
Abstract
m6A, m5C and m7G are common types of RNA methylation modifications that are widely involved in key mechanisms regulating malignancy. However, the role of RNA methylation-related genes in the immune microenvironment of bladder cancer (BLCA) remains elusive. In this study, we established RNA methylation molecular subtypes by analyzing the TCGA and GEO datasets. Risk model and nomogram were constructed by LASSO and multivariate Cox regression analysis and validated by external datasets. Genetic variations, functional enrichment analysis and immune cell infiltration were analyzed. The expression levels of hub genes were detected by real-time polymerase chain reaction (qRT-PCR) and immunohistochemistry (IHC). The effect of FN1 on cellular function was determined using experimental assays. Finally, we identified a 7-gene signature associated with BLCA prognosis. GSE19423 validated the predictive value of the risk model. The IMvigor210 data showed the model had promising predictive efficacy for BLCA immunotherapy. Significant differences in biological function, immune cell infiltration and drug sensitivity were observed between high- and low-risk groups. Furthermore, FN1 was upregulated in BLCA, as determined by qRT-PCR and IHC. Depletion of FN1 using siRNA impaired cell motility in T24 and 5637 cells. In conclusion, RNA methylation-related risk model can predict the prognosis, immune landscape and response to immunotherapy in BLCA. Among the 7-gene signature, FN1 is a pivotal gene that promotes the migration of bladder cancer cells.
Similar content being viewed by others


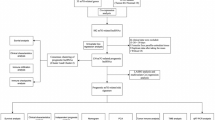
Introduction
Bladder cancer (BLCA) is a prevalent urinary malignancy globally and contributes to significant morbidity and mortality worldwide1. According to statistics, over 500,000 new cases of bladder cancer are reported annually, and approximately 210,000 cases result in death each year2. BLCA is divided into non-muscle invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC) according to their invasiveness, with most cases being NMIBC3. Despite adequate surgical intervention and intravesical chemotherapy, NMIBC patients are at risk of recurrence, with 50% of cases relapsing within a year, and 30% eventually progressing to MIBC4. The 5-year survival rate for patients with MIBC is less than 50%, while metastasis decreases the survival rate to 6% within the same period5. The existing treatment strategies for BLCA are inadequate, as they frequently encounter obstacles such as limited effectiveness of chemotherapy and the emergence of drug resistance. Consequently, the identification of novel biomarkers that can predict the prognosis of BLCA patients is of paramount importance. This is essential for not only developing more effective treatment approaches but also improving clinical outcomes for individuals with BLCA.
Over 170 distinct RNA modifications have been identified, and among them, RNA methylation has been shown to be pivotal in these processes6. The development of RNA methylation profiling technologies and high-throughput sequencing has enabled the characterization of different types of RNA methylation modifications including N1-methyladenosine (m1A), N6-methyladenosine (m6A), 5-methylcytosine (m5C), and 7-methylguanosine (m7G), which are involved in several essential biological properties7,8,9. Notably, m6A modification is the most frequent RNA modification type in eukaryotic cells and has been extensively studied10. All the findings highlight the potential of RNA methylation as a promising target for diagnostic and prognostic biomarkers for cancer patients.
This study aimed to assess the potential significance of m6A/m5C/m7G-related genes in the diagnosis and prognosis of BLCA. The transcriptome and mutation landscape, and clinical follow-up data were obtained from the Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. RNA methylation-associated risk models were constructed for prognostic prediction using univariate, multivariate, and a least absolute shrinkage and selection operator (LASSO) Cox regression analyses. The predictive accuracy of the model in terms of survival and immunotherapy response was then validated using the GEO dataset (GSE19423) and the IMvigor210 cohort, respectively. To test the adaptability of the model, we also assessed whether the model was able to distinguish differences in immune cell subpopulations in the tumor microenvironment (TME). The expression and distribution of seven genes in bladder cancer cells were analyzed by Single-cell RNA sequencing (scRNA-seq) analysis. Quantitative real-time polymerase chain reaction (qRT-PCR) was employed to ascertain the mRNA expression levels of the four hub genes. Additionally, in vitro assays including Transwell and scratch assays were performed to investigate the biological functions of FN1 in bladder cancer cells. The results of our study emphasize the noteworthy associations between the risk model, prognosis, immune cell infiltration, and response to immunotherapy. These findings hold potential implications for the development of more effective diagnostic and therapeutic approaches for BLCA patients.
Methods
Data collection and processing
Transcriptome data, somatic mutation data, and clinical data for BLCA patients were retrieved from the TCGA-BLCA cohort via the Cancer Genomics Data Portal (https://portal.gdc.cancer.gov/). Transcriptome matrix and clinical data from the GSE1350711 and GSE1942312 datasets were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). For the survival analysis, only tumor samples with complete clinical information were included, while patients with a survival period of less than 30 days were excluded. To ensure compatibility, the TCGA downloaded genes’ fragments per kilobase (FPKM) data were transformed into per million (TPM) data and merged with the GSE13507 data. To account for batch effects between the two datasets, the “ComBat” algorithm available in the “sva” package was applied. A total of 70 m6A/m5C/m7G regulatory genes were collected from relevant literature and previous studies and shown in Supplementary Tables 113,14.
Identification of molecular cluster by consensus unsupervised clustering analysis
In order to enhance the identification of distinct molecular subtypes in BLCA, we conducted consensus unsupervised clustering analysis using the expression data of the m6A/m5C/m7G genes. This clustering process was iterated for 1000 times to ensure robustness and reproducibility of the classification outcomes. Subsequently, the Kaplan-Meier survival analysis was performed on the clustering results to investigate potential differences in prognosis among the identified clusters. Moreover, to gain insights into the underlying biological functions associated with different clusters, we conducted Gene Set Variation Analysis (GSVA) to determine potential biological pathways enriched in each cluster. To further elucidate differences in immune function among the clusters, Single-sample Gene Set Enrichment Analysis (ssGSEA) was employed. Additionally, the CIBERSORT algorithm was used to evaluate variations in the extent of immune cell infiltration between the subclusters15.
Recognition of m6A/m5C/m7G‑related differentially expressed genes (DEGs)
Adjusted P < 0.05 and |log2 Fold-change| ≥ 1 were considered as significance criteria for m6A/m5C/m7G regulatory DEGs. Subsequently, to gain further insights into potential biological functions or signaling pathways, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted. Additionally, in order to identify DEGs with prognostic value, a univariate Cox regression analysis was performed to screen for genes that exhibited a close association with prognosis. To determine the optimal k-value, a consensus clustering algorithm was utilized to divide all samples into distinct clusters. Subsequently, the Kaplan-Meier survival curves were employed to compare the differences in prognosis between these different clusters. Moreover, in order to evaluate the clinical relevance of the clustering results, a comparison was made by integrating clinical features with the molecular subtypes.
Construction and validation of the prognostic risk model
Patients were randomly divided equally into a training set and a testing set, and the LASSO regression analysis was performed with the data from the training set to obtain representative gene set. A multivariate Cox regression analysis was conducted to develop a prognostic prediction risk model. The risk score formula was calculated as follows: risk score = ∑(the expression of the genes * coefi), where coefi represents the correlation coefficient of each gene. Subsequently, the patients from both the training and testing sets were categorized into high- and low-risk groups based on the median risk scores obtained from the training set. The accuracy of the risk model was evaluated by survival and receiver-operating characteristic (ROC) curves. External validation of the risk model was performed using the GSE19423 dataset. Finally, by integrating clinical characteristics with the risk scores, a nomogram scoring system was developed to facilitate digital predictions of 1-, 3-, and 5-year survival rates. This nomogram scoring system aims to assist clinicians in evaluating patient outcomes more effectively.
Assessing the predictive power of risk models for immunotherapy responses
Risk scores were computed for a cohort of 195 bladder cancer patients who received immunotherapy within the IMvigor210 dataset16. Based on the median risk scores, the patients were stratified into high- and low-risk groups. The prediction accuracy of the risk model was evaluated using the ROC curve. Furthermore, we investigated the potential correlation between the risk scores and the responsiveness to immunotherapy. This analysis aimed to provide insights into the association between the risk scores and the clinical outcomes and therapeutic responses of bladder cancer patients treated with immunotherapy.
Immune landscape analysis and tumor mutation burden (TMB)
The immune-related algorithm was used to calculate the characteristics of immune cell infiltration between different molecular types, immune types and the high- and low- risk groups, and the single sample Gene Set Enrichment Analysis (ssGSEA) method was used to calculate the immune cell activity and immune function of each sample. The ESTIMATE algorithm was used to calculate the immune score, the stromal score, the estimated score, and the tumor purity based on the fraction of stromal and immune cells. For immunotherapy, we compared four common immune checkpoints (PD-1, PD-L1, CTLA4, and LAG3) according to different clusters and risk groups. In addition, the potential clinical efficacy of immunotherapy in different groups was evaluated by calculating the Tumor Immune Dysfunction and Exclusion (TIDE) score. The higher the TIDE score, the higher the risk of immune evasion, indicating that patients are less likely to benefit from immunotherapy. Similarly, the higher the TMB, the more types and quantities of neoantigens are produced by the tumor cells, and the easier they are to recognize by the immune system and thus eliminated by the body. Therefore, we mapped gene mutations and compared the differences in TMB between the high- and low-risk groups by analyzing all gene mutation data in the sample. Finally, the mutation frequencies of the genes included in the prognostic model were further analyzed using the cBioPortal database (http://www.cbioportal.org/).
Drug sensitivity analysis
To identify potential chemotherapeutic agents, we used the R package “pRRophetic” to calculate the half maximal inhibitory concentration (IC50) to evaluate the response to chemotherapeutic agents in the high - and low-risk groups.
Single-cell RNA sequencing (scRNA-seq) analysis
The scRNA-seq data for two bladder cancer samples were retrieved from the GSE130001 cohort17. Initially, the expression matrix of the single-cell transcriptome data was acquired, and the “Seurat” R package was employed to construct a Seurat object. Subsequently, the data was normalized using the NormalizeData function, and dimensionality reduction clustering was performed using RunPCA and RunTSNE. Cell type annotation was then executed using the SingleR package. Finally, at the single-cell level, the expression signatures of genes associated with prognosis were analyzed. This comprehensive approach enabled the exploration of the prognostic-related gene expression patterns within the bladder cancer scRNA-seq dataset.
Cell culture, reagents and small interfering RNA transfection
T24, 5637, TCCSUP, UMUC-3 and SV-HUC-1 cells were purchased from the American Type Culture Collection (ATCC). T24, 5637 and UMUC-3 cells were cultured in recommended DMEM medium (Corning) with 10% fetal bovine serum (FBS), 100 mg/ml penicillin and 100 mg/ml streptomycin at 37℃ in 5% CO2. TSSSUP cells were cultured in completed RPMI-1640 medium (Corning). SV-HUC-1 cells were cultured in completed F-12 K medium (Sciencecell). GLPG0187 was purchased from Selleck.cn. Recombinant human fibronectin protein (His Tag) was purchased from SinoBiological.
Small interfering RNA (siRNA) was purchased from GenePharma Biological Technology (Shanghai, China). T24 and 5637 were transfected with siRNA (siFN1: 5′-GCAGCACAACUUCGAAUUA-3′; siFN2:5′-GGAGUUGAUUAUACCAUCA-3′) or negative control (5′-CUUACGCUGAGUACUUCGA-3′) to block FN1 expression using lipofectamine 2000 reagent (Invitrogen, USA) according to the manufacturer’s instructions.
Ethics approval
This study received approval from the Ethics Committee of the first affiliated Hospital of Chongqing Medical University (Approval number: 20182101). A written informed consent of each individual was obtained before the study. All procedures were performed in accordance with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Quantitative real‑time PCR (qRT‑PCR)
Total RNA of fresh clinical tissues (15 cases) and cells were extracted using TRIzol reagent (Invitrogen; ThermoFisher Scientific) according to the manufacturer’s instructions. The collected fresh samples were stored at − 80 °C until used for RNA isolation.
qRT‑PCR was performed using SYBR Green assays (Takara), which were operated on a Bio-Rad CFX96 real-time fluorescent quantitative PCR system. The relative expression of the genes to internal control (ACTB) was calculated using the 2−ΔCt method. The primer sequences were listed in Supplementary Table 2.
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded (FFPE) tissue sections collected from the Department of Pathology were deparaffinized and rehydrated in different concentration gradients of ethanol, immersed in sodium citrate buffer for antigen retrieval, incubated with 3% hydrogen peroxide, blocked with goat serum, incubated with primary antibody to FN1 (Proteintech, 66042-1-Ig) at 4 ℃ overnight; the samples were next incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at 37℃. Then signals were visualized using a diaminobenzidine kit (ZSGB‑BIO) and slides were counterstained with hematoxylin.
The IHC scores were assessed by two experienced pathologists and calculated based on the staining intensity and extent; the score criterion was described specifically in previous study18. The clinical information of twenty patients was recorded in Supplementary Table 3.
Cell scratch and transwell migration assays
The treated T24 and 5637 cells were cultured in six-well plates. A linear scratch was made using a pipette tip and the healing distance was measured at different times with a comparative microscope.
T24 (5 × 104 cells/per well) and 5637 (1 × 105 cells/per well) cells were cultured for 48 h in the upper chamber of 24-well plates with 200 µl serum-free medium. Then, 700 µl culture medium with 10% FBS was added to the lower chamber. T24 and 5637 cells were cultured at 37 °C with 5% CO2 for 24 h and 48 h, respectively. After fixing with 4% paraformaldehyde, staining with crystal violet, and washing with double distilled water, the upper chamber cells were calculated with five random areas per chamber.
Statistical analysis
All calculations and statistical analyses in this study were performed using the R software (version 4.2.0). SPSS20.0 software (Chicago, IL) was used for statistical analyses. All data are presented as the mean ± SD. Student’s t-test and one-way analysis of variance (ANOVA) was used to analyze the differences between two groups and multiple groups, respectively. The significance level was *P < 0.05, **P < 0.01, ***P < 0.005.
Results
The m6A/m5C/m7G regulatory genes are highly associated with the prognosis of BLCA
The detailed flow chart of this study was shown in Fig. 1. Seventy m6A/m5C/m7G regulatory genes were included in this study (Fig. 2A). The expression levels of 70 methylation genes were analyzed between normal and bladder tumor tissues in TCGA and GEO cohorts (Fig. S1A). Figure 2B showed the mutations spectrum of 70 genes in bladder cancer. A total of 201 out of 414 samples were mutated, and the top 5 mutated genes were EIF4G3, VIRMA, PRRC2A, METTL3, ZC3H13. Univariate Cox analysis showed that 49 m6A/m5C/m7G genes were associated with the prognosis of BLCA patients. A methylation network was generated based on these prognostic genes. As shown in Fig. 2C, the interaction and prognostic values of these methylation regulators were analyzed. The majority of the genes showed strong positive correlations with others, except for NSUN6, IGF2BP2, EIF4E2. We therefore propose that the crosstalk among m6A/m5C/m7G-regulated genes may play a crucial role in significantly influencing the RNA modification pattern of BLCA.

The work flow of this study.

Molecular clustering of methylation-related genes in BLCA. (A) 70 m6A/m5C/m7G-related genes involved in this research. (B) The mutation frequency of 70 methylation-related genes in BLCA patients from TCGA cohort. (C) The network of methylation-related gene interactions and prognostic value in BLCA. The line connecting two methylation-related genes represents their interactions; red lines represent positive correlations, blue lines represent negative correlations. (D) Unsupervised clustering of methylation-related genes and consensus matrix heat maps at k = 2. (E) Kaplan-Meier curve between two molecular clusters in patients with BLCA. (F) PCA analysis. (G,H) The immune-related function and the immune cell infiltration in the TME between the two molecular clusters. (I–L) Expression levels of CTLA4, PD-1, PD-L1 and LAG3 in cluster A and cluster B. *P < 0.05, **P < 0.01, ***P < 0.001.
Identification of m6A/m5C/m7G-related molecular clusters
Based on the expression profiles of the 70 m6A/m5C/m7G genes, we utilized consensus clustering method to categorize the BLCA samples into two distinct clusters, with 51.4% of the cases in cluster A and 48.6% of cases belonging to cluster B (Fig. 2D). We performed Kaplan-Meier analysis and identified that patients in cluster A exhibited a significantly higher overall survival (OS) than those in cluster B (Fig. 2E). In addition, Principal Component Analysis (PCA) result validated the accuracy of the clustering and confirmed the distinguishing ability of this classification algorithm (Fig. 2F). To investigate whether biological pathways and functions differ between the two clusters, GSVA analysis was carried out. Interestingly, cluster A was primarily characterized by metabolism-related pathways, such as “ARACHIDONIC_ACID_METABOLISM”, “FATTY_ACID_METABOLISM” and “RETINOL_METABOLISM” (Fig. S1B). We further investigated the relationship between the clusters and TME. Through the ssGSEA algorithm, we found that the immune-related functions or pathways were mainly enriched in cluster B, with higher immune infiltration (Fig. 2G). Also, CIBERSORT results showed that cluster A had more adaptive immune cells, such as T and B cells; whereas cluster B was infiltrated by NK cells and macrophages (Fig. 2H). Compared with cluster A, immune checkpoints (PD-1, PD-L1, CTLA4 and LAG3) were highly expressed in cluster B (P < 0.001), suggesting a subgroup where the patients were more likely to benefit from immunotherapy (Fig. 2I–L).
Identification of DEGs and classification of m6A/m5C/m7G regulatory gene clusters
After differential analysis between cluster A and cluster B, we identified 241 DEGs, including 96 down-regulated and 145 up-regulated genes (Fig. 3A). To investigate the biological processes and pathways associated with BLCA progression, GO and KEGG analyses were performed. GO analysis showed that these methylated genes are associated with the cell development and differentiation (Fig. 3B). The KEGG analysis exhibited a significant enrichment of several signaling pathways, such as PI3K-Akt pathway, cytokine-cytokine receptor interaction, IL-17 pathway and metabolism, demonstrating an important regulatory role of methylation in TME and carcinogenesis (Fig. 3C). In order to identify the prognostic factors, univariate Cox regression analysis was performed, and 119 OS-related genes were identified. Based on the prognostic genes, we utilized a consensus clustering algorithm to separate the BLCA samples into two gene clusters: I and II (Fig. 3D). Kaplan-Meier curves showed that patients in gene cluster I had a more favorable prognosis than gene cluster II (Fig. 3E). Combining clinical characteristics, molecular clusters, and gene clusters, we generated a heat map to visually show the differences in each subgroup (Fig. S1C). GSVA results displayed some pathways, such as NOD-like and Toll-like receptor signaling, are enriched in cluster II, suggesting a immune activation subgroup. In contrast, cluster I is predominantly associated with lipid metabolism-related pathways (Fig. S1D). To further explore the difference between the clusters and TME, we conducted ssGSEA and CIBERSORT algorithm. The ssGSEA results showed cluster II was involved in multiple immune processes or pathways (Fig. 3F). CIBERSORT data showed a significant difference in immune cell infiltration between the two gene clusters, with more M2 macrophages and neutrophils infiltrated in cluster II, while cluster I was enriched with more B cells, monocytes and plasma cells (Fig. 3G). Moreover, CTLA4, LAG3, PD-1 and PD-L1 were highly expressed in cluster II (Fig. 3H–K). This finding supports that different subgroups based on the m6A/m5C/m7G gene expression pattern exhibit different immune cell compositions, thereby affecting the immune response.

Identifying gene clusters based on the characteristics of DEGs of molecular cluster. (A) Based on the difference analysis of molecular clusters, a total of 241 DEGs were identified according to the criteria of adjusted P < 0.05 and |log2 Fold-change| ≥ 1. (B) GO enrichment analysis of DEGs between two gene clusters. (C) KEGG enrichment analysis of DEGs between two gene clusters. (D) Consensus matrix heatmap defining two gene clusters (k = 2). (E) Kaplan-Meier curve between two gene clusters. (F,G) The immune-related function and the immune cell infiltration in the TME between cluster I and cluster II. (H–K) The expression levels of CTLA4, LAG3, PD-1 and PD-L1 in gene cluster II were significantly higher than that in gene cluster I. *P < 0.05, **P < 0.01, ***P < 0.001.
Constructing and verifying the prognostic risk model
To further explore the prognostic value of the 119 DEGs obtained by univariate analysis, all patients were randomly divided into training and testing sets. LASSO and multivariate Cox regression analyses were performed on the training set to identify the most representative prognostic gene model (Fig. 4A,B). This risk model was composed of seven genes, including six high-risk genes (HMGA2, SERPINB7, HES2, FN1, UCHL1 and S100A9) and one low-risk gene (AIM2). Fig. S2A illustrates the coefficients for each gene in the prognostic risk model. Figure S2B graphically shows the internal distribution relations of the m6A/m5C/m7G molecular clusters, gene clusters, risk scores and survival status. Additionally, we observed a similar distribution of risk scores between molecular clusters and gene clusters, with higher risk in cluster B and cluster II (Fig. S2C,D). Most of the 70 m6A/m5C/m7G regulatory genes were highly expressed in high-risk group (Fig. S2E). According to the median risk score of the training set, the entire and testing sets were divided into high- and low-risk groups to validate the accuracy of the model. Kaplan-Meier analysis revealed that patients in the low-risk group had better OS (Fig. 4C–E). Figure S2F–H presents the distribution of risk scores, survival status and model gene expression profiles for the training set, testing set and entire set that patients with higher risk scores had shorter survival and higher mortality rates. The ROC curves showed that the 7-gene signature had high sensitivity and specificity for predicting clinical outcome. The area under the curve for the 5-year predictions for the training set, test set, and entire set were 0.727, 0.631, and 0.680, respectively (Fig. 4F–H).

Construction and validation of the risk model. (A,B) Construction of prognostic risk model by LASSO regression analysis. (C–E) Survival analysis of training set, testing set and entire set. (F–H) ROC curves for predicting performance of training set, testing set and entire set. (I,J) External validation set (GSE19423) were used to verify the accuracy and specificity of our prognostic risk model.
To further evaluate the predictive ability of our risk model, we calculated the risk scores of external validation sets (GSE19423) according to the calculation formula and divided them into high- and low-risk groups based on the median values. Consistent with our previous results, the survival analysis showed better outcomes in the low-risk group (Fig. 4I). The ROC curves showed that AUC remained over 0.6 when the model was utilized to predict 2- and 3-year OS (Fig. 4J), suggesting that the risk model performs well in predicting patient survival and might be a useful tool for clinicians.
Establishment of an integrated nomogram based on the risk model
We attempted to determine whether risk scores could be served as an independent prognostic factor in conjunction with other traditional clinical characteristics. After combining the results of univariate and multivariate Cox regression analyses, we confirmed that risk score, age, gender, T stage, and M stage were independent prognostic factors for BLCA patients (Fig. S3A,B). In addition, the relationship between these clinical parameters and the risk score was analyzed, showing that elder, high grade, and advanced T stage but not gender and metastasis, correlated with higher risk score (Fig. S3C–H).
Due to the inadequacy of gene signature in predicting the prognosis of BLCA patients in clinical practice, we designed a nomogram scoring system that combines risk scores with other traditional clinical characteristics to predict survival (Fig. 5A). This tool demonstrated a gradual decline in survival over time, which corresponds with clinical practice. Calibration curves were utilized to illustrate the precision and reliability of our predictive model in contrast to an ideal model (Fig. 5B). The decision curve analysis (DCA) graphically depicted the relationship between the threshold probability on the x-axis and the net benefit on the y-axis. As demonstrated in Fig. 5C,D, the integrated nomogram system and risk model outperformed other clinical parameters, which substantiates their accuracy and reliability in predicting patient outcomes.

Construction of a nomogram scoring system. (A) Construction of a prognostic nomogram based on several clinical parameters and risk scores to predict 1-year, 3-year, and 5-year outcomes. (B) Calibration curves of the nomogram for prediction of the accuracy of 1-, 3- and 5-year survival. (C,D) DCA and ROC curves were used to evaluate the accuracy of clinical parameters and risk scores as independent prognostic factors.
Biological function and mutation analyses in the high- versus low-risk groups
We performed GSEA on the basis of risk scores to investigate the potential biological processes involved in high- and low-risk groups. The results showed that the high-risk group had strong associations with cancer-promoting processes, including epithelial-mesenchymal transition (EMT), inflammatory response, hypoxia, and cell cycle (Fig. S4A). In order to further examine the relationship between risk score and somatic mutation, we compared the differences in frequent genetic mutations between high- and low-risk groups. The total mutation rate of the high-risk group was 93.97%, with the Top 3 mutated genes being TP53 (54%), TTN (41%), and KMT2D (28%) (Fig. S4B). In contrast, the low-risk group had TTN (46%), TP53 (42%), and MUC16 (27%) as the Top 3 mutated genes (Fig. S4C). Comparisons displayed that the TMB in both groups are similar, with only slight deviations in mutation frequencies. We also examined the interaction of somatic mutations between the two risk groups. The majority of genes co-occurred in both groups. Gene mutual exclusion was found in RB1-KDM6A in the low- risk group (Fig. S4D,E).
Immunotherapy response and drug sensitivity in different risk groups
Immune checkpoint inhibition therapy has the potential to provide rapid and durable outcomes in cancer patients. According to TCGA and GEO datasets, we calculated stromal and immune scores using the ESTIMATE algorithm and found that all scores were higher in the high-risk group (Fig. 6A). We next utilized the TIDE algorithm to investigate whether our constructed risk model could predict immunotherapy response. While the results indicated that the patients in the high-risk group had higher TIDE scores than that in the low-risk group (Fig. 6B). We also assessed the association between risk score and immune checkpoints, and found a positive correlation in the majority of checkpoints (Fig. 6C). Besides, patients in the high-risk group showed high expression levels of CD274, CTLA4, LAG3, PDCD1, CD44, and CD276 (Fig. 6D). To further explore the potential value of the risk model in predicting the response to immunotherapy, we verified its efficiency and sensitivity using the IMvigor210 cohort. BLCA patients with complete clinical data in the IMvigor210 were divided into the high-risk and low-risk groups based on median risk score. Survival curves revealed that patients in the low-risk group had a better prognosis than the high-risk group (P = 0.017), and the AUC under the 1-year ROC curve was 0.715 (Fig. 6E,F), suggesting that the model can be used to predict the efficacy of immunotherapy in BLCA patients. The type and abundance of infiltrating immune cells in TME play a vital role in the immune response. We therefore analyzed the difference of immune cell infiltration between high and low-risk groups. The majority of immune cells were observably related to the 7-gene signature (Fig. 6G). These genes exhibited positive correlations with activated memory T cells, NK cells, neutrophils and macrophages, while showing negative correlations with regulatory T cells, B cells, and monocytes. Additionally, Fig. 6H–O illustrated a positive correlation between risk score and M0 macrophages, neutrophils, and activated mast cells (P < 0.05).

Immunotherapy and the TME analysis. (A) Relationship between risk score and TME. (B) Relationship between risk score and TIDE score. (C) Correlation between risk score and common immune checkpoint. (D) Correlation between immune checkpoint and risk groups. (E,F) Validation of the predictive value of the risk model for immunotherapy response in the IMvigor 210 dataset. (G) The association between the seven prognostic genes and immune cell abundance. (H–O) The correlation between risk scores and immune cell infiltration. *P < 0.05, **P < 0.01, ***P < 0.001.
For NMIBC patients, in addition to transurethral resection of the bladder tumor (TURBT), intravesical perfusion therapy is required. For MIBC patients, in addition to radical surgery, adjuvant chemotherapy or neoadjuvant chemotherapy is indispensable. However, current chemotherapy drugs are generally less reactive, lack efficacy, leading to drug resistance and quantitative side effects. It is thus crucial to find more effective candidates for cancer therapy. The pRRophetic algorithm was used to evaluate the difference in sensitivity of patients in high- and low-risk groups to commonly used chemotherapeutic agents and targeted therapies. We screened a total of 81 chemotherapy drugs with significant differences in IC50 values. These drugs, such as cisplatin, docetaxel, doxorubicin, gemcitabine, paclitaxel, dasatinib, imatinib, and sunitinib, demonstrated higher sensitivity towards high-risk patients (Fig. 7). Guidelines for BLCA recommend cisplatin, gemcitabine, and paclitaxel as first-line drugs, and dasatinib, imatinib, and sunitinib as novel targeted therapy compounds.

Drug sensitivity analysis between the high- and low-risk groups. (A–I) Most drugs had lower IC50 values in the high-risk group, indicating that patients in the high-risk group were more sensitive to these drugs.
Distribution of prognosis-related genes on the single-cell level
We further investigated the distribution and expression of these seven prognosis-related genes at the single-cell level using scRNA-seq. As shown in Fig. 8A–C, the cells from GSE130001 were classified into 8 clusters containing 4 cell types, including epithelial cells, chondrocytes, endothelial cells and tissue stem cells, among which epithelial cells accounted for the largest cell type. Except for HMGA2, all six genes were expressed in epithelial cells, with FN1 having the highest expression level (Fig. 8D). Bladder tumors are primarily composed of urothelial cells. The expression and distribution of the six genes in epithelial cells indicate their potential significance in the onset and development of bladder cancer. Finally, we tested the mutation frequency of the seven genes in the model and found that these genes had low mutation rates, with the highest rate of 9% for the AIM2 gene (Fig. 8E).

Analysis of seven prognostic genes on the single-cell level. (A) The results of PCA in GSE130001 dataset, different colors represent cells from different samples. (B,C) Cells in the GSE130001 dataset were divided into 8 clusters and 4 cell types. (D) Expression and distribution of the seven genes in cells. (E) Mutation rates of the seven genes in BLCA patients from the cBioPortal database.
Verification of hub genes expression at both mRNA and protein levels
Combining the results of our single-cell results, we selected four hub genes, namely HMGA2, FN1, S100A9 and UCHL1, to verify their expression at both the mRNA and protein levels. The Human Protein Atlas (HPA) database (https://www.proteinatlas.org/) was used to observe the expression patterns of these four genes (FN1, HMGA2, S100A9 and UCHL1) between the normal and tumor tissues (Fig. 9A). Based on the TCGA-BLCA cohort, we analyzed the differential expression of the four genes in 19 bladder cancer tissues and corresponding adjacent tissues. The results showed a significant increase of HMGA2 mRNA expression in BLCA (Fig. 9B). Furthermore, the mRNA expression of the four genes in 15 bladder cancer and adjacent tissue samples were determined using qRT-PCR, and we observed a significant increase in the expression of FN1 mRNA in bladder cancer tissues (Fig. 9C). IHC staining showed that FN1 proteins were predominantly distributed in the tumor stroma and were more abundant in tumors than in normal tissues. (Fig. 9D). In addition, we measured FN1 mRNA expression in 5637, T24, UMUC-3, TCCSUP and SV-HUC-1. qRT-PCR results showed that 5637, T24 and TCCSUP cells had a higher mRNA expression of FN1 compared with the normal SV-HUC-1 cells (Fig. 9E).

Expression validation of hub genes using qRT-PCR and IHC. (A) Analysis of four hub genes in normal and tumor tissues from the HPA database. (B) Expression levels of HMGA2, FN1, S100A9 and UCHL1 mRNA in 19 pairs of BLCA and paired para-tumor samples from TCGA cohort. (C) The expression levels of HMGA2, FN1, S100A9 and UCHL1 mRNA in 15 BLCA and normal tissues were examined by qRT-PCR. (D) Validation of FN1 protein expression by IHC in 20 cases of BLCA and adjacent normal tissues. (E) Using qRT-PCR to detect FN1 expression levels between SV-HUC-1 and bladder cancer cells. *P < 0.05, ***P < 0.001.
FN1 facilitates BLCA cell motility through integrin-dependent manner
To investigate the role of FN1 on the BLCA phenotype, we performed GSEA analysis with the use of TCGA cohort. GSEA results displayed that several phenotype, like ‘CELL_MIGRATION’, ‘MULTICANCER_INVASIVENESS_SIGNATURE’, ‘WOUND_HEALING’ and ‘EPITHELIAL_MESENCHYMAL_TRANSITION’ that are involved in cell motility and aggressiveness, were highly enriched in the patients with high risk (Fig. 10A). Kaplan-Meier curves showed that OS and cancer-specific survival (CSS) were shorter in patients with high FN1 expression than in patients with low FN1 expression in the TCGA (HR = 1.67, 95% CI = 1.19–2.34, P = 0.003) and GSE13507 [For OS, HR = 1.76, 95% CI = 1.06–2.93, P = 0.027; for CSS, HR = 2.70, 95% CI = 1.28–5.72, P = 0.007] datasets (Fig. 10B).

FN1 participates in cell migration of BLCA. (A) GSEA (B) Survival curves showing that patients with high-FN1 had worse outcomes than those with low-FN1, according to TCGA and GEO datasets. (C) The expression levels of FN1 in T24 and 5637 cells were decreased after transfection with siRNAs. (D) Wound healing assay indicating impaired cell movement after depletion of FN1. (E) Transwell assay showing that FN1 loss suppressed cell migration. *P < 0.05, ***P < 0.001.
We then verified the role of FN1 in cell migration of BLCA. Two siRNAs against FN1 were transefected into T24 and 5637 cells, and the mRNA expression of FN1 was determined by qRT-PCR. The results demonstrated that, compared with the control groups, the expression of FN1 was decreased by over 50% in T24 and 5637 cells after transfection with siRNAs (Fig. 10C). Functional assay indicated showed that deletion of FN1 impaired wound healing and cell migration in T24 and 5637 cells (Fig. 10D,E), suggesting that FN1 may participate in the progression of BLCA. Furthermore, FN1 often regulates aggressive phenotypes through ligand- and receptor-dependent binding (e.g., to integrins). We then performed experiments using the broad-spectrum integrin antagonist GLPG0187 (1 µM) and exogenous rFN1 (10 µg/mL), treating the cells for 24 h before completing the Transwell assay. The results showed that GLPG0187 blocked exogenous FN1-induced migration in 5637 and T24 cells (Fig. S5), suggesting that integrins are indispensabe in FN1-droven cell movement. However, which member of the integrin family is involved in mediating this process remains to be further investigated.
Discussion
Accumulating evidence indicates that RNA methylation plays an indispensable role in tumorigenesis and immune response. However, most studies have focused on a single type of RNA modification, particularly m6A19,20, while the relationship between other types of RNA modifications and malignancy is not yet fully understood. We therefore investigated the role of m6A/m5C/m7G-related genes in BLCA and their possible influence on TME and immunotherapy. Initially, the expression of seventy m6A/m5C/m7G related genes between bladder cancer and normal tissues was analyzed. Among them, 31 genes including METTL1, METTL3, WDR4, were upregulated in cancer samples. METTL3 could accelerate pri-miR221/222 maturation in an m6A-dependent manner to promote the proliferation of bladder cancer cells21. Overexpressed METTL1 mediates m7G tRNA modification, activates the EFGR pathway by upregulating epidermal growth factor receptor and EFEMP1 levels, and promotes bladder tumorigenesis22. m5C promotes the proliferation and invasion of bladder cancer cells by cross-regulating ALYREF and NSUN223. These studies suggested that methylation-related genes are required in the development of BLCA.
We here constructed a risk prediction model based on RNA methylation-related genes by LASSO and multivariate Cox regression analysis. Patients were divided into high- and low-risk groups based on the median value of the risk score. Survival curves showed that patients in the high-risk group had a significantly worse prognosis than those in the low-risk group, which is further verified by an external GSE19423 dataset. There were significant differences in clinical features, TME scores, immune checkpoint expression and drug sensitivity between the two groups. By integrating clinical parameters and risk scores, we constructed a nomogram to improve applicability in clinical practice and provide a new approach for prognostic assessment and individualized therapeutic strategy for patients with BLCA. GSEA analysis showed that several important biological processes, including EMT, hypoxia, inflammatory response, cell cycle, ECM signaling pathways, were enriched in the high-risk group. The ECM signaling pathway not only promotes tumor cell growth, invasion, metastasis, and angiogenesis, but is also closely related to the composition of TME and anti-tumor immunotherapy24. The IMvigor210 cohort was used to predict the response to immunotherapy of patients in different risk groups. In addition, we analyzed the expression and distribution of seven genes at the single-cell level. The results showed that most genes were expressed in epithelial cells. Given that urothelial cells are the predominant cell type involved in bladder carcinogenesis, the gene expression patterns observed in bladder epithelial cells may imply that they participate in the pathogenesis and progression of BLCA, which is worth investigating.
Previous study has shown that HMGA2 is a prognostic marker for prediction of recurrence and progression of BLCA25. Increased expression of FN1 in cadmium-treated urothelial cells and BLCA cell lines suggests that it is a potential prognostic marker for BLCA26. Co-expression of S100A9 and EGFR, proved to be an independent prognostic factor for disease progression after chemotherapy in patients with locally recurrent or metastatic MIBC and could be used as a marker to predict chemotherapy response in these patients27. Combining with our research and published literatures, we finally identified four hub genes for next verification, namely HMGA2, FN1, S100A9 and UCHL1. The HPA database revealed that the four hub genes expressed higher in bladder cancer than normal tissues. 19 bladder cancer and paired adjacent samples from the TCGA-BLCA cohort showed that the expression of HMGA2 mRNA was increased in bladder cancer tissues. Then, qRT-PCR was conducted to analyze the their expression levels in 15 paired bladder cancer and adjacent normal tissue samples collected from the local hospital, and the results indicated that the expression of FN1 is enhanced in BLCA tissues. IHC staining demonstrated its high abundance in the tumor stroma.
FN1 is a key member of the extracellular matrix (ECM) glycoprotein family, primarily mediating interactions between cells and the ECM and participating in ECM remodeling through integrin signaling. Binding of FN1 to integrins results in activation of the integrin, subsequently leading in Rho-mediated acto-myosin contractility that in turn promotes assembly of fibronectin into a fibrillar matrix, thus mediating cell movement28. In general, it is highly expressed in some solid cancers, promotes adhesion, differentiation, epithelial-mesenchymal transition (EMT), invasion, metastasis, as well as predicts a poor prognosis29,30,31. In addition, FN1 impairs the efficacy of immune checkpoint inhibitors by upregulating immunosuppressive molecules that impede the killing function of T cells32,33. Notably, FN1 has been reported to facilitate polarization and function of tumor-associated macrophage34. However, it was also found that high expression of FN1 inhibited melanoma development and metastasis35, showing FN1’s dual effects on malignant behaviors. To clarify the specific role of FN1 in the progression of BLCA, we performed RNA interfering, wound healing and transwell assays, and found that FN1 mediated cell migration in bladder cancer cells, whereas blockade of integrins using GLPG0187 reversed exogenous FN1-droven cell motility, suggesting that integrins are indispensabe in FN1-induced malignant transition.
Nevertheless, our study has some limitations. Firstly, we only validated the model using the GEO database and the IMvigor210 dataset, and its clinical value requires investigation. Further verification with data from more centers and larger sample sizes is also needed. Secondly, the effects of other members of 7-gene signature on cell phenotype are still unknown. Further research is needed to understand how these genes affect the specific molecular mechanisms underlying the development of BLCA, and how they are modulated by RNA methylation.
Conclusion
This study systematically evaluated the expression of m6A/m5C/m7G-related genes in bladder cancer, and investigated their potential biological functions and immune functions. A seven-gene prognostic model was constructed, and the reliability of the model was verified using the GEO dataset. Additionally, we explored the potential value of this prognostic model in predicting immunotherapy using the IMvigor210 dataset. The immune landscape, gene mutation status and drug sensitivity between different risk groups were also analyzed. Furthermore, the expression of four hub genes was verified at mRNA and protein expression levels. Among the signature, FN1 is a pivotal gene that promotes the movement of bladder cancer cells. In conclusion, the risk model provides evidence for prognostic prediction, immunotherapy, and drug sensitivity for patients with bladder cancer, and FN1 may play a crucial role in the pathogenesis of bladder cancer.
Data availability
All datasets used in the present study are publicly available: GEO repository (http://www.ncbi.nlm.nih.gov/geo/, accession number: GSE13507, GSE19423 and GSE130001), TCGA (https://portal.gdc.cancer.gov/), Tumor Immune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/), The cBio Cancer Genomics Portal (cBioPortal, http://www.cbioportal.org/), Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp), and The Human Protein Atlas repository (HPA, https://www.proteinatlas.org/).
Abbreviations
- BLCA:
-
Bladder cancer
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- LASSO:
-
Least absolute shrinkage and selection operator
- qRT-PCR:
-
Quantitative reverse transcriptase polymerase chain reaction
- IHC:
-
Immunohistochemistry
- TME:
-
Tumor microenvironment
- NMIBC:
-
Non-muscle‐invasive bladder cancer
- MIBC:
-
Muscle‐invasive bladder cancer
- m1A:
-
N1-methyladenosine
- m6A:
-
N6-methyladenosine
- m5C:
-
5-Methylcytosine
- m7G:
-
7-Methylguanosine
- FPKM:
-
Fragments per kilobase
- GSVA:
-
Gene set variation analysis
- ssGSEA:
-
Single-sample gene set enrichment analysis
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PCA:
-
Principal component analysis
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under curve
- TIDE:
-
Tumor immune dysfunction and exclusion
- TMB:
-
Tumor mutation burden
- IC50:
-
Half maximal inhibitory concentration
- ATCC:
-
American type culture collection
- DCA:
-
Decision curve analysis
- TURBT:
-
Transurethral resection of the bladder tumor
- HPA:
-
Human Protein Atlas
- OS:
-
Overall survival
- CSS:
-
Cancer-specific survival
- HR:
-
Hazard ratio
- CI:
-
Confdence interval
References
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953 (2019).
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Witjes, J. A. et al. European association of urology guidelines on Muscle-invasive and metastatic bladder cancer: Summary of the 2020 guidelines. Eur. Urol. 79, 82–104 (2021).
Mitra, A. P. et al. Management trends and outcomes of patients undergoing radical cystectomy for urothelial carcinoma of the bladder: Evolution of the university of Southern California experience over 3,347 cases. J. Urol. 207, 302–313 (2022).
Saginala, K. et al. Epidemiology of bladder cancer. Med. Sci. (Basel) 8, 15 (2020).
Frye, M., Harada, B. T., Behm, M. & He, C. RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018).
Shi, H., Chai, P., Jia, R. & Fan, X. Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the Bridge between transcription and translation. Mol. Cancer 19, 78 (2020).
Zhang, Q. et al. The role of RNA m(5)C modification in cancer metastasis. Int. J. Biol. Sci. 17, 3369–3380 (2021).
Li, K. & Wang, W. Establishment of m7G-related gene pair signature to predict overall survival in colorectal cancer. Front. Genet. 13, 981392 (2022).
He, L. et al. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18, 176 (2019).
Lee, J. S. et al. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 28, 2660–2667 (2010).
Kim, Y. J. et al. Gene signatures for the prediction of response to bacillus calmette-guerin immunotherapy in primary pT1 bladder cancers. Clin. Cancer Res. 16, 2131–2137 (2010).
Tomikawa, C. 7-Methylguanosine modifications in transfer RNA (tRNA). Int. J. Mol. Sci. 19, 4080 (2018).
Ye, X. et al. m(6)A/ m(1)A /m(5)C/m(7)G-related methylation modification patterns and immune characterization in prostate cancer. Front. Pharmacol. 13, 1030766 (2022).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Necchi, A. et al. Atezolizumab in platinum-treated locally advanced or metastatic urothelial carcinoma: Post-progression outcomes from the phase II IMvigor210 study. Ann. Oncol. 28, 3044–3050 (2017).
Wang, L. et al. A reference profile-free deconvolution method to infer cancer cell-intrinsic subtypes and tumor-type-specific stromal profiles. Genome Med. 12, 24 (2020).
Yin, H. et al. EGF-induced nuclear translocation of SHCBP1 promotes bladder cancer progression through inhibiting RACGAP1-mediated RAC1 inactivation. Cell. Death Dis. 13, 39 (2022).
Chen, L. et al. The roles and mechanism of m(6)A RNA methylation regulators in cancer immunity. Biomed. Pharmacother. 163, 114839 (2023).
Zhao, Y. et al. Loss of m6A demethylase ALKBH5 promotes post-ischemic angiogenesis via post-transcriptional stabilization of WNT5A. Clin. Transl. Med. 11, e402 (2021).
Han, J. et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol. Cancer 18, 110 (2019).
Ying, X. et al. METTL1-m(7) G-EGFR/EFEMP1 axis promotes the bladder cancer development. Clin. Transl. Med. 11, e675 (2021).
Wang, N. et al. m(5)C-dependent cross-regulation between nuclear reader ALYREF and writer NSUN2 promotes urothelial bladder cancer malignancy through facilitating RABL6/TK1 mRNAs splicing and stabilization. Cell. Death Dis. 14, 139 (2023).
Yuzhalin, A. E., Lim, S. Y., Kutikhin, A. G. & Gordon-Weeks, A. N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta Rev. Cancer. 1870, 207–228 (2018).
Yang, G. L. et al. Overexpression of HMGA2 in bladder cancer and its association with clinicopathologic features and prognosis HMGA2 as a prognostic marker of bladder cancer. Eur. J. Surg. Oncol. 37, 265–271 (2011).
Zhang, L. et al. Fibronectin 1 as a key gene in the genesis and progression of cadmium-related bladder cancer. Biol. Trace Elem. Res. 201, 4349–4359 (2023).
Kim, W. T. et al. S100A9 and EGFR gene signatures predict disease progression in muscle invasive bladder cancer patients after chemotherapy. Ann. Oncol. 25, 974–979 (2014).
Efthymiou, G. et al. Shaping up the tumor microenvironment with cellular fibronectin. Front. Oncol. 10, 641 (2020).
Cai, X. et al. Down-regulation of FN1 inhibits colorectal carcinogenesis by suppressing proliferation, migration, and invasion. J. Cell. Biochem. 119, 4717–4728 (2018).
Yan, J., Wu, L., Zheng, M. & Pan, F. Exploring programmed cell death-related biomarkers and disease therapy strategy in nasopharyngeal carcinoma using transcriptomics. Front. Biosci. (Landmark Ed). 29, 240 (2024).
Lin, Y. et al. YY1 mediated DCUN1D5 transcriptional activation promotes triple-negative breast cancer progression by targeting FN1/PI3K/AKT pathway. Biol. Direct. 19, 42 (2024).
Xu, H. et al. Single-cell RNA sequencing identifies a subtype of FN1+ tumor-associated macrophages associated with glioma recurrence and as a biomarker for immunotherapy. Biomark. Res. 12, 114 (2024).
Sheng, S. et al. Aberrant methylation and immune microenvironment are associated with overexpressed fibronectin 1: A diagnostic and prognostic target in head and neck squamous cell carcinoma. Front. Mol. Biosci. 8, 753563 (2021).
Zhou, W. H. et al. The overexpression of fibronectin 1 promotes Cancer progression and associated with M2 macrophages polarization in head and neck squamous cell carcinoma patients. Int. J. Gen. Med. 15, 5027–5042 (2022).
Liu, W., Cheng, S., Asa, S. L. & Ezzat, S. The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res. 68, 8104–8112 (2008).
Acknowledgements
We would like to thank the contributors of TCGA, GEO, Kaplan-Meier plotter, TIMER, cBioPortal, Molecular Signatures Database, and The Human Protein Atlas database for data sharing.
Funding
This study was supported by the National Natural Science Foundation of China (NO. 82303655); the Chongqing Municipal Science and Technology Commission Doctoral Through Train Project (CSTB2022BSXM-JCX0037); the China Postdoctoral Science Foundation (2023M740452); the First Clinical College of Clinical Medicine First-class Discipline Construction Program (472020320230017); Chongqing Young and Middle-aged High-end Medical Talents Project Fund (2023GDRC013); Innovative Program for Doctoral Students of the First Affiliated Hospital of Chongqing Medical University (CYYY-BSYJSCXXM-202328).
Author information
Authors and Affiliations
Contributions
Conceptualization, H.Y., and J.L.; methodology, B.L, Y.L.; software B.L., J.C., T.L.; validation, H.Y. and J.L.; formal analysis, B.L., J.C.; investigation H.Y., Y.K. and J.L.; resources, B.L.; data curation, B.L.; writing-original draft preparation, B.L.; writing—review and editing, H.Y. and J.L.; visualization, B.L., T.L and J.C.; supervision, H.Y.; project administration, H.Y.; funding Acquisition, H.Y., J. L and Y.K; revision, H.Y and J.G. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and informed consent
The present study was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (Approval number: 20182101), and written informed consent was obtained from all patients.
Consent for publication
All listed authors have actively participated in the study and have read and.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, B., Gan, J., Li, T. et al. Comprehensive analysis of RNA methylation-related genes to identify molecular cluster for predicting prognosis and immune profiles in bladder cancer. Sci Rep 15, 9147 (2025). https://doi.org/10.1038/s41598-025-93674-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93674-2
