
Abstract
The extracellular matrix (ECM) in the brain can be divided into aggregated ECM, such as perineuronal nets (PNNs) around neurons, and diffuse ECM, which is present throughout the brain parenchyma. Both aggregated and diffuse ECM restrict synaptic plasticity and stabilize neural circuits in the adult brain. Hyaluronan (HA) acts as a scaffold for the brain ECM, and multiple proteoglycans, such as aggrecan, bind to HA to form a macromolecular complex. Recent evidence suggests that microglia, the resident immune cells of the brain, play a crucial role in ECM homeostasis. However, it remains unclear how microglia influence the molecular composition of the ECM. Using a tissue-clearing technique and histochemical analysis, we found that microglial depletion increased the staining intensity of aggrecan and HA in both PNNs and diffuse ECM. Biochemical analyses further confirmed the accumulation of the aggrecan core protein and HA following microglial depletion. Our findings highlight the essential role of microglia in regulating the ECM composition and provide new insights into the mechanisms by which microglia influence neuronal function.
Similar content being viewed by others

Introduction
The extracellular matrix (ECM) in the brain supports tissue integrity and regulates neuronal differentiation, maturation, and synaptic function. The brain ECM can be divided into two distinct groups: aggregated and diffuse ECM1,2,3. Aggregated ECM can only be extracted from tissue using denaturants such as urea. A prominent example of an aggregated ECM is perineuronal net (PNN), which preferentially forms around parvalbumin-expressing inhibitory neurons in the adult cerebral cortex. Enzymatic and genetic disruption of PNNs in the adult brain reactivates juvenile-like brain plasticity4,5,6. PNNs are thought to facilitate the transition from juvenile to adult neural circuits by promoting the maturation of parvalbumin-expressing inhibitory neurons. In contrast, diffuse ECM is present throughout the brain parenchyma and results in the formation of loose, water- and detergent-soluble structures. Degradation of perisynaptic diffuse ECM increases dendritic spine motility, suggesting that diffuse ECM limits the structural plasticity of synapses7,8. A recent study also reported that perisynaptic ECM dynamics contribute to long-term potentiation and memory9.
Both diffuse and aggregated ECMs share similar molecular compositions. Hyaluronan (HA) is a long linear glycosaminoglycan that serves as a scaffold for the brain ECM. A family of HA-associated chondroitin sulfate proteoglycans (CSPGs), such as aggrecan, bind to HA and assemble into a large molecular complex1,2. Although the two types of ECMs display similarities, they also exhibit differences. We previously reported that the size of HA chains contributes to the properties of ECM structures: longer HA chains found in PNNs facilitate the formation of insoluble aggregates, whereas shorter HA chains contribute to the formation of diffuse ECM10.
The brain ECM is compromised in various pathological conditions. For instance, mouse models of neurodegenerative diseases, such as Alzheimer’s disease, exhibit a reduction in the density of PNNs coupled with changes in their molecular composition11,12,13,14,15. Similar decreases in PNN density and degradation of PNN components have been observed in aged brains16,17,18. PNNs protect neurons against oxidative stress and various insults19,20,21,22; decreasing PNNs may increase susceptibility to damage in neurons under pathological conditions.
Microglia are the resident immune cells in the brain that elaborate neural circuits by engulfing and eliminating excess synapses during brain development23. In addition, proinflammatory responses by chronically activated microglia, as observed in neurodegenerative diseases, can lead to progressive neuronal damage. Microglia are involved in the maintenance of PNN homeostasis under normal conditions and in PNN degradation under pathological conditions24,25. Colony-stimulating factor 1 signaling is essential for microglial survival, and oral administration of its inhibitor PLX3397 results in microglial depletion without affecting other cell types26,27. Microglial depletion by PLX3397 in healthy adult brains increases the density and intensity of PNNs and alters their microstructure27,28,29. Notably, microglial elimination can reverse the loss of PNNs observed in mouse models of neurodegenerative disease11,13. These studies indicate that microglia actively remodel PNNs in healthy and pathological brains.
Emerging evidence indicates that microglial depletion leads to increased staining of PNNs; however, the specific molecular changes in PNNs underlying this phenomenon have not been determined. This may be due, in part, to the limitations of the current histochemical method used to detect PNNs. Many studies rely on histochemical staining with a single probe, Wisteria floribunda agglutinin (WFA), a lectin that binds to the glycan moieties of CSPGs, to detect PNNs. Because WFA specifically recognizes only certain glycans within PNNs30,31,32, caution should be exercised in interpreting changes in WFA staining as indicative of broader changes in PNNs. It is also unclear whether microglial depletion affects diffuse ECM in addition to PNNs. In the present study, we aimed to investigate the effects of microglial depletion on the molecular composition of diffuse and aggregated ECM, particularly its major components, aggrecan and HA. Histochemical analysis using a tissue clearing technique revealed the proximity of PNNs and microglia in the adult mouse brain. We found that microglial depletion increased aggrecan and HA levels in both diffuse and aggregated ECM. These findings highlight the role of microglia in maintaining the molecular composition of PNNs.
Materials and methods
Ethics information
All animal experimental protocols were approved by the Committee on Animal Experiments of Tokyo University of Agriculture and Technology. All methods were performed in accordance with the relevant guidelines and regulations. The study was conducted in accordance with ARRIVE guidelines.
Animals
Eight-week-old male C57BL/6JJmsSlc (RRID:MGI:5,488,963) mice were purchased from Japan SLC. Mice were randomly assigned to the control or PLX3397-treated (PLX) group. All mice were housed under a 12-h light and dark cycle with ad libitum access to water and chow. The PLX groups were fed chow containing 290 mg/kg of PLX3397 for four weeks13,27. PLX3397 was provided by ChemGood and formulated in AIN-76A chow by Research Diets.
Brain sectioning
The 12-week-old control and PLX groups were deeply anesthetized with isoflurane and transcardially perfused with 50 ml of ice-cold phosphate-buffered saline (PBS), followed by 50 ml of 4% paraformaldehyde (PFA) in PBS. The brains were fixed in 4% PFA in PBS at 4 °C overnight and separated sagittally into left and right hemispheres. The right hemispheres were sectioned coronally on a vibratome at 100 µm thickness and stored in PBS containing 0.02% sodium azide at 4 °C for later use in a tissue-clearing protocol. The left hemispheres were sectioned at 80 µm thickness, mounted on glass slides, and stored at − 30 °C until use for standard histochemical staining.
Tissue clearing and histochemical analysis
Brain sections (100 µm thick) were tissue-cleared with a clear, unobstructed brain imaging cocktail (CUBIC) protocol with slight modifications33,34,35. Briefly, brain sections were washed with PBS and immersed in 10% w/v N-butyl diethanolamine and 10% w/v Triton X-100 for 1 h at room temperature. After washing with PBS until no bubbles formed, the sections were blocked with 2% bovine serum albumin in PBS for 1 h at room temperature. The sections were incubated with primary probes in blocking solution for 3–6 days at 4 °C. The following primary probes were used: biotinylated WFA (1:500 dilution; Vector Laboratories), biotinylated HA-binding protein (bHABP; 1:500 dilution; Hokudo), anti-ionized calcium binding adaptor molecule 1 (IBA1) antibody (1:500 dilution,;Fujifilm Wako Pure Chemical), and anti-aggrecan antibody (1:200 dilution; Sigma-Aldrich). After washing with PBS containing 0.1% Tween 20 (PBST), the sections were incubated with Alexa Fluor 594-conjugated streptavidin and Alexa Fluor 488-conjugated anti-rabbit IgG (Thermo Scientific) at 4 °C overnight. The sections were then immersed in 4 M urea, 40% w/v D-sorbitol, 10% w/v glycerol, and 20% v/v dimethyl sulfoxide for 1 h and mounted in the same solution on a glass slide.
Standard histochemical staining was performed without the tissue-clearing process. Brain sections (80 µm thick) were permeabilized with PBS containing 0.2% Triton X-100 for 20 min and blocked with 2% bovine serum albumin in PBS for 1 h at room temperature. The sections were incubated with primary probes overnight at 4 °C and Alexa Fluor-conjugated secondary probes for 2 h at room temperature.
Images were captured using a Nikon Ti2 confocal laser microscope. We obtained a single image or Z-stack from the same cortical region of each mouse. Z-stack images were reconstructed using maximum intensity projection in Imaris software. The regions inside and outside PNNs were distinguished and analyzed using ImageJ FIJI software as follows: Binary masks for WFA or aggrecan staining were created using the “moments” autothresholding method. The mask was applied to the original image to select the “PNN region”, and the reverse mask was applied to select the “outside PNN region”. To analyze HA intensity within and outside of PNNs, we used double-labeled images of HA and aggrecan. PNN regions were defined based on aggrecan staining using binarization analysis in ImageJ as described above. The extracted PNN regions were then applied to HA-stained images to calculate HA staining intensities within and outside the PNN regions. The number of WFA- and aggrecan-positive PNNs was quantified using the automated particle analysis function in ImageJ FIJI, applied to the same binary mask used to define the PNN regions.
Extraction of soluble and insoluble components from the cerebral cortex
Twelve-week-old control and PLX mice were deeply anesthetized with isoflurane and transcardially perfused with 50 ml of PBS. The brains were chilled in ice-cold PBS for 3 min and sliced into 1 mm thick slices using a vibratome. Cerebral cortices were dissected from the brain slices. Detergent-soluble and insoluble fractions were prepared according to previous reports with minor modifications3,10,18. Tissues were homogenized in PBS containing 0.5% Triton X-100, 2 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (buffer 1). After incubating on ice for 30 min, the homogenate was centrifuged at 20,000 × g for 30 min at 4 °C, after which the supernatant was collected as the soluble fraction. The pellet was resuspended in buffer 1 containing 6 M urea, incubated on ice for 30 min, and centrifuged at 20,000 × g for 30 min at 4 °C. The supernatant was dialyzed against PBS and collected as the insoluble fraction. To determine the percentage of aggrecan in the soluble and insoluble fractions, we added PBS to adjust the total volume of both fractions to 480 μl per 100 mg of brain tissue. Protein quantification using the BCA assay kit confirmed that there was no significant difference in protein concentration between the control and PLX groups.
Preparation of whole brain extracts
The brains were dissected and homogenized in buffer 1 containing 6 M urea. After incubating on ice for 1 h, the homogenate was centrifuged at 12,000 × g for 30 min at 4 °C. The supernatant was dialyzed against PBS at 4 °C overnight. PBS was added to adjust the total volume to 1300 μl per 100 mg of brain tissue. Protein quantification using the BCA assay confirmed that there was no significant difference in protein concentration between the control and PLX groups.
Immunoblotting
Fifty microliters of whole brain extract was digested with 1 mU of chondroitinase ABC (Seikagaku Corp.). Fifty microliters of the soluble and insoluble fractions were digested with 17 mU of chondroitinase ABC (Sigma-Aldrich). After digestion for 1.5 h at 37 °C, the samples were denatured with 2.5% sodium dodecyl sulfate and 5% 2-mercaptoethanol for 20 min at 60 °C. Total protein loaded per lane was as follows: Whole brain extract: control group; 119 ± 5.8 μg; PLX group; 114 ± 10.1 μg. Soluble fraction: control group; 113 ± 5.6 µg; PLX group; 108 ± 3.6 µg. Insoluble fraction: control group; 96 ± 11.7 µg; PLX group; 89 ± 8.9 µg. Proteins were separated by electrophoresis on a 4.5% polyacrylamide gel at 20 mA per gel for 1.5–2 h, and subsequently transferred onto a polyvinylidene difluoride membrane (0.45 μm pore size, Millipore) using a semidry transfer cell (Bio-Rad) in Tris–glycine buffer containing 10% methanol at 10 V for 2 h. The membranes were blocked with 2% skim milk and incubated with anti-aggrecan antibody (1:2,000 dilution; Sigma-Aldrich) at 4 °C overnight. After washing with PBST, the membranes were incubated with peroxidase-conjugated anti-rabbit IgG (1:2000 dilution; Cell Signaling Technology) for 1 h at room temperature and developed with a chemiluminescent peroxidase substrate (Millipore). Chemiluminescence signals were visualized using a LuminoGraph I (ATTO), and band intensity was analyzed using ImageJ FIJI software.
HA quantification by competitive enzyme-linked immunosorbent assay (ELISA)
An ELISA-like assay for HA quantification was performed as previously described10,36,37. Briefly, a 96-well plate (Iwaki) was coated overnight with 0.25 µg/ml HABP (Hokudo) in 0.5% Block Ace (Snow Brand Milk Products) and then washed with PBST. The wells were blocked with 1% Block Ace and coated with 1 µg/ml HA sodium salt (Fujifilm Wako Pure Chemicals). After dilution to the appropriate concentration with 0.5% Block Ace, the samples were added to the wells, incubated with 0.125 μg/ml bHABP in 0.5% Block Ace for 1 h at room temperature, and subsequently washed with PBST. Peroxidase-conjugated streptavidin (Thermo Scientific) was added to the wells, which were subsequently incubated for 30 min at 37 °C. After washing with PBST, the peroxidase substrate 3, 3, 5, 5-tetramethylbenzidine (Nacalai Tesque) was added and incubated in the dark until proper coloration was achieved. The reaction was stopped by adding 1 N H2SO4, and the absorbance was measured at 450 nm.
Purification of HA from soluble and insoluble fractions
HA was purified from the soluble and insoluble fractions as described previously10. The samples were digested with 1 mg/ml of actinase E (Kaken Pharmaceutical) at 40 °C overnight to release HA chains from the proteins, followed by heat inactivation of actinase E for 5 min at 95 °C. After centrifugation at 20,000 × g for 5 min at room temperature, the HA concentration in the supernatant was quantified by ELISA. Samples containing 0.2 μg of HA were incubated with 2.5 μg of bHABP for 1 h at room temperature, followed by incubation with 25 μl (250 μg) of streptavidin-conjugated magnetic beads (Thermo Scientific) for 1 h at room temperature with constant mixing. Magnetic beads were collected with a magnet and washed with PBST. HA was then released by boiling at 95 °C for 10 min and stored at − 30 °C until use.
Agarose gel electrophoresis and staining of HA
HA purified from the soluble and insoluble fractions was separated by agarose gel electrophoresis10,38. Briefly, purified HA and the molecular mass standards of HA (Select-HA HiLadder, Hyalose) were loaded on a 0.5% SeaKem gold agarose gel (Lonza) and separated for 30 min at 100 V in Tris–acetate-EDTA buffer. The gels were stained with 20 μg/ml Stains-all (Sigma-Aldrich) in 30% ethanol in the dark with gentle shaking overnight and destained by submerging in 30% ethanol. Images were captured using a LuminoGraph I (ATTO). Band intensity and mobility were analyzed using ImageJ FIJI software. The molecular weight of HA was estimated based on its mobility.
mRNA extraction and quantitative reverse transcription (RT)-PCR
Total RNA was prepared from the cerebral cortex using RNeasy Plus kits (Qiagen). cDNA was synthesized using PrimeScript RT Master Mix (Takara). Real-time quantitative RT‒PCR was performed on a Thermal Cycler Dice Real Time System (Takara) using TB Green Premix Ex Taq II (Takara). The primers used are listed in Table 1. The expression levels of the target genes were calculated using the improved delta-delta Ct (cycle threshold) method (Pfaffl method), with beta-actin (Actb) expression used as an internal reference.
Statistical analysis
The statistical significance of differences was assessed by an independent Student’s t-test. P-values less than 0.05 were considered to indicate statistical significance.
Results
Microglial depletion increases the staining intensity of WFA in the mouse cerebral cortex
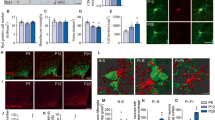
We utilized the tissue-clearing technique to explore the spatial relationship between microglia and PNNs in the adult mouse cerebral cortex. Histochemical analysis revealed that IBA1-positive microglial cell bodies and processes were in close contact with WFA-positive PNNs (Fig. 1a, b). To investigate the effect of microglia on PNN maintenance, we depleted microglia by oral administration of PLX3397 for 4 weeks, starting at 8 weeks of age, when PNN formation was complete (Fig. 1c).4,6. IBA1 staining confirmed that most microglia were depleted from the cerebral cortex by PLX3397 administration, consistent with the finding of previous reports (Fig. 1d, e)13,27,28. Microglial depletion increased the intensity of WFA staining compared to the control group (Fig. 1f, g). A separate analysis of WFA staining intensity in PNNs around neuronal cell bodies and in diffuse ECM uniformly observed outside PNNs showed increased WFA staining in both PNNs and diffuse ECM (Fig. 1f, h). Although microglial depletion increased the intensity of WFA staining, the number of PNNs did not differ between the two groups (Fig. 1i).

Microglial depletion increases the intensity of WFA staining in the mouse cerebral cortex. (a) Tissue-cleared mouse brain slices (100 µm thickness) were stained to visualize WFA-positive PNNs (red) and IBA1-positive microglia (green). Nuclei were counterstained with DAPI (blue). Left: maximum intensity projection of a 20 µm Z-stack. Right: single confocal section showing contact between PNNs and microglia. (b) Fluorescence intensity profiles of WFA (red) and IBA1 (green) along white lines 1 and 2 in panel (a). (c) Schematic of the microglial depletion experiment. (d) IBA-1 staining of microglia in the control and PLX cerebral cortex. (e) Microglial cell density in the control and PLX groups. n = 5 mice. Control 46.4 ± 10.04, PLX 0 ± 0. (f) WFA staining of the control and PLX cerebral cortices. (g) Relative fluorescence intensity of WFA staining in a 550 × 550 μm area in the control and PLX groups. n = 5 mice. Control 1.000 ± 0.138, PLX 1.634 ± 0.376. (h) Relative fluorescence intensity of WFA staining in PNNs (left) and outside PNNs (right). n = 5 mice. PNNs: Control 1.000 ± 0.271, PLX 1.467 ± 0.306; Outside PNNs: Control 0.370 ± 0.049, PLX 0.601 ± 0.142. (i) Number of WFA-positive PNNs in the control and PLX groups. n = 5 mice. Control 24.6 ± 3.97, PLX 25.8 ± 3.83. The error bars represent the SD. Statistical analysis was performed by an independent t-test; * indicates p < 0.05.
Increased levels of the aggrecan core protein in microglia-depleted brains
Since WFA lectin is known to bind to glycan moieties on aggrecan30,39,40, we hypothesized that microglial depletion might cause an increase in aggrecan. Histochemical analysis showed that microglial depletion increased the staining of aggrecan core proteins (Fig. 2a, b). Similar to WFA staining, microglial depletion elevated aggrecan staining in both PNNs and diffuse ECM (Fig. 2a, c). Our analysis showed no significant difference in the number of aggrecan-positive PNNs between the control and PLX groups (Fig. 2d). Furthermore, Western blot analysis of the aggrecan core protein showed a 30% increase in the PLX group compared to the control group (Fig. 2e, f). It has been reported that diffuse ECM is recovered in the soluble fraction, whereas PNN is recovered in the insoluble fraction3,10,18. Therefore, we analyzed the amount of aggrecan in the soluble and insoluble fractions separately. The amount of aggrecan in both fractions was greater in the PLX group than in the control group, although these differences did not reach statistical significance (Fig. 2g, h). Immunohistochemistry and Western blotting showed that the aggrecan core protein level was increased by microglial depletion; however, quantitative RT‒PCR showed no difference in aggrecan mRNA expression between the control and PLX groups (Fig. 2i).

Increased levels of the aggrecan core protein in microglia-depleted brains. (a) Immunostaining of the aggrecan core protein (green) in the control and PLX cerebral cortices. (b) Relative fluorescence intensity of aggrecan staining in a 300 × 300 μm area in the control and PLX groups. n = 5 mice for the control; n = 4 mice for the PLX group. Control 1.000 ± 0.140, PLX 1.607 ± 0.124. (c) Relative fluorescence intensity of aggrecan in PNNs (left) and outside PNNs (right). n = 5 mice for the control; n = 4 mice for the PLX group. PNNs: Control 1.000 ± 0.098, PLX 1.393 ± 0.377; Outside PNNs: Control 0.309 ± 0.047, PLX 0.452 ± 0.084. (d) Number of aggrecan-positive PNNs in the control and PLX groups. n = 5 mice. Control 11.4 ± 3.0; PLX 14.2 ± 4.4 (e) Western blot analysis of the aggrecan core protein in the control and PLX groups. Brain extracts were predigested with chondroitinase ABC to digest the glycan moieties of the CSPGs. PC: positive control. (f) The total amount of aggrecan core protein was assessed by measuring the band intensity and is expressed relative to the control group. n = 5 mice. Control 1.000 ± 0.144, PLX 1.306 ± 0.103. (g) Western blot analysis of aggrecan in the soluble (left) and insoluble (right) fractions prepared from the control and PLX cerebral cortices. (h) The levels of aggrecan in the soluble (left) and insoluble (right) fractions are shown relative to the control group. n = 4 mice. Soluble: Control 1.000 ± 0.418, PLX 1.594 ± 0.300; Insoluble: Control 1.000 ± 0.257, PLX 1.181 ± 0.442. (i) Aggrecan mRNA expression levels in the control and PLX groups. n = 4 mice. Control: 1.000 ± 0.103, PLX: 0.906 ± 0.205. The error bars represent the SD. Statistical analysis was performed by an independent t-test; * indicates p < 0.05.
Increased staining of HA in PNNs and diffuse ECM following microglial depletion
Next, using the tissue clearing technique, we examined the relationship between microglia and HA, another major ECM component. In the control cortex, a clear accumulation of HA is observed around the cell bodies of a subpopulation of neurons, which corresponds to PNNs as we previously reported (Fig. 3a)10,41. In addition, weak diffuse HA staining was observed outside PNNs. Confocal z-stack images showed that microglial cell bodies were closely associated with and partially overlapped with HA accumulation in PNNs (Fig. 3b, c). Microglial depletion significantly increased HA staining compared to the control group (Fig. 3a, d). There was a trend toward greater HA staining intensity in the PLX group than in the control group for both PNNs and diffuse ECM, but these increases did not reach statistical significance (Fig. 3e). Double labeling of HA and aggrecan revealed that regions with increased HA staining in the PLX-treated group also showed a corresponding increase in aggrecan staining (Supplementary Fig. 1). These results suggest that microglial depletion increases both HA and aggrecan levels in the same regions of the ECM.

Increased staining of HA in PNNs and diffuse ECM following microglial depletion. (a) HA staining of the control and PLX cerebral cortices. HA accumulated in PNNs or was diffusely distributed outside PNNs. (b) Spatial relationship between HA stained with bHABP (red) and microglia stained with IBA1 antibody (green). Top: maximum intensity projection of a 20 µm Z-stack. Bottom: single confocal section showing contact between HA and microglia. (c) Fluorescence intensity profiles of HA (red) and microglia (green) along the white line in panel (b). (d) Relative fluorescence intensity of HA staining in a 300 × 300 μm area in the control and PLX groups. n = 5 mice for the control; n = 4 mice for the PLX group. Control: 1.000 ± 0.259, PLX: 1.480 ± 0.180. (e) Relative fluorescence intensity of HA staining in PNNs (left) and outside PNNs (right). n = 5 mice for the control; n = 4 mice for the PLX group. PNNs: Control 1.000 ± 0.216, PLX 1.274 ± 0.341; Outside PNNs: Control 0.545 ± 0.143, PLX 0.736 ± 0.171. The error bars represent the SD. Statistical analysis was performed by an independent t-test; * indicates p < 0.05.
Microglial depletion increases the amount of HA in the cerebral cortex
To investigate whether microglial depletion alters the amount and molecular weight of HA, we isolated HA from soluble and insoluble fractions using bHABP according to a method we previously developed10. Quantification by ELISA showed that the total amount of HA in the cerebral cortex was greater in the PLX group than in the control group (Fig. 4a), in agreement with our histochemical analysis. In the control group, the majority (85%) of HA was recovered in the insoluble fraction (Fig. 4b). We found that the amount of soluble HA was significantly greater in the PLX group than in the control group (Fig. 4a). The amount of insoluble HA increased only slightly in the PLX group, but this increase was not statistically significant. This result highlights the simultaneous increase in both soluble and insoluble HA in the PLX group, with a more pronounced increase in the soluble fraction. Consequently, the proportion of soluble HA was greater in the PLX group than in the control group (Fig. 4b). These results suggest that microglial depletion has a greater effect on soluble HA present in diffuse ECM than on insoluble HA in PNNs.

Microglial depletion increases the amount of HA in the cerebral cortex. (a) ELISA-based quantification of HA in the soluble (Sol) and insoluble (Ins) fractions prepared from the control and PLX groups. n = 5 mice. Control: Sol 4.17 ± 0.85, Ins 23.94 ± 1.63; PLX: Sol 6.16 ± 0.53, Ins 26.45 ± 0.25. (b) Percentage of HA in the Sol and Ins fractions. n = 5 mice. Control: Sol 14.73 ± 1.95, Ins 85.27 ± 1.95; PLX: Sol 18.87 ± 1.32, Ins 81.13 ± 1.32 (c) Agarose gel electrophoresis and Stains-all staining of HA isolated from the soluble and insoluble fractions of the control and PLX groups. HA std: molecular mass standards of HA. (d) Normalized staining intensity profiles and mobilities of HA std (top), soluble HA (middle), and insoluble HA (bottom). The size distribution peaks of soluble and insoluble HA are indicated. Mean ± SD (shaded area). n = 4 mice. (e) mRNA expression levels of HA synthases and HA degrading enzymes in the control and PLX groups. n = 4 mice. Has1: Control 1.00 ± 0.20, PLX 1.08 ± 0.20; Has2: Control 1.00 ± 0.42, PLX 0.51 ± 0.19; Has3: Control 1.00 ± 0.20, PLX 1.01 ± 0.16; Hyal1: Control 1.00 ± 0.61, PLX 2.00 ± 0.45; Hyal2: Control 1.00 ± 0.22, PLX 0.93 ± 0.14; Hyal3: Control 1.00 ± 0.23, PLX 0.89 ± 0.10; Hybid: Control 1.00 ± 0.14, PLX 0.98 ± 0.28; Tmem2: Control 1.00 ± 0.18, PLX 1.12 ± 0.13. The error bars represent the SD. Statistical analysis was performed by an independent t-test; * indicates p < 0.05.
Agarose gel electrophoresis followed by Stains-all staining showed that the molecular weight of soluble HA was lower than that of insoluble HA, consistent with our previous report (Fig. 4c)10. Normalized intensity profiles confirmed that the peak sizes of soluble and insoluble HA in the control group were 500 kDa and 1560 kDa, respectively (Fig. 4d). When comparing the control and PLX groups, the size distributions of soluble and insoluble HA showed little difference. The peaks in the HA size distribution were only slightly shifted toward a lower molecular weight in the PLX group. Since the amount of HA increased in the PLX group, we analyzed the mRNA expression levels of enzymes involved in HA metabolism. The expression level of HA synthase 2 (Has2) tended to decrease in the PLX group, while the expression level of hyaluronidase 1 (Hyal1) tended to increase in the PLX group; however, neither change was statistically significant (Fig. 4e). Thus, the increased amount of HA in the PLX group could not be explained by the gene expression levels of HA metabolic enzymes (Supplementary information 1).
Discussion
Previous studies have focused primarily on the involvement of microglia in PNNs and reported that microglial depletion increases the staining intensity of PNNs detected by WFA11,13,27,28. However, the involvement of microglia in diffuse ECM has not been investigated. In the present study, we examined the effect of microglial depletion on the molecular composition of diffuse and aggregated ECM. We demonstrated that microglial depletion leads to increased levels of aggrecan and HA in diffuse and aggregated ECM, suggesting that microglia play a broader regulatory role in brain ECM homeostasis beyond their established functions in synapse refinement and neuroinflammation. In addition, the effects of microglial removal on the brain ECM may vary by brain region. For example, it has been reported that WFA staining is not increased in the hippocampus of PLX-treated mice, but rather another ECM component, brevican, is increase29.
Histochemical staining with WFA is widely used for PNN detection, but it is important to note that WFA recognizes glycan moieties of CSPGs30,39,40. In our study, immunohistochemical and Western blot analyses revealed an increase in the aggrecan core protein due to microglial depletion. This result suggested that the increased staining intensity of WFA can be attributed to the increase in the aggrecan core protein. Previous studies have reported that microglia depletion by PLX diet enhances the staining intensities of both WFA and aggrecan core protein in the cerebral cortex11,42. Our study reproduced these results, further confirming the effects of microglial depletion on the ECM components in the cerebral cortex. In the microglia-depleted brain, aggrecan gene expression remained unchanged, whereas aggrecan protein levels increased. This finding suggests that under steady-state conditions, aggrecan may be continuously degraded by matrix metalloproteinases and other enzymes expressed by microglia. Previous studies have reported that certain microglial populations in the mouse spinal cord express matrix metalloproteinase 9 and contribute to the degradation of aggrecan-positive PNNs in multiple sclerosis43,44. Further studies are needed to elucidate the precise molecular mechanisms by which microglia mediate aggrecan degradation.
Our study also highlighted the impact of microglial depletion on HA, a critical scaffolding component of the brain ECM. The levels of both soluble and insoluble HA increased after microglial depletion, with a more pronounced increase in soluble HA. The molecular mechanisms by which microglia affect HA metabolism have not been determined, as there were no significant differences in the expression levels of genes encoding enzymes involved in HA synthesis and degradation. In a previous study, we reported an accumulation of HA in the brain with aging, with a particular increase in soluble HA18. The present study showed that soluble HA increases with microglial depletion, suggesting the involvement of microglia in the accumulation of HA in the aged brain. However, further studies are needed to determine how microglia contribute to the degradation of the brain ECM during aging. We have reported that the molecular weight of insoluble HA is significantly greater than that of soluble HA10. In this study, we did not observe dramatic changes in the molecular weight of HA upon microglial depletion, suggesting that microglia are unlikely to be responsible for the size differences between soluble and insoluble HA.
Histochemical staining using the tissue-clearing technique revealed that microglia are in close proximity to PNNs and HA, consistent with the finding of previous studies suggesting that microglia phagocytose and degrade PNNs. In mouse models of neurodegenerative disease, microglia have been shown to internalize PNNs and HA and degrade them in lysosomes13,45. However, the molecular mechanisms by which microglia contribute to brain ECM homeostasis remain unclear. In particular, it is unclear whether microglia recognize specific ECM components to phagocytose PNNs. Immune cells, including macrophages, are known to express hyaluronan receptors such as CD4446. These receptors may play a role in the phagocytosis of the brain ECM by microglia.
In the adult brain, both diffuse and aggregated ECM restrict neuronal plasticity by stabilizing synaptic contacts and limiting dendritic spine motility4,5,6,7,8. The observed increase in PNN components due to microglial depletion may result in altered synaptic dynamics and neural circuit stability. Moreover, given that PNNs provide neuroprotective functions, the regulation of PNN components by microglia may be critical in disease contexts where PNN degradation is observed, such as in Alzheimer’s disease11,12,13,14,15.
In conclusion, our findings underscore the pivotal role of microglia in maintaining the molecular integrity of PNNs and highlight their broader impact the composition of the brain ECM. Investigating the mechanisms by which microglia modulate PNN components may reveal novel therapeutic targets for modulating neural plasticity and protecting neurons from neurodegeneration.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Miyata, S. & Kitagawa, H. Formation and remodeling of the brain extracellular matrix in neural plasticity: Roles of chondroitin sulfate and hyaluronan. Biochim. Biophys. Acta Gen. Subj. 1861, 2420–2434 (2017).
Fawcett, J. W., Oohashi, T. & Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat. Rev. Neurosci. 20, 451–465 (2019).
Deepa, S. S. et al. Composition of perineuronal net extracellular matrix in rat brain: A different disaccharide composition for the net-associated proteoglycans. J. Biol. Chem. 281, 17789–17800 (2006).
Miyata, S., Komatsu, Y., Yoshimura, Y., Taya, C. & Kitagawa, H. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat. Neurosci. 15, 414–422 (2012).
Carulli, D. et al. Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain 133, 2331–2347 (2010).
Pizzorusso, T. et al. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298, 1248–1251 (2002).
Orlando, C., Ster, J., Gerber, U., Fawcett, J. W. & Raineteau, O. Perisynaptic chondroitin sulfate proteoglycans restrict structural plasticity in an integrin-dependent manner. J. Neurosci. 32, 18009–18017 (2012).
de Vivo, L. et al. Extracellular matrix inhibits structural and functional plasticity of dendritic spines in the adult visual cortex. Nat. Commun. 4, 1484 (2013).
Chelini, G. et al. Focal clusters of peri-synaptic matrix contribute to activity-dependent plasticity and memory in mice. Cell Rep. 43, 114112 (2024).
Egorova, D., Nomura, Y. & Miyata, S. Impact of hyaluronan size on localization and solubility of the extracellular matrix in the mouse brain. Glycobiology 33, 615–625 (2023).
Arreola, M. A. et al. Microglial dyshomeostasis drives perineuronal net and synaptic loss in a CSF1R +/− mouse model of ALSP, which can be rescued via CSF1R inhibitors. Sci. Adv. 7, eabg1601 (2021).
Cattaud, V. et al. Early disruption of parvalbumin expression and perineuronal nets in the hippocampus of the Tg2576 mouse model of Alzheimer’s disease can be rescued by enriched environment. Neurobiol. Aging 72, 147–158 (2018).
Crapser, J. D. et al. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 58, 102919 (2020).
Kudo, T. et al. Selective dysfunction of fast-spiking inhibitory interneurons and disruption of perineuronal nets in a tauopathy mouse model. iScience 26, 106342 (2023).
Logsdon, A. F. et al. Perineuronal net deglycosylation associates with tauopathy-induced gliosis and neurodegeneration. J. Neurochem. 168, 1923–1936 (2024).
Brewton, D. H., Kokash, J., Jimenez, O., Pena, E. R. & Razak, K. A. Age-related deterioration of perineuronal nets in the primary auditory cortex of mice. Front. Aging Neurosci. 8, 270 (2016).
Gray, D. T. et al. Retrosplenial cortex microglia and perineuronal net densities are associated with memory impairment in aged rhesus macaques. Cereb Cortex 33, 4626–4644 (2023).
Sugitani, K. et al. Hyaluronan degradation and release of a hyaluronan-aggrecan complex from perineuronal nets in the aged mouse brain. Biochim. Biophys. Acta Gen. Subj. 1865, 129804 (2021).
Brückner, G. et al. Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer’s disease. Neuroscience 92, 791–805 (1999).
Cabungcal, J.-H. et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA. 110, 9130–9135 (2013).
Morawski, M., Brückner, G., Jäger, C., Seeger, G. & Arendt, T. Neurons associated with aggrecan-based perineuronal nets are protected against tau pathology in subcortical regions in Alzheimer’s disease. Neuroscience 169, 1347–1363 (2010).
Morawski, M., Brückner, M. K., Riederer, P., Brückner, G. & Arendt, T. Perineuronal nets potentially protect against oxidative stress. Exp. Neurol. 188, 309–315 (2004).
Wu, Y., Dissing-Olesen, L., MacVicar, B. A. & Stevens, B. Microglia: Dynamic mediators of synapse development and plasticity. Trends Immunol. 36, 605–613 (2015).
Venturino, A. et al. Microglia enable mature perineuronal nets disassembly upon anesthetic ketamine exposure or 60-Hz light entrainment in the healthy brain. Cell Rep. 36, 109313 (2021).
Nguyen, P. T. et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388–403 (2020).
Elmore, M. R. P. et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397 (2014).
Liu, Y.-J. et al. Microglia elimination increases neural circuit connectivity and activity in adult mouse cortex. J. Neurosci. 41, 1274–1287 (2021).
Crapser, J. D. et al. Microglial depletion prevents extracellular matrix changes and striatal volume reduction in a model of Huntington’s disease. Brain 143, 266–288 (2020).
Strackeljan, L. et al. Microglia depletion-induced remodeling of extracellular matrix and excitatory synapses in the hippocampus of adult mice. Cells 10, 1862 (2021).
Miyata, S., Nadanaka, S., Igarashi, M. & Kitagawa, H. Structural variation of chondroitin sulfate chains contributes to the molecular heterogeneity of perineuronal nets. Front. Integr. Neurosci. 12, 3 (2018).
Nadanaka, S. et al. Reconsideration of the semaphorin-3A binding motif found in chondroitin sulfate using Galnac4s-6st-knockout mice. Biomolecules 10, 1499 (2020).
Härtig, W., Meinicke, A., Michalski, D., Schob, S. & Jäger, C. Update on perineuronal net staining with wisteria floribunda agglutinin (WFA). Front. Integr. Neurosci. 16, 851988 (2022).
Susaki, E. A. et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739 (2014).
Tsuji, Y. et al. Diffusion magnetic resonance tractography-based evaluation of commissural fiber abnormalities in a heparan sulfate endosulfatase-deficient mouse brain. Magn. Reson. Imaging 88, 123–131 (2022).
Tainaka, K. et al. Chemical landscape for tissue clearing based on hydrophilic reagents. Cell Rep. 24, 2196–2210 (2018).
Fosang, A. J., Hey, N. J., Carney, S. L. & Hardingham, T. E. An ELISA plate-based assay for hyaluronan using biotinylated proteoglycan G1 domain (HA-binding region). Matrix 10, 306–313 (1990).
Haserodt, S., Aytekin, M. & Dweik, R. A. A comparison of the sensitivity, specificity, and molecular weight accuracy of three different commercially available Hyaluronan ELISA-like assays. Glycobiology 21, 175–183 (2011).
Lee, H. G. & Cowman, M. K. An agarose gel electrophoretic method for analysis of hyaluronan molecular weight distribution. Anal. Biochem. 219, 278–287 (1994).
Giamanco, K. A., Morawski, M. & Matthews, R. T. Perineuronal net formation and structure in aggrecan knockout mice. Neuroscience 170, 1314–1327 (2010).
Rowlands, D. et al. Aggrecan directs extracellular matrix-mediated neuronal plasticity. J. Neurosci. 38, 10102–10113 (2018).
Takechi, M. et al. A pericellular hyaluronan matrix is required for the morphological maturation of cortical neurons. Biochim. Biophys. Acta Gen. Subj. 1864, 129679 (2020).
Crapser, J. D., Arreola, M. A., Tsourmas, K. I. & Green, K. N. Microglia as hackers of the matrix: Sculpting synapses and the extracellular space. Cell Mol. Immunol. 18, 2472–2488 (2021).
Cheung, S. W., Willis, E. F., Simmons, D. G., Bellingham, M. C. & Noakes, P. G. Phagocytosis of aggrecan-positive perineuronal nets surrounding motor neurons by reactive microglia expressing MMP-9 in TDP-43Q331K ALS model mice. Neurobiol. Dis. 200, 106614 (2024).
Cheung, S. W., Bhavnani, E., Simmons, D. G., Bellingham, M. C. & Noakes, P. G. Perineuronal nets are phagocytosed by MMP-9 expressing microglia and astrocytes in the SOD1G93A ALS mouse model. Neuropathol. Appl. Neurobiol. 50, e12982 (2024).
Soria, F. N. et al. Synucleinopathy alters nanoscale organization and diffusion in the brain extracellular space through hyaluronan remodeling. Nat. Commun. 11, 3440 (2020).
Johnson, L. A. & Jackson, D. G. Hyaluronan and its receptors: Key mediators of immune cell entry and trafficking in the lymphatic system. Cells 10, 2061 (2021).
Acknowledgements
We thank members of the Smart-Core-Facility Promotion Organization of Tokyo University of Agriculture and Technology for technical assistance.
Author information
Authors and Affiliations
Contributions
D.E., S.M. Conceptualization; D.E., A.K., E.A. Methodology; D.E. Data curation; D.E. Formal analysis; D.E., S.M. Validation; D.E., S.M. Investigation. D.E. Writing original draft; D.E., A.K., M.I., Y.I., E.A., S.M., Review and editing; S.M. Supervision; D.E., S.M. Funding acquisition. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Egorova, D., Kerever, A., Inada, M. et al. Microglial depletion increases aggrecan and hyaluronan levels in the diffuse and aggregated extracellular matrix of the mouse brain. Sci Rep 15, 9376 (2025). https://doi.org/10.1038/s41598-025-94224-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94224-6
