistry staining of PKM2 in brain tissues of the four groups. D. Western blot analysis of PKM2, H4K12la, and H4 protein expression in brain tissues of the four groups. E. ELISA detection of serum lactate levels and lactate levels in brain tissue homogenates of the four groups. F. Western blot analysis of IL-1β, TNFα, PCG1α, MFN1 and DRP1 protein expression in hippocampal tissues of the four groups. G. ELISA detection of IL-1β and TNFα levels in brain tissue homogenates of the four groups. (*P<0.05, **P<0.01, ns indicates no difference).
PKM2 promotes glycolysis, lactate accumulation, and h4k12la/nf-κb-mediated neuroinflammation
In the in vitro Western blot analysis (Fig. 4A), compared to the NC group, the lactate group showed increased expression of PKM2, LDHB, H4K12la, P-P65, IL-1β, and TNFα proteins. Compared to the lactate group, the PKM2-OE group showed higher expression of these proteins. Compared to the PKM2-OE group, the PKM2-OE + 3-BP group showed no significant change in PKM2, but decreased expression of LDHB, H4K12la, P-P65, IL-1β, and TNFα. Compared to the PKM2-OE + 3-BP group, the PKM2-OE + 3-BP + SN-50 group showed no significant changes in PKM2, LDHB, or H4K12la, but decreased expression of P-P65, IL-1β, and TNFα. Compared to the PKM2-OE + 3-BP group, the PKM2-OE + 3-BP + activator-1 group showed no significant changes in PKM2, LDHB, or H4K12la, but increased P-P65, IL-1β, and TNFα expression. In the ELISA assay (Fig. 4B), compared to the NC group, the lactate group showed increased levels of IL-1β and TNFα; these levels were higher in the PKM2-OE group compared to the lactate group. IL-1β and TNFα levels decreased in the PKM2-OE + 3-BP group compared to the PKM2-OE group and further decreased in the PKM2-OE + 3-BP + SN-50 group compared to the PKM2-OE + 3-BP group. IL-1β and TNFα levels increased in the PKM2-OE + 3-BP + activator-1 group compared to the PKM2-OE + 3-BP group. In glycolysis and lactate level assays (Fig. 5A and B), the lactate group showed increased glycolysis and lactate levels compared to the NC group; these levels were even higher in the PKM2-OE group compared to the lactate group, while they decreased in the PKM2-OE + 3-BP group. There were no significant differences in glycolysis or lactate levels among the PKM2-OE + 3-BP, PKM2-OE + 3-BP + SN-50, and PKM2-OE + 3-BP + activator-1 groups, but all were lower than in the lactate group.
In immunofluorescence analysis (Fig. 5C), the lactate group showed increased green fluorescence compared to the NC group; the PKM2-OE group had even more green fluorescence than the lactate group. The PKM2-OE + 3-BP group had less green fluorescence compared to the PKM2-OE group, and the PKM2-OE + 3-BP + SN-50 group had even less green fluorescence compared to the PKM2-OE + 3-BP group. The PKM2-OE + 3-BP + activator-1 group showed increased green fluorescence compared to the PKM2-OE + 3-BP group.

PKM2 Enhances Glycolysis, Lactate Accumulation, and H4K12la/NF-κB-Mediated Neuroinflammation. A. Western blot analysis of PKM2, LDHB, H4K12la, P-P65, IL-1β, and TNFα protein expression in six groups of hippocampal cells: NC group (①), Lactate group (②), PKM2-OE group (③), PKM2-OE+3-BP group (④), PKM2-OE+3-BP+SN-50 group (⑤), and PKM2-OE+3-BP+activator-1 group (⑥).B. ELISA detection of IL-1β and TNFα levels in the supernatants of the six groups of hippocampal cells. (*P<0.05, **P<0.01, ns indicates no difference).

A. Glycolysis level detection using a glycolysis assay kit in the six groups. B. ELISA detection of lactate levels in the six groups.C. JC-1 assay to detect mitochondrial function in hippocampal cells of the six groups. (*P<0.05, **P<0.01, ns indicates no difference).
Discussion
The accelerated work pace and increased labor intensity in modern society have made prolonged excessive physical exertion a common phenomenon for many individuals. However, the potential impact of prolonged overexertion on human health, especially on brain function, has not received sufficient attention. Existing studies suggest that excessive physical exertion not only leads to the production of large amounts of lactic acid in muscles but that long-term lactic acid accumulation may, through mechanisms such as epigenetic regulation and neuroinflammation, impair neuronal function and subsequently affect cognitive function25. This paper explores how lactic acid metabolism, H4K12la histone lactylation, enhanced glycolysis, the NF-κB signaling pathway, and neuroinflammation contribute to cognitive impairment and accelerate the progression of chronic fatigue syndrome (CFS).
During intense physical labor, the body primarily generates energy through anaerobic glycolysis, a process that produces large amounts of lactic acid. Lactic acid accumulation not only causes muscle fatigue but can also cross the blood-brain barrier and enter brain tissue26. In the brain, lactic acid serves as both an energy source and a metabolic signaling molecule27. However, when lactic acid accumulates in the brain, its negative effects begin to manifest. Prolonged lactic acid buildup increases the brain’s acidic environment, thereby affecting neuronal function and the brain’s metabolic balance28.
The role of lactic acid is not limited to metabolism; it also mediates epigenetic regulatory mechanisms, particularly histone lactylation. Lactylation is a novel post-translational modification that allows lactic acid to directly influence gene expression29. In the brain, lactic acid accumulation promotes the production of H4K12la (histone H4 lysine 12 lactylation)30. H4K12la is an important epigenetic marker that regulates the expression of metabolism-related genes, especially the pyruvate kinase M2 (PKM2) gene16. PKM2 is a key enzyme in the glycolysis pathway, accelerating the glycolysis process by catalyzing the conversion of phosphoenolpyruvate to pyruvate31. The H4K12la-mediated enhancement of PKM2 expression is one of the key mechanisms by which excessive physical exertion leads to lactic acid accumulation16. The upregulation of PKM2 not only accelerates the glycolysis pathway but also, through the synergistic actions of LDHA (lactate dehydrogenase A) and LDHB (lactate dehydrogenase B), further increases lactic acid production32. Additionally, HK-2 (hexokinase-2) may play an important role in this process by initiating glucose metabolism and further driving lactic acid production33. This positive feedback loop in the metabolic pathway leads to further lactic acid accumulation in the brain, exacerbating the acidic environment and causing an imbalance in brain energy metabolism34.
Excessive lactic acid accumulation affects not only metabolism but also activates a series of inflammatory signaling pathways, among which the NF-κB pathway is particularly important35. In renal tissues, H4K12la enhances NF-κB activity, leading to the upregulation of inflammatory factors such as IL-1β (interleukin-1β) and TNF-α (tumor necrosis factor-α)36. We speculate that modification of H4K12la in the brain also promotes the secretion of inflammatory factors. These inflammatory factors are major drivers of neuroinflammation, and prolonged neuroinflammation disrupts normal neuronal function37. Mitochondria are crucial for maintaining normal neuronal function, as they are responsible for energy production, as well as regulating calcium balance and apoptosis38. During the neuroinflammatory response, the continuous release of inflammatory factors triggers oxidative stress, increasing the production of reactive oxygen species (ROS). This oxidative stress not only directly damages the mitochondrial membrane but also disrupts mitochondrial DNA, leading to mitochondrial dysfunction38. The weakening of mitochondrial function directly affects the energy supply to nerve cells, further reducing the efficiency of neural signal transmission38. There is a close relationship between mitochondrial damage and cognitive decline39. Research indicates that mitochondrial dysfunction is a common pathological feature of various neurodegenerative diseases, including Alzheimer’s and Parkinson’s38. In chronic fatigue syndrome induced by excessive physical labor, the combined effects of lactic acid accumulation and neuroinflammation lead to mitochondrial damage, subsequently impairing hippocampal neuronal function17. The hippocampus, as the brain’s center for memory and learning, is directly affected by its damage, leading to a decline in memory and cognitive abilities39.
Chronic fatigue syndrome (CFS) is a syndrome characterized by persistent fatigue, often accompanied by cognitive impairments such as reduced attention and memory40. Lactic acid accumulation, H4K12la histone lactylation, and NF-κB-mediated neuroinflammation caused by excessive physical labor may be one of the core mechanisms in the development of CFS. Enhanced glycolysis and excessive lactic acid production lead to metabolic disorders, while sustained inflammation further damages mitochondrial function, exacerbating the state of energy deficiency41. Based on the above mechanisms, we can speculate that long-term excessive physical exertion not only affects physical strength but also has the potential to negatively impact cognitive function. After crossing the blood-brain barrier and entering brain tissue, lactic acid regulates PKM2 expression through H4K12la and activates the NF-κB signaling pathway, triggering neuroinflammation and mitochondrial damage, ultimately leading to neuronal apoptosis and cognitive impairment. This mechanism is particularly evident in chronic fatigue syndrome, where cognitive function may gradually decline in individuals who engage in prolonged excessive physical labor, manifesting as memory loss, difficulty concentrating, and slowed thinking.
In summary, PKM2 in hippocampal cells can lead to mitochondrial damage and accelerate the progression of chronic fatigue syndrome by enhancing glycolysis, lactic acid accumulation, and H4K12la/NF-κB-mediated neuroinflammation (Fig. 6). This study systematically analyzes the biological mechanisms of lactic acid metabolism, histone lactylation, enhanced glycolysis, neuroinflammation, and mitochondrial dysfunction, elucidating the potential impact of excessive physical labor on brain function and the progression of chronic fatigue syndrome. Lactic acid accumulation not only triggers metabolic disorders but also causes neuronal damage through epigenetic regulation and inflammatory signaling pathways. These mechanisms, acting together, may lead to a decline in cognitive function in individuals who engage in long-term excessive labor. Future research should further explore these mechanisms to provide effective prevention and treatment strategies for those affected by excessive labor.

PKM2 in Hippocampal Cells Leads to Mitochondrial Damage and Accelerates the Progression of Chronic Fatigue Syndrome by Enhancing Glycolysis, Lactate Accumulation, and H4K12la/NF-κB-Mediated Neuroinflammation.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Xie, F. et al. Effects of the prolong life with nine turn method (Yan Nian Jiu Zhuan) qigong on brain functional changes in patients with chronic fatigue syndrome in terms of fatigue and quality of life. Front. Neurol. 13, 866424 (2022).
Morris, G. & Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab. Brain Dis. 29 (1), 19–36 (2014).
Tomas, C. et al. The effect of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) severity on cellular bioenergetic function. PloS One. 15 (4), e0231136 (2020).
Wang, Y. et al. PKM2 functions as a histidine kinase to phosphorylate PGAM1 and increase Glycolysis shunts in cancer. EMBO J. 43 (12), 2368–2396 (2024).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Sci. (New York NY). 324 (5930), 1029–1033 (2009).
Rusin, A. et al. Commonalities in the features of Cancer and chronic fatigue syndrome (CFS): evidence for Stress-Induced phenotype instability?? Int. J. Mol. Sci. 23(2). 691 (2022).
Naviaux, R. K. Metabolic features of the cell danger response. Mitochondrion 16, 7–17 (2014).
Wei, Y. et al. Progress in research on the effect of melatonin on postoperative cognitive dysfunction in older patients. Front. Aging Neurosci. 14, 782358 (2022).
Fluge, Ø. et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. 1 (21), e89376 (2016).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574 (7779), 575–580 (2019).
Li, K. et al. Netrin-1 ameliorates postoperative Delirium-Like behavior in aged mice by suppressing neuroinflammation and restoring impaired Blood-Brain barrier permeability. Front. Mol. Neurosci. 14, 751570 (2021).
Hornig, M. et al. Cytokine network analysis of cerebrospinal fluid in myalgic encephalomyelitis/chronic fatigue syndrome. Mol. Psychiatry. 21 (2), 261–269 (2016).
Sun, H. et al. The role of cancer-associated fibroblasts in tumorigenesis of gastric cancer. Cell Death Dis. 13 (10), 874 (2022).
Wei, Y., Giunta, S. & Xia, S. Hypoxia in aging and aging-Related diseases: mechanism and therapeutic strategies. Int. J. Mol. Sci. 23(15). 8165 (2022).
Lian, B. et al. SIRT1 improves lactate homeostasis in the brain to alleviate parkinsonism via deacetylation and Inhibition of PKM2. Cell. Rep. Med. 5 (8), 101684 (2024).
Pan, R. Y. et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metabol. 34 (4), 634–648e636 (2022).
Jiang, L. et al. TAK–242 exerts a neuroprotective effect via suppression of the TLR4/MyD88/TRIF/NF–κB signaling pathway in a neonatal hypoxic–ischemic encephalopathy rat model. Mol. Med. Rep. 22 (2), 1440–1448 (2020).
Castro-Marrero, J. et al. Does coenzyme Q10 plus selenium supplementation ameliorate clinical outcomes by modulating oxidative stress and inflammation in individuals with myalgic encephalomyelitis/chronic fatigue syndrome?? Antioxid. Redox. Signal. 36 (10–12), 729–739 (2022).
He, J. et al. Upregulated mitochondrial dynamics is responsible for the procatabolic changes of chondrocyte induced by α2-Adrenergic signal activation. Cartilage 15 (4), 440–452 (2024).
Fu, J. et al. Regeneration of glomerular metabolism and function by podocyte pyruvate kinase M2 in diabetic nephropathy. JCI Insight 7(5). e155260 (2022).
Rahman, A. & Fazal, F. Blocking NF-κB: an inflammatory issue. Proc. Am. Thorac. Soc. 8 (6), 497–503 (2011).
Singh, A. et al. Effect of natural and synthetic antioxidants in a mouse model of chronic fatigue syndrome. J. Med. Food. 5 (4), 211–220 (2002).
Chiba, S. et al. Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog. Neuro-psychopharmacol. Biol. Psychiatry. 39 (1), 112–119 (2012).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–677 (2024).
Wu, J. et al. Regular exercise ameliorates high-fat diet-induced depressive-like behaviors by activating hippocampal neuronal autophagy and enhancing synaptic plasticity. Cell Death Dis. 15 (10), 737 (2024).
Cai, M. et al. Lactate is answerable for brain function and treating brain diseases: energy substrates and signal molecule. Front. Nutr. 9, 800901 (2022).
Zhang, F. et al. Lactylation of histone by BRD4 regulates astrocyte polarization after experimental subarachnoid hemorrhage. J. Neuroinflamm. 21 (1), 186 (2024).
Chen, X. et al. The regulatory effects of lactic acid on neuropsychiatric disorders. Discover Mental Health. 2 (1), 8 (2022).
Sun, Z. et al. Comprehensive analysis of lactate-related gene profiles and immune characteristics in lupus nephritis. Front. Immunol. 15, 1329009 (2024).
Li, F. et al. Positive feedback regulation between Glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma. Mol. Cancer. 23 (1), 90 (2024).
Cheng, Q. et al. Current Advances on Nanomaterials Interfering with Lactate Metabolism for Tumor Therapy. Advanced science (Weinheim, Baden-Wurttemberg, Germany) 11(3):e2305662. (2024).
Huang, M. et al. The immune checkpoint TIGIT/CD155 promotes the exhaustion of CD8 + T cells in TNBC through glucose metabolic reprogramming mediated by PI3K/AKT/mTOR signaling. Cell. Communication Signaling: CCS. 22 (1), 35 (2024).
Li, J. et al. Upregulated hexokinase 2 expression induces the apoptosis of dopaminergic neurons by promoting lactate production in Parkinson’s disease. Neurobiol. Dis. 163, 105605 (2022).
Jiang, M. et al. Multimodal imaging evaluation of early neurological deterioration following acute ischemic stroke. Quant. Imaging Med. Surg. 14 (7), 4763–4778 (2024).
Yang, Y. et al. Lactate and lysine lactylation of histone regulate transcription in cancer. Heliyon 10 (21), e38426 (2024).
Wang, Y. et al. The glycolytic enzyme PFKFB3 drives kidney fibrosis through promoting histone lactylation-mediated NF-κB family activation. Kidney Int. 106 (2), 226–240 (2024).
Latham, A. S. et al. Gliosis, misfolded protein aggregation, and neuronal loss in a guinea pig model of pulmonary tuberculosis. Front. NeuroSci. 17, 1157652 (2023).
Zhao, J. et al. Protopanaxadiols eliminate behavioral impairments and mitochondrial dysfunction in Parkinson’s disease mice model. Neurochem. Res. 49 (7), 1751–1761 (2024).
Armstrong, C. W. et al. Metabolism in chronic fatigue syndrome. Adv. Clin. Chem. 66, 121–172 (2014).
Robinson, L. J. et al. Impairments in cognitive performance in chronic fatigue syndrome are common, not related to co-morbid depression but do associate with autonomic dysfunction. PloS One. 14 (2), e0210394 (2019).
Xia, N. et al. Protective effects of Lycium ruthenicum Murray against acute alcoholic liver disease in mice via the Nrf2/HO-1/NF-κB signaling pathway. Pharmaceuticals (Basel Switzerland) 17(4). 497 (2024).
Acknowledgements
none
Funding
none.
Author information
Authors and Affiliations
Contributions
Meng Sun and Xinwen Zhang participated in the conception and design of the study, organized the database, and wrote the first draft of the manuscript. Xinli Feng performed a statistical analysis of the data. Meng Sun, Xinwen Zhang and Lu Liang conducted the experiment. All authors participated in the revision of the manuscript, read and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Patient consent for publication
Not involve
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This study was guided and approved by the Ethics Committee of the Affiliated Hospital of Hebei University (Ethics approval number: HBDX202310030005).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, M., Zhang, X., Feng, X. et al. PKM2 accelerated the progression of chronic fatigue syndrome via promoting the H4K12la/ NF-κB induced neuroinflammation and mitochondrial damage. Sci Rep 15, 10772 (2025). https://doi.org/10.1038/s41598-025-93313-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93313-w
