
Abstract
In recent years, nanoparticle (NP) systems have demonstrated significant promise in pharmaceutical applications. This study focused on the development of donepezil hydrochloride-loaded PLGA-NPs, prepared using the ‘Double Emulsion Solvent Evaporation’ method. The impact of varying concentrations of polyvinyl alcohol-(PVA) in the aqueous phase and sonication time on NP characteristics was comprehensively examined. Results showed that increasing PVA concentration and sonication time resulted in a reduction in NP size, with an optimal formulation (I-DNP) achieving a particle size of 136.37 nm ± 0.93 and a PDI of 0.122 ± 0.011, indicating uniformity. The zeta potential was measured at − 24.17mV ± 1.21, confirming the electrostatic stability of the formulation, essential for long-term stability. Trehalose was incorporated to enhance stability, and gastrointestinal stability testing revealed that I-DNP degraded faster in acidic environments. The encapsulation efficiency reached 69.22 ± 4.84%, suggesting effective drug loading, and release studies exhibited a sustained release profile, with a Fickian and non-Fickian release mechanism. DSC, FT-IR, and 1H-NMR analyses confirmed the encapsulation and structural integrity of the formulation. In biological activity studies, I-DNP exhibited potent anti-AChE and anti-BuChE activities, with Chorioallantoic Membrane (CAM) assays showing significant inhibition of angiogenesis. These findings highlight the potential of I-DNP as a promising therapeutic strategy for Alzheimer’s disease, demonstrating its ability to enhance drug stability, controlled release, and potential blood-brain barrier (BBB) penetration. Future studies will focus on long-term stability testing and in vivo Alzheimer’s models to further validate its clinical applicability. This research contributes to the advancement of nanoparticle-based drug delivery systems for neurodegenerative diseases, paving the way for innovative therapeutic approaches.
Similar content being viewed by others

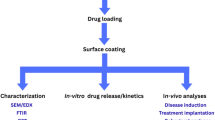
Introduction
Cognitive decline is a prevalent condition among the aging population and a significant cause of death. According to the World Health Organization, it is estimated that more than 55 million people worldwide are affected by severe cognitive impairment, commonly known as dementia. This condition significantly impacts the lives of patients and their families, placing a burden on the healthcare system, society, and the global economy. While there are many different types of dementia, Alzheimer’s disease (AD) is one of the most common cognitive decline disorders. As a result, numerous clinicians, scientists, and researchers in academic, institutional, and industrial settings worldwide are dedicating significant efforts to find a treatment for AD. Unfortunately, despite these extensive efforts, a complete solution has yet to be found, and the underlying reasons for this gap continue to be investigated1,2.
The World Health Organization’s projections indicate that by 2030, one in six people worldwide will be over the age of 60, and the number of people over 60 will double by 2050. This suggests an increase in age-related, non-communicable diseases such as dementia and AD, the leading contributors to dementia. AD is the seventh leading cause of death globally and is a condition that significantly increases healthcare costs3.
At the beginning of the 20th century, Alois Alzheimer, Oskar Fischer, and other researchers examined histochemical observations of neurotic plaque accumulation (amyloid), neurofibrillary tangles/aggregate formation (tau), neuroinflammation, synaptic/neural loss, and brain atrophy as pathological events that define AD. It was discovered that abnormal amyloid accumulation occurs years before the onset of AD (approximately 10 years), while tau aggregation and accumulation begin only a few years before the onset of cognitive impairment in the neocortex. The areas in the brain where tau accumulates and deposits are strongly associated with regional atrophy and related cognitive deficits4,5. As a result, research suggests that biomarkers for AD are progressive neuronal and synaptic loss and the accumulation of two pathognomonic protein aggregates. These biomarkers are commonly represented in the literature by amyloid-β (Aβ) plaques and phosphorylated tau neurofibrillary tangles6. The synaptic levels of acetylcholine decrease as a result of cholinergic neuron involvement. The depletion of other neurotransmitters, loss of neural synapses, mitochondrial dysfunction, oxidative stress, inflammation, ischemia, insulin signaling, and cholesterol metabolism are among many other factors that can contribute to the pathophysiology of AD. The treatment of AD is shaped based on these factors7. The exact pathogenesis of AD, including neuronal degeneration and cognitive impairment, is still not fully understood. However, pharmacological and neurochemical evidence associated with AD indicates a deficiency in central cholinergic neurotransmission. The activity of choline acetyltransferase (ChAT), which synthesizes acetylcholine (ACh), is known to be significantly reduced in the brains of AD patients, particularly in the cortex and hippocampus. The depletion of ChAT is also correlated with the severity of cognitive impairment. Cholinesterase inhibitors (ChEI) increase ACh concentrations by preventing the hydrolysis of acetylcholine in the synaptic cleft, thereby enhancing cholinergic transmission8.
Postmortem analyses of the brain tissue of individuals with AD typically reveal damage and abnormalities in the basal and rostral cholinergic systems. The loss of cholinergic neurons leads to a decline in awareness, attention, learning, sleep, and working memory, emphasizing the importance of therapeutic interventions targeting cholinergic pathways. As a direct result of these approaches, acetylcholinesterase inhibitors (AChEIs) have been developed. Many of these inhibitors serve as clinically effective cholinergic substitutes and are widely used in the treatment of cognitive dysfunction associated with AD9. Despite the urgent need for AD treatment, no fully effective drug has been developed so far. The medications approved by the U.S. Food and Drug Administration (FDA) for clinical use are limited to rivastigmine, donepezil, galantamine, and memantine. However, due to the complex pathological features of AD, these drugs are only effective in alleviating symptoms10 In the modern era, AD is expected to spread globally. Therefore, various drug modifications have been developed aimed at crossing the blood-brain barrier (BBB). AD consists of three stages: mild, moderate, and severe. Donepezil, a piperidine derivative approved by the FDA in 1986, is the only drug used in the treatment of all stages of AD11. Donepezil hydrochloride (DNP) [((±)-2-[(1-benzyl-piperidin-4-yl)ethyl]-5,6-dimethoxyindan-1-one hydrochloride], commercially available as Aricept®, is a potent, selective, and reversible acetylcholinesterase inhibitor and is prescribed worldwide for the treatment of AD12.
In recent years, research on Alzheimer’s disease (AD) therapy has focused on specific targets such as glycogen synthase kinase-3 (GSK-3) and Fyn kinase. GSK-3 regulates key targets associated with neuronal degeneration, and its inhibition has been linked to the reduction of tau hyperphosphorylation, the prevention of β-amyloid formation, and neuroprotective effects. Therefore, GSK-3 inhibitors are considered a potential strategy for AD treatment. Similarly, Fyn kinase, a member of the Src family kinases, plays a crucial role in processes such as myelination and synaptic transmission in the central nervous system. Fyn is associated with two major pathological features of AD due to its interaction with tau protein and activation via amyloid-beta (Aβ). It mediates Aβ-induced neurotoxicity and synaptic dysfunction while also playing a role in the regulation of Aβ synthesis. Consequently, therapeutic inhibition of Fyn kinase is emerging as a promising target for AD management. Additionally, traditional medicinal formulations are also being explored for AD treatment. For instance, the neuroprotective mechanisms of the Bushen Tiansui formula have been investigated in a rat model induced with Aβ25–35, using untargeted metabolomic analyses in the hippocampus and cerebral cortex. Such studies highlight the potential of alternative therapeutic approaches. These findings emphasize the significance of new targets and strategies in AD treatment, providing valuable insights into understanding the complex pathophysiology of the disease and guiding the development of effective therapeutic interventions. There are important studies like this in the literature that aim to shed light on AD disease13,14,15,16,17,18.
Nano carrier systems (NCSs), with unique physicochemical and biological properties, are easily taken up by cells compared to larger molecules, making them effective drug delivery systems. NCSs used in medical applications must be biocompatible and non-toxic. Nanoparticles (NPs) are widely accepted as NCSs for medical use19. Key advantages of using NPs as drug delivery systems include the ability to modify particle size and surface properties for active or passive drug targeting, controlled and/or sustained drug release, high drug loading capacity, enhanced therapeutic efficacy, reduced side effects, and the use of various administration routes such as oral and nasal20.
Oral administration remains the most suitable, convenient, and optimal method for dosing most pharmaceutical agents. However, a major challenge for many orally administered drugs and drug delivery systems is overcoming several barriers before reaching their target sites. Research continues on approaches to improve the oral bioavailability of low-permeability and low-solubility compounds. The use of NPs as a drug delivery system has been recognized as one of these strategies in the literature. By applying nanotechnology to drugs, NPs are created to mimic or alter biological processes. While NPs range from 10 to 1000 nanometers (nm), those between 100 and 500 nm are preferred in medical applications21.
Poly(lactic-co-glycolic acid) (PLGA) consists of endogenous and easily biodegradable lactic and glycolic acid monomers, which is the main reason why NPs prepared with PLGA exhibit low toxicity or no toxicity at all. PLGA is a gold-standard polymer approved by the FDA and the European Medicines Agency (EMA). PLGA exists in different forms depending on the ratio of the monomers, and these forms can provide sustained release profiles over days to years. Additionally, both hydrophilic and hydrophobic active ingredients can be encapsulated in NPs prepared with PLGA. PLGA is one of the most commonly used biologically degradable polymers22,23,24.
Various approaches can be used to produce/synthesize PLGA-based NPs. Two commonly used methods for preparing drug-loaded PLGA-based NPs are the double emulsion solvent/evaporation technique and the nanoprecipitation technique. The difference between these techniques is that hydrophilic drugs are more easily loaded into PLGA-based NPs using the double emulsion solvent/evaporation technique, while hydrophobic drugs are more easily loaded into PLGA-based NPs using the nanoprecipitation technique. DNP is a water-soluble active compound, and therefore, in this study, PLGA-based NPs were preferred to be produced using the double emulsion solvent/evaporation technique25.
In this study, DNP-loaded PLGA-based NPs were successfully synthesized using the “Double Emulsion Solvent Evaporation” method. The main reason for choosing this method was its ability to control the NP size, as well as provide high loading capacity. To optimize the properties of the prepared NPs, the effects of the PVA (polyvinyl alcohol) concentration in the aqueous phase and sonication time on NP size, surface charge, and stability were thoroughly examined. Experimental results showed that certain combinations of PVA concentration and sonication time had significant effects on NP properties, and the optimal formulation was determined based on these data. To enhance the stability of the selected optimal formulation, trehalose was added, and its protective effects were evaluated. Additionally, appropriate storage conditions for the formulation were identified. Stability tests were conducted in gastrointestinal (GIS) fluids over a 24-hour period to assess the formulation’s stability and its ability to maintain effectiveness in a biological environment. Furthermore, in vitro dissolution profiles, release kinetics, and solid-state characterization analyses were performed. These analyses were conducted to determine whether the NPs provided controlled drug release and whether their physicochemical properties were suitable. In the final phase of the study, biological activity tests were carried out.
As a result, this stufy provides important findings for the development of NP-based carrier systems with the potential to overcome the blood-brain barrier, which could be promising for AD treatment. These systems are expected to have significant potential for future clinical applications.
Materials and methods
Materials
Donepezil hydrochloride (DNP) was provided by DEVA (Istanbul, Türkiye). Resomer RG 502 H (PLGA), sodium hydroxide, and trehalose dihydrate were obtained from Sigma-Aldrich (USA). PVA (polyvinyl alcohol), hydrochloric acid, ethyl acetate, and potassium phosphate monobasic were also obtained from Sigma-Aldrich (Germany). Acetone and deuterated chloroform were sourced from Merck (Germany). Deionized water was used in all experiments (Milli-Q Academic, Millipore, Molsheim, France). All other chemicals were of analytical grade. The following equipment was used in the study: Refrigerator (Arçelik, Turkey), freezer (Liebherr LGEX 3410 Medline, Switzerland), analytical balance (Ohaus E1 2140, USA), lyophilizer (Scanvac-Coolsafe Pro, Denmark), magnetic stirrer (Jeiotech MS-53 M, Korea), micropipette set (Eppendorf, Germany), pH meter (WTW Profi Lab pH 597, Germany), probe sonicator (Sonics, USA), ultrasonic bath (Wisd Laboratory Instruments, Korea), UV spectrophotometer (Shimadzu, Japan), vortex (Jeo Tech, Korea), zetasizer (Malvern Nano ZS, UK).
Preparation of NP formulations
To prepare blank NPs, a 20 mg mL− 1 concentration of PLGA solution was first prepared by dissolving 60 mg of PLGA (Resomer RG 502 H) in 3 mL of ethyl acetate. This process was carried out with sufficient mixing time to ensure complete dissolution of the PLGA in the ethyl acetate. Then, 200 µL of polyvinyl alcohol (PVA) solution was gradually added to the prepared 3 mL PLGA solution, using an ultrasonic bath for 1 min. This step resulted in the formation of the W1/Y emulsion. The ultrasonic bath helped achieve a homogeneous emulsion by applying high-frequency sound waves to the solution. The obtained W1/Y emulsion was then gradually added to a 10 mL aqueous PVA solution containing the same concentration of PVA as the initial 200 µL PVA, using a probe sonicator operating at 60% amplitude. This step ensured that the emulsion was uniformly distributed, and the nanoparticle size was maintained at the desired level. As a result, the W1/Y/W2 emulsion was obtained. After preparing the W1/Y/W2 emulsion, the ethyl acetate solvent was evaporated. This process was carried out by stirring the emulsion on a magnetic stirrer (250 rpm) at room temperature, which evaporated the ethyl acetate. After the ethyl acetate was evaporated, centrifugation was applied to collect the nanoparticles from the remaining aqueous solution. This process was performed at 11.000 rpm, 4 °C for 30 min. After centrifugation, the supernatant was carefully removed, and the collected nanoparticles were resuspended in 15 mL of distilled water. To ensure complete washing of the NPs and removal of any residues, this process was repeated three times. After each centrifugation step, the supernatant was carefully discarded, and the NPs were redistributed in distilled water. In this way, clean and pure NPs were obtained. This detailed preparation and washing process ensured the production of high-quality nanoparticles with consistent properties. The same process was used when preparing the formulations with the active ingredient. The only difference from the blank NPs was the addition of 6 mg of DNP to the 200 µL polyvinyl alcohol solution26,27,28. The formulation contents, PVA concentrations, and probe sonication times are presented in Table 1.
Particle size, polydispersity index (PDI), and zeta potential
The particle size (PS) and polydispersity index (PDI) of NPs were analyzed using dynamic light scattering (DLS) on a Zetasizer Nano (Zetasizer Nano ZS, Malvern Instruments, Malvern, UK). The NPs were dispersed in distilled water for PS and PDI measurements. The zeta potential (ZP) was also measured using the same instrument in a disposable folded capillary zeta cell, with the samples diluted with distilled water at a room temperature of 25 °C. All measurements were performed in triplicate, and the results were expressed as mean values with standard deviation (Mean ± SD)29,30,31,32.
Cryoprotectant effect and determination of storage conditions
To determine the cryoprotectant effect and storage conditions, the optimum NP, designated as I-DNP, was prepared. Initially, the PS, PDI, and ZP of the freshly prepared I-DNP formulation were measured. These measurements were performed to establish the initial physicochemical properties of the formulation. Subsequently, the prepared I-DNP formulation underwent centrifugation (11,000 rpm, 4 °C, 30 min). At this stage, a 5.0% (w/v) trehalose solution was prepared to be used as the cryoprotectant and was filtered through a 0.22 μm membrane filter. After centrifugation, the collected NPs were subjected to a washing process, followed by the addition of 1 mL of the 5.0% (w/v) trehalose solution and vortexing. This step ensured the uniform distribution and coating of the NP with trehalose. The formulation with added trehalose was divided into five equal volumes (approximately 1 mL each) in eppendorf tubes. Different volumes of the 5.0% (w/v) trehalose solution were added to each tube: 200 µL to the first tube, 300 µL to the second tube, 500 µL to the third tube, 600 µL to the fourth tube, and 800 µL to the fifth tube. This resulted in five formulations with varying concentrations and amounts of trehalose. All tubes were frozen at − 20 °C and then lyophilized. The lyophilization process converted the formulations into a suitable powder form for long-term storage. Prior to analysis, 1 mL of distilled water was added to each lyophilized nanoparticle formulation, which was then vortexed to resuspend the nanoparticles. After resuspension, the PS, PDI, and ZP were re-measured for each tube. These measurements were conducted to evaluate the cryoprotectant effect on the NPs and to assess the storage conditions. The results obtained from all the tubes were compared to determine the optimal storage conditions and the most suitable trehalose concentration. This study is crucial for ensuring the long-term stability of the formulations and identifying the ideal storage conditions33.
Short-term gastrointestinal stability evaluation
The stability of NPs, especially those made from hydrolytically degradable polymers like PLGA, is known to deteriorate over time. pH and temperature are important factors for long-term stability. In this study, an experiment was designed to evaluate the short-term stability of the I-DNP coded formulation. Before starting the experiment, solutions mimicking gastrointestinal (GI) fluids were prepared. The following solutions were prepared: pH 1.2 HCl buffer (Solution 1), pH 6.8 phosphate buffer (Solution 2), pH 7.4 phosphate buffer (Solution 3).
These three buffer solutions, along with distilled water used as a control, were placed in a shaking water bath at 37 ± 1 °C and 50 rpm to simulate the gastrointestinal environment (total of 4 Falcon tubes). The optimum I-DNP coded formulation was prepared and, after washing procedures, the volume was adjusted to 4 mL. From this NP dispersion, 1 mL was added to the three different incubated buffer solutions and distilled water. The stability study was carried out at 37 ± 1 °C in each environment. Samples were taken from each tube at specific time intervals (1 h, 2 h, 3 h, 4 h, 5 h, 6 h, 7 h, and 24 h) and PS and PDI measurements were performed. Based on the obtained measurement results, the effect of different GI environments on the stability of PLGA NPs was evaluated. This study aims to determine in which pH and temperature conditions the NPs are most stable. The results provide important information regarding the GI environment in which PLGA NPs are most stable, which is crucial for the bioavailability and efficacy of the formulations34.
UV–visible spectrophotometric method
For in vitro studies, the quantification of DNP was performed using a UV spectrophotometer. Two different validation studies were conducted for the encapsulation efficiency (EE %) and in vitro dissolution studies.
UV–visible spectrophotometric method for EE % test
The UV technique was carried out at 325 nm using a quartz cell and a UV-160 A UV/vis recording spectrophotometer (Shimadzu). In this study, analytical validation studies were performed for DNP. A precisely weighed 25 mg of DNP was added to a 25 mL volumetric flask, and a standard solution with a final concentration of 1000 µg.mL− 1 was prepared by dissolving it in a mixture of acetone and distilled water (1:1, v/v). A calibration curve was generated using six concentrations of the standard solution (5–50 µg.mL− 1). To evaluate linearity, a linear regression analysis calculated by the least squares regression approach was used. To calculate the accuracy of the method, repeatability (within-day) and intermediate precision (between-day) were assessed. Three sample solutions (15, 25, and 35 µg mL− 1) were prepared and analyzed for each concentration. At the beginning of the procedure, the amount of known DNP reference standards added to the samples was recovered to assess accuracy. For this purpose, 10 mg of DNP was accurately weighed and placed into a 100 mL volumetric flask and dissolved in a mixture of acetone and distilled water (1:1, v/v). To evaluate accuracy, solutions with concentrations of 15, 25, and 35 µg mL− 1 were prepared from this stock solution. Regarding the selectivity and specificity of the method, spectra obtained from the UV spectrophotometer were compared by observing the overlap of the spectra of DNP, acetone: water (1:1, v/v) mixture, and I-Blank (blank formulation) in the range of 200–800 nm.
UV–visible spectrophotometric method for in vitro dissolution test
The UV technique was performed at 315 nm using a quartz cell and a UV-160 A UV/vis recording spectrophotometer (Shimadzu). In this study, analytical validation studies were conducted for DNP. A precisely weighed 25 mg of DNP was added to a 25 mL volumetric flask, and a standard solution with a final concentration of 1000 µg.mL− 1 was prepared by dissolving it in PBS (pH 6.8). A calibration curve was generated using six concentrations of the standard solution (5–40 µg.mL− 1). To evaluate linearity, linear regression analysis calculated using the least squares regression approach was applied. To calculate the accuracy of the method, repeatability (within-day) and intermediate precision (between-day) were assessed. Three sample solutions (15, 25, and 35 µg.mL− 1) were prepared and analyzed for each concentration. At the beginning of the procedure, the amount of known DNP reference standards added to the samples was recovered to assess accuracy. For this purpose, 10 mg of DNP was accurately weighed and placed into a 100 mL volumetric flask and dissolved in PBS (pH 6.8). To evaluate accuracy, solutions with concentrations of 15, 25, and 35 µg.mL− 1 were prepared from this stock solution and analyzed. Regarding the selectivity and specificity of the method, spectra obtained from the UV spectrophotometer were compared by observing the overlap of the spectra of DNP, PBS (pH 6.8), and I-Blank (blank formulation) in the range of 200–800 nm35,36,37,38,39,40.
Encapsulation efficiency
The DNP content of the DNP-loaded PLGA-based NP formulation was evaluated by extracting the drug directly from the NP formulation. The lyophilized NP was carefully weighed to 5 mg. Then, 1 mL of acetone: water (1:1, v/v) was added, and the NPs were vortexed for 5 min to dissolve them. After vortexing, the prepared solution was filtered through a 0.22 μm membrane filter, and after appropriate dilutions, the DNP content was analyzed using a UV spectrophotometer at 325 nm24,41. Encapsulation efficiency was expressed as EE % and calculated using the following Eq. (1):
Dissolution
Briefly, 5 mg of the formulation containing an amount equivalent to 5 mg of DNP and 5 mg of pure DNP were separately transferred into dialysis bags containing 2 mL of PBS (pH 6.8). The dialysis bags used were cellulose membrane tubes with an average flat width of 33 mm (1.3 inches) and a molecular weight cutoff (MWCO) of 14,000 (D9652, Sigma-Aldrich).
The dialysis bags were then placed into a beaker containing 80 mL of dissolution medium and maintained at 37 ± 1 °C. To prevent evaporation of the dissolution medium, the receptor compartment/beaker was sealed. Samples (3 mL) were withdrawn from the medium at 1, 2, 3, 4, 5, 6, 7, 9, 12, and 24 h, and an equal volume of fresh medium (PBS, pH 6.8) was replenished each time. The DNP concentration in the samples was analyzed using a UV spectrophotometer at 315 nm.
The in vitro dissolution / release study was conducted in triplicate for both I-DNP and pure DNP, and the results were calculated as mean ± SD. Subsequently, the results were plotted as cumulative dissolution data38,42,43.
Release kinetics
The DDSolver software was employed to analyze the dissolution/release kinetics. After determining the dissolution results and profiles, the data were uploaded to the DDSolver application to evaluate four significant and widely used criteria. These criteria and coefficients include the correlation coefficient (Rsqr, R2, or COD), the adjusted correlation coefficient (Rsqr_adj or R2 adjusted), the Akaike Information Criterion (AIC), and the Model Selection Criterion (MSC). To assess different models, the highest R2, R2 adjusted, and MSC values, along with the lowest AIC values, were used as benchmarks for selecting the best-fitting model44,45,46.
Solid-state characterization
Thermal analysis
Thermal properties were analyzed using a differential scanning calorimeter (DSC-60, Shimadzu Scientific Instruments, Columbia, MI). Approximately 5 mg of each sample was placed in an aluminum crucible and subjected to a temperature range of 30–300 °C, with a heating rate of 10 °C/min and an air flow of 50 mL min− 1. Alongside the I-DNP sample, reference samples including pure DNP, I-Blank, and a physical mixture were also tested for comparison47,48.
Fourier-transform infrared spectroscopy (FT-IR) analysis
FT-IR spectra were recorded using a Shimadzu IR Prestige-21 (Shimadzu Corporation, Kyoto, Japan) over the wavelength range of 4000–500 cm–1. The analysis included PLGA, DNP, the physical mixture, and the blank formulation, which served as reference samples49.
Nuclear magnetic resonance 1H-NMR) analysis
1H-NMR analysis was performed using an Ultra Shield CPMAS NMR spectrometer (Brucker, Rheinstetten, Germany). Samples were prepared by dissolving formulations in deuterated chloroform. Reference analyses were also conducted on the blank formulation, physical mixture, DNP, and PLGA50,51.
Biological activity studies
In vitro ache and BuChE inhibitor activity assay
The acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) enzyme inhibition activities of I-DNP and I-Blank were evaluated using a modified Ellman Test to enhance colorimetric detection. The test was conducted with key materials such as BuChE (from horse serum), AChE (from electric eel), Ellman reagent, buffer solutions (potassium dihydrogen phosphate and potassium hydroxide), 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB), sodium bicarbonate, acetylthiocholine iodide (ATC), butyryl thiocholine iodide (BTC), gelatin, dimethyl sulfoxide (DMSO), and DNP. Measurements were performed at 412 nm using a BioTek-Synergy H1 Microplate Reader on 96-well plates. Enzyme solutions (2.5 µg mL− 1), substrate solutions (0.075 M ATC and BTC), DTNB solution (0.01 M), and phosphate buffer (pH 8.0, 0.1 M) were prepared alongside test solutions containing DNP concentrations of 100, 200, and 400 µg mL− 1.
The assay involved two test solutions: the first contained phosphate buffer, DTNB, and enzyme solution (AChE or BuChE), while the second included phosphate buffer and substrate (ATC or BTC). Each well was loaded with 20 µL of the test solution and 110 µL of the first solution, mixed for 5 min, and incubated at 25 °C for 15 min. Following incubation, 80 µL of the second solution was added, mixed for 30 s, and the initial absorbance measurement was taken at 412 nm. The reaction was allowed to proceed for 5 min, and a second spectrophotometric measurement was performed33. The absorbance differences were calculated to determine the percentage inhibition rates (Eq. 2). The abbreviations used in the formula are as follows: B: Blank (the well where the test solution and substrate were not added), C: Control (the well where only the test solution was not added), A(B): Absorbance difference of the blank well, A(C): Absorbance difference of the control well, A(I): Absorbance difference of the test solution well.
CAM (chorioallantoic membrane assay) analysis
For the CAM (chorioallantoic membrane) assay, the protocol suggested by Kıyan and colleagues was modified and applied52,53. Fertilized chicken eggs used for the CAM method were obtained from Bursa HAS Tavuk A.Ş. (Sivrihisar). Due to the period of the experimental process takes place in the first (early) embryonic growth section, between the days 3–7, the test procedure does not call for any ethical committee approval indeed20.
Pellet preparation
The I-DNP, I-Blank, and test substances were suspended in a 2.5% agarose (w/v) solution at a concentration of 1 mg mL− 1. From these suspensions, 10 µL samples were taken and placed on 5 mm diameter stainless steel cylinder supports, then solidified at room temperature to form pellets. The prepared pellets were applied to the CAM surface. A 2.5% agarose (w/v) solution was used as the control.
CAM analysis
Before starting the experiment, fertilized chicken eggs were incubated horizontally in an incubator set at 36.5 °C and 80% relative humidity for 72 h. During the incubation, the eggs were carefully rotated. After the incubation period, 10–15 mL of albumin (egg white) was slowly extracted from the bottom of each egg using sterile syringes. The eggshell and membrane at the top of each egg were then carefully removed. The developing embryo and CAM were covered with stretch film and incubated again under the same conditions (36.5 °C and 80% relative humidity) for another 72 h. When the CAM reached a diameter of approximately 2 cm, freshly prepared sample pellets were placed on the capillaries of the CAM in each egg. For each test substance, 10–15 pellets were applied to an equal number of eggs, and the openings were sealed with stretch film. Before evaluation, the eggs with applied pellets were incubated for an additional 24 h under the specified conditions. A total of 15 eggs were used for each test substance.
Results and discussion
Particle size, polydispersity index (PDI), and zeta potential
The Double Emulsion Solvent Evaporation technique was selected for encapsulation due to the hydrophilic nature of DNP, which makes it more suitable for this method. This technique facilitates the efficient encapsulation of hydrophilic drugs within PLGA NPs by forming a water-in-oil-in-water (W/O/W) emulsion, ensuring better drug entrapment and a controlled release profile. Additionally, it allows for precise PS control, high encapsulation efficiency, and improved stability, which are crucial for optimizing NP-based drug delivery systems. Several studies have demonstrated the effectiveness of this method in encapsulating hydrophilic drugs within PLGA-based NPs, further supporting its selection for this study.
In this study, polyvinyl alcohol (PVA) was used as a stabilizer and emulsifier in the Double Emulsion Solvent Evaporation method to facilitate the formation of homogeneous and stable nanoparticles. PVA plays a critical role in reducing surface tension, thereby stabilizing the water-in-oil-in-water (W/O/W) emulsion and preventing particle aggregation. This leads to a narrow size distribution with a low PDI, which is essential for achieving consistent drug delivery and enhanced bioavailability. Additionally, PVA improves encapsulation efficiency by forming a protective barrier around the DNP-loaded NPs, contributing to a sustained and controlled drug release profile. Given these advantages, PVA was chosen as the surfactant in alignment with previous studies that have demonstrated its effectiveness in PLGA-based NP formulations.
In this study, the PS, PDI, and ZP values of all formulations are presented in Table 2. According to the PS analysis results, it was observed that the DNP-loaded NP formulations had a larger PS compared to the blank NP formulations. When reviewing previous studies in the literature, it is noted that drug-loaded NP formulations are generally larger than blank formulations. This is explained by the fact that large molecular substances, such as polymers, encapsulate the active ingredient to form NPs, which can lead to an increase in the size of the resulting NP54. The smallest PS was obtained in the I-Blank and I-DNP formulations by using the highest PVA concentration (1.0% w/v) and the longest probe sonication time (120 s). These results indicate that increasing concentrations of the surfactant PVA used during the formulation process and longer probe sonication times reduce the PS. This suggests that the presence of the surfactant in the aqueous phase reduces surface tension, allowing newly formed surfaces to be more easily stabilized, thereby facilitating the production of smaller NPs55,56,57,58.
In the literature, PDI values that are generally considered optimal for evaluating the monodispersity of NPs are those smaller than 0.3, while values smaller than 0.5 are also considered acceptable. The monodisperse nature of the I-DNP formulation plays a crucial role in enhancing its therapeutic effectiveness by ensuring a uniform PS distribution, which directly impacts drug release, stability, and bioavailability. The low PDI observed in the formulation indicates a homogeneous NP population, leading to consistent pharmacokinetics and predictable drug release profiles. This uniformity is particularly beneficial for blood-brain barrier (BBB) penetration, a key challenge in the treatment of AD, as it improves cellular uptake and drug transport efficiency. Additionally, the stabilizing effect of trehalose further supports the structural integrity and stability of the NP, preventing aggregation and degradation during storage. These combined factors contribute to the overall therapeutic potential of the I-DNP formulation, making it a promising candidate for NP-based drug delivery in neurodegenerative diseases59. Based on the information in the literature, it can be concluded that all PLGA NPs prepared in this study were produced of high quality and monodisperse.
There are several studies indicating that PS is a critical factor for the successful passage of nanosystems across the blood-brain barrier. For example, a study by Pinheiro et al. found that all NPs were below 250 nm, emphasizing that this PS is important for the ability to cross the blood-brain barrier (BBB)60. Similarly, Revdekar and Shende suggested that systems suitable for diseases such as AD should typically be below 150 nm61. Another study by Madhu, Komala, and Pandian indicated that the optimal PS for nanosystems used in AD treatment is 121 nm62. In general, the literature recommends nanosystems with smaller PSs for the ability to cross the blood-brain barrier. The ability of the I-DNP formulation to cross the BBB is primarily attributed to its nano-sized structure and surface charge properties, which facilitate passive diffusion and endocytosis-mediated transport. NPs with a size below 200 nm have been reported to enhance BBB permeability by utilizing transcytosis pathways, such as adsorptive-mediated and receptor-mediated transport mechanisms. The negative zeta potential of the I-DNP formulation further contributes to its stability in circulation, preventing aggregation and ensuring efficient drug delivery to the brain. These characteristics suggest that the developed PLGA-based NP system may enhance the bioavailability of DNP, thereby improving its therapeutic potential in the treatment of AD63,64,65,66. In line with this literature information, the I-DNP coded formulation was chosen as the optimal formulation due to its smallest PS.
Upon examining Table 2, the prepared formulations have a negative ZP. PLGA has a negative surface potential due to its terminal carboxyl groups in neutral environments. This property explains the negative ZP values obtained for the formulations67. As noted by Öztürk and Aygül, a ZP value in the range of +/− 30 mV is considered acceptable for a stable colloidal system68. Studies by Müller, Mäder and Gohla have shown that zeta potential values between − 5.0 and − 15.0 mV are on the threshold of flocculation in nanosystems, with values between − 5.0 and − 3.0 mV indicating maximum flocculation69. Based on this information, the I-DNP coded formulation with a zeta potential of -24.17 mV ± 1.21 can be concluded to be stable.
Cryoprotectant effect and storage condition determination
The results for this study are presented in Table 3. Lyophilization (freeze-drying) is widely used to enhance the stability of pharmaceutical products and nanosystems and to facilitate their storage70. However, PS may increase during the lyophilization of NP formulations, so cryoprotectant agents are added. The most common cryoprotectants are trehalose, sucrose, glucose, and mannitol71. In this study, trehalose solution was used in different concentrations to prevent changes in PS after lyophilization of the I-DNP coded formulation. Trehalose was exclusively used as the stabilizing agent due to its well-established cryoprotective and stabilizing properties in NP formulations. Trehalose plays a crucial role in preventing aggregation and degradation during the freeze-drying process by forming a protective glassy matrix around the NPs, thereby preserving their PS, PDI, and zeta ZP. Its biocompatibility, non-toxicity, and widespread use in pharmaceutical applications further support its selection as an ideal stabilizer. Given these advantages, trehalose was chosen to ensure the long-term stability and structural integrity of the developed NPs. After centrifugation and washing, the NPs were processed with a 5.0% (w/v) trehalose solution. Trehalose solution was added to five tubes in the following amounts: 200 µL, 300 µL, 500 µL, 600 µL, and 800 µL, and then freeze-dried. After lyophilization, PS, PDI, and ZP were measured for each tube. The results showed that 800 µL of trehalose solution was sufficient to maintain a stable PS. It was found that this amount preserved the properties of the fresh formulation.
Short-term gastrointestinal stability evaluation
In this study, the stability of the I-DNP coded formulation, selected as the optimal formulation, was analyzed at 37 °C in different GI environments to better understand its short-term GI stability and in vitro dissolution profiles. These analyses were carried out for 24 h in pH 1.2 HCl buffer, pH 6.8 phosphate buffer, pH 7.4 phosphate buffer, and distilled water. It is known that NPs made from hydrolytically degradable polymers degrade over time. Temperature and pH are reported to have significant effects on the long-term stability of drugs72. Upon reviewing the analysis results (see Table 4), the behavior of NPs in different pH environments was observed to differ significantly. In the pH 6.8 phosphate buffer, pH 7.4 phosphate buffer, and distilled water, the PS showed a slower and less significant increase over time. In these environments, the PS increased minimally compared to its initial value. However, in the pH 1.2 HCl buffer, the PS increased rapidly. The PS showed a significant increase from zero time, reaching 514.8 nm ± 20.3 after 24 h. Literature suggests that the I-DNP coded NP formulation prepared in this study undergoes rapid degradation in the pH 1.2 HCl buffer, while it degrades more slowly in the other environments21,72. In conclusion, these analyses show that the I-DNP coded NP formulation exhibits different stability profiles in GI environments and undergoes rapid degradation in acidic environments (pH 1.2 HCl buffer). This information is crucial for evaluating the in vivo performance of the formulation and its potential applications.
UV–visible spectrophotometric method
The UV-visible spectrophotometric method designed for quantitative determination in EE % studies was validated in an acetone: distilled water (1:1, v/v) medium. As a result of the linearity study, the regression equation was found to be y = 0.0186x + 0.0177 (r² = 0.9992) in the concentration range of 5–50 µg.mL− 1. The %RSD calculated for each precision level (15, 25, and 35 µg.mL− 1) was found to be less than 2%, indicating that the method is precise. For the UV-visible spectrophotometric method, accuracy values were determined as 100.124 ± 0.207%, 99.770 ± 0.431%, and 99.925 ± 0.387% for the concentrations of 15, 25, and 35 µg.mL− 1, respectively. In the selectivity study, blank formulations (I-blank) and the acetone: water (1:1 v/v) mixture were analyzed photometrically between 200 and 800 nm and did not produce an absorbance peak at 325 nm. The LOD and LOQ were obtained as 1.275 µg.mL− 1 and 3.863 µg.mL− 1, respectively, which were lower than the lowest concentration studied in linearity, proving the suitability of the method.
In in vitro dissolution studies, the UV-visible spectrophotometric method designed for quantitative determination was validated in PBS pH 6.8 medium. The regression equation was found to be y = 0.0227x + 0.0253 (r² = 0.9991) in the concentration range of 5–40 µg.mL− 1. The %RSD calculated for each precision level (15, 25, and 35 µg.mL− 1) was found to be less than 2%, confirming the precision of the method. For the UV-visible spectrophotometric method, accuracy values were determined as 100.306 ± 0.449%, 99.850 ± 1.275%, and 100.074 ± 0.126% for concentrations of 15, 25, and 35 µg.mL− 1, respectively. In the selectivity study, blank formulations (I-blank) and PBS pH 6.8 were analyzed photometrically between 200 and 800 nm and did not produce an absorbance peak at 315 nm. The LOD and LOQ were obtained as 1.220 µg.mL− 1 and 3.694 µg.mL− 1, respectively, which were lower than the lowest concentration studied in linearity, proving the suitability of the method73,74,75,76.
As a result, the two methods proposed in this study can be used for routine and simultaneous DNP determination using simple and cost-effective approaches.
Encapsulation efficiency
EE % is a critical parameter for the prepared NP formulations, as higher encapsulation and loading efficiency allows for the administration of fewer NPs to the patient for a given drug dose. In this study, the EE % of the I-DNP coded formulation was determined to be 69.22 ± 4.84%. According to the literature, this result can be considered ideal, especially for oral applications34,77.
Dissolution
In previous studies, it has been observed that pH 6.8 PBS is a preferred medium for release and dissolution tests for DNP in the literature25,78. Based on GI stability evaluation and existing literature information, it was decided to perform the dissolution test in a pH 6.8 PBS environment for this study. The in vitro dissolution results are shown in Fig. 1. In the dissolution test conducted in this study, pure DNP showed a release rate of 79.3 ± 6.8% at the end of the first hour, and 97.6 ± 2.3% at the end of the second hour in pH 6.8 PBS. By the third hour, the release rate reached 99.8 ± 0.1%, indicating almost complete release. When the release profile of DNP from the I-DNP coded NP formulation was examined, the following release values were observed at 1, 2, 3, 4, 5, 6, 7, 9, 12, and 24 h: 19.7 ± 3.5%, 22.6 ± 6.2%, 34.6 ± 3.4%, 40.3 ± 2.9%, 47.4 ± 3.1%, 52.6 ± 2.6%, 54.2 ± 4.8%, 60.4 ± 2.7%, 68.4 ± 4.5%, and 83.7 ± 5.2%. Based on the literature, it is clear that the I-DNP coded NP formulation shows a slower and extended release compared to pure DNP. This release profile, continuing for 24 h, indicates that the formulation may potentially provide extended release effects79,80,81.

In vitro dissolution results graph.
Release kinetics
The release kinetics results are presented in Table 5. Upon examining Table 5 and according to the defined criteria, it was determined that the DNP release from the I-DNP formulation follows the Weibull model. In the literature, the Weibull model is reported to be a highly suitable model for matrix-type nano drug delivery systems82,83,84. When all kinetic results were examined, it was found that the R², R² adjusted, MSC, and AIC values were highest for the Weibull model. However, for some models, the obtained values, although not as high as the Weibull model, were quite close. In other words, a high correlation was observed between some models and the Weibull model. Such situations have also been previously reported in the literature85,86. As a result, all obtained data and literature review suggest that the release of DNP from the I-DNP NP formulation occurs through a combination of both Fickian (pure diffusion phenomenon) and non-Fickian (due to polymer chain relaxation) release mechanisms, rather than a single mechanism86.
Solid-state characterization
Thermal analysis
In this study, thermal analyses were performed for DNP, PLGA, I-Blank, I-DNP, and the physical mixture, and Differential Scanning Calorimetry (DSC) curves/thermograms were obtained (see Fig. 2). Pure DNP exhibited two distinct melting peaks at 133.69 °C and 198.06 °C (see Fig. 2A). According to literature reviews, these two characteristic endothermic melting peaks are typical for the pure form of DNP and confirm its crystalline structure25.
In the blank formulation (I-Blank) and DNP-loaded I-DNP formulation, endothermic peaks were observed at 43.48 °C and 44.39 °C, respectively (see Fig. 2C and D). These peaks represent the glass transition temperature of PLGA, indicating the amorphous nature of the polymer27. In the thermogram of the DNP-loaded I-DNP formulation, no clear melting peak was observed. This suggests a possible reduction in the crystallinity of the drug and/or the dissolution of the drug in the melted carrier. In the thermogram of the physical mixture (see Fig. 2E), both the characteristic melting peaks of DNP and the glass transition temperature peaks of PLGA were observed. This indicates that the components of the physical mixture maintain their individual existence and characteristic properties. However, in the thermogram of the I-DNP formulation, the disappearance of DNP’s characteristic melting peaks suggests that the drug is encapsulated in an amorphous form and is molecularly dispersed within the polymer matrix, without undergoing any chemical changes during the formulation preparation process28.
As a result, thermal analyses confirm the crystalline structure of pure DNP, the glass transition temperature of PLGA, and the encapsulation efficiency and physical state of the DNP-loaded formulation. Specifically, the loss of the crystalline structure of DNP in the I-DNP formulation indicates that the drug is encapsulated in its amorphous form and is uniformly distributed within the polymer matrix. This suggests that the drug is effectively encapsulated within the polymer and maintains its chemical integrity during the formulation process.

Thermograms obtained from thermal analysis. *(A) DNP, (B): PLGA, (C) I-Blank, (D) I-DNP, (E) physical mixture.
Fourier-transform infrared spectroscopy (FT-IR) analysis
In this study, FT-IR analyses were performed for pure DNP, PLGA, I-Blank, I-DNP, and the physical mixture, and FT-IR spectra were obtained (See Fig. 3). In the FT-IR spectrum of pure DNP, sharp absorption bands observed at 1681.93 cm− 1 and 1313.5 cm− 1 correspond to the characteristic peak of the carbonyl (C= O) group and the stretching vibrations of carbon–nitrogen (C–N), respectively (See Fig. 3A). In the FT-IR analysis, the carbon–nitrogen (C–N) stretching vibrations were observed within the expected spectral range, with aliphatic C-N stretching typically occurring between 1200 and 1350 cm− 1 and aromatic C-N stretching around 1650–1700 cm− 1. These findings are consistent with literature values, confirming the structural integrity. These bands provide important clues for determining the chemical structure of DNP10. In the FT-IR spectra of PLGA, I-Blank, and I-DNP coded formulations (See Fig. 3B, C, and D, respectively), medium-intensity bands observed in the range of 1450 –1200 cm− 1 reflect the asymmetric and symmetric carbonyl-carbon (C-C(= O)-O) extensions of the PLGA polymer. Additionally, C-O stretching and C-H bending vibrations were observed in the range of 1200 –1050 cm− 1 and 1450–850 cm− 1. These vibrations and bends can be attributed to the structural features of PLGA. The presence of these bands reveals the characteristic features of the polymer in the spectrum87. The absence of new and distinct peaks in the FT-IR spectrum of the I-DNP coded formulation suggests that there is no chemical interaction between PLGA and DNP. This indicates that PLGA forms a physical mixture with DNP, without the formation of chemical bonds. In the I-DNP coded formulation (See Fig. 3D), the reduced intensity of the characteristic DNP peaks suggests a decrease in the crystallinity of DNP and its transition to an amorphous form. These results demonstrate that DNP is distributed at the molecular level within the polymer matrix and encapsulated. FT-IR analyses confirm that DNP exhibits a homogeneous distribution within the formulation and is effectively encapsulated in the PLGA polymer matrix. Furthermore, when evaluated together with the DSC results, this supports the notion that DNP exists in an amorphous form and retains its chemical integrity within the polymer matrix88. In conclusion, when all FT-IR analyses are collectively considered, it is clearly evident that DNP is molecularly distributed within the formulation and encapsulated within the polymeric structure. This demonstrates that effective encapsulation of DNP has occurred and that its chemical properties have been preserved during the formulation process. The FT-IR spectra support the structural integrity and stability of the physical mixture between DNP and PLGA, thereby supporting the effectiveness and reliability of the formulation.

FT-IR analysis results. *(A) DNP, (B) PLGA, (C) I-Blank, (D) I-DNP, (E) physical mixture.
Nuclear magnetic resonance 1H-NMR) analysis
In this study, 1H-NMR analyses were performed for pure DNP, PLGA, I-Blank, I-DNP, and physical mixture, and 1H-NMR spectra were obtained (see Fig. 4). As a result of the analyses, specific peaks for pure DNP were observed at the ranges of 7.43–7.64 ppm, 7.09 ppm, 6.82 ppm, 3.95 ppm, 3.82 ppm, 3.66 ppm, 2.65-3.00 ppm, 1.84–2.06 ppm, and 1.54–1.85 ppm89. The 1H-NMR spectrum of pure DNP obtained in this study was found to be consistent with the values reported in the literature (see Fig. 5A). This compatibility confirms the accuracy and purity of the chemical structure of pure DNP. In the 1H-NMR spectrum of the blank formulation (I-Blank) without the active substance (see Fig. 4C), a similarity with the spectrum of pure polymer (see Fig. 4B) was observed, and no peaks corresponding to the specific DNP peaks were detected. This finding verifies that the I-Blank formulation consists only of PLGA polymer and does not contain DNP21. In the 1H-NMR spectrum of DNP-loaded I-DNP coded formulation, characteristic peaks of DNP were observed (see Fig. 4D). However, a decrease in the intensity of these peaks was noted. This reduction in peak intensity at characteristic ppm values in proportion to the molecular distribution and concentration of DNP indicates that DNP is molecularly distributed within the polymeric structure. This suggests that DNP has been successfully loaded into the nanoparticles and is homogeneously distributed within the polymer matrix55. These results demonstrate that DNP has been successfully encapsulated within the PLGA polymer matrix and exhibits a molecularly homogeneous distribution in this structure. The 1H-NMR analyses have played a critical role in determining the state and distribution of DNP within the polymer matrix. The obtained data show that DNP is in an amorphous form within the polymer matrix and that its crystalline form has largely disappeared. These findings confirm that DNP has been effectively loaded into the nanoparticles and that no changes have occurred in its chemical structure during the formulation process. Furthermore, these results demonstrate that the formulation maintains its physicochemical properties and performs effectively as a drug carrier system.

1H-NMR analysis results. *(A): DNP, (B) PLGA, (C) I-Blank, (D) I-DNP, (E) physical mixture.
Biological activity studies
In vitro ache and BuChE inhibitor activity assay
Acetylcholinesterase (AChE) inhibitor activity is an important component of this study and has been evaluated with the results presented in Table 6. According to the obtained data, the I-DNP coded formulation demonstrated 96.88 ± 0.16% inhibition at a concentration of 400 µg mL− 1, indicating strong anti-AChE activity compared to DNP. On the other hand, no activity was observed in the Blank formulation (I-Blank), confirming that the formulation has no effect on AChE. AChE inhibitor activity is an important potential strategy in the pharmacological treatment of AD. The activity of the I-DNP coded formulation provides a valuable example of how NP carrier systems can be used in therapeutic applications. These findings represent an important step in understanding the biological efficacy of the formulation and evaluating its potential clinical applications.
Another important component of this study is the BuChE inhibitor activity, which is evaluated with the results presented in Table 7. According to the obtained data, the I-DNP formulation at a concentration of 400 µg mL− 1 showed 82.96 ± 0.57% inhibition. This result demonstrates that the formulation exhibits a strong anti-BuChE activity compared to DNP. On the other hand, no activity was observed in the blank formulation (I-Blank), confirming that the formulation has no effect on BuChE. BuChE inhibitor activity may be a potential target in the pharmacological treatment of AD. The activity of the I-DNP formulation provides insight into the role of nanoparticle carrier systems in AD treatment. These findings support the biological efficacy of the formulation and its potential therapeutic applications.
According to the anti-Alzheimer activity results of this study, the I-DNP formulation has been found to exhibit strong anti-AChE and anti-BuChE activities compared to DNP. AChE and BuChE inhibitor activities are important as potential strategies in the pharmacological treatment of AD. The strong activities of the I-DNP formulation emphasize the potential of NP carrier systems in AD treatment. AChE and BuChE inhibitor activities have the potential to protect nerve cell health and alleviate the symptoms of AD90,91. These findings represent an important step in understanding the biological efficacy of the I-DNP formulation and its potential clinical application.
CAM (chorioallantoic membrane assay) analysis
Angiogenesis was defined by the English surgeon Dr. John Hunter at the end of the 17th century as the “formation of new blood vessels from existing ones”92. This process can occur in both physiological and pathological contexts in the human body. Physiological angiogenesis is observed in healthy individuals during conditions such as embryogenesis, wound healing, and the female reproductive system, whereas pathological angiogenesis is associated with various diseases. Pathological angiogenesis occurs particularly in conditions such as psoriasis, atherosclerosis, chronic inflammation, rheumatoid arthritis, inflammatory bowel syndrome, tumor development, and metastasis. Additionally, age-related conditions like diabetic retinopathy and macular degeneration can also involve angiogenesis in cardiovascular and ocular diseases. It is well known that angiogenesis plays a crucial role in the development of these diseases, and research on the molecular mechanisms of this process contributes to the development of new therapeutic strategies92,93,94,95,96.
Angiogenesis, or neovascularization, is a complex process that involves the activation, adhesion, migration, and proliferation of endothelial cells from existing blood vessels97,98. This process appears differently in both neoplastic and non-neoplastic diseases. Neoplastic diseases, particularly tumors, are characterized by significant neovascularization. In these diseases, the formation of new blood vessels supports tumor growth and contributes to metastasis. On the other hand, in non-neoplastic diseases, angiogenesis processes can occur without new blood vessel formation. In these diseases, the reorganization of existing blood vessels and the other effects of angiogenesis can influence the course of the disease. For example, non-neoplastic diseases like AD and diabetes involve angiogenesis as an important part of the pathophysiological process. Understanding and targeting the molecular mechanisms of angiogenesis in the development of these diseases can contribute to the development of potential therapeutic strategies99,100,101.
According to the angiogenic effect results of this study, the effects of the pure DNP, I-DNP, and I-Blank formulations on the CAM model are presented in Table 8. The results show that the I-DNP formulation significantly inhibited capillary vessel development at a concentration of 10 µg pellet− 1, compared to the control (%2.5 agarose) and I-Blank formulations. This finding indicates the anti-angiogenic effect of I-DNP. The ranking of activity based on the reduction in capillary area shows that I-DNP is the most effective formulation, while DNP is more effective than I-Blank but not as strong as I-DNP. These findings support the potential use of the I-DNP NP formulation in anti-angiogenic therapeutic strategies. The anti-angiogenic effect images are shown in Fig. 5.

Anti-angiogenic effect results. *(A) Anti-angiogenic effect of DNP (10 µg pellet− 1) (total capillary area = 17.982 ± 2.595), (B) anti-angiogenic effect of I-DNP formulation (10 µg pellet− 1) (total capillary area = 14.702 ± 3.395), (C) effect of I-Blank formulation (10 µg pellet− 1) (total capillary area = 20.362 ± 3.353), (D) normal embryo development with agarose (%2.5, a/h) (total capillary area = 37.023 ± 1.230).
Conclusions
This study successfully developed and characterized donepezil hydrochloride-loaded PLGA-based nanoparticles (I-DNP) using the Double Emulsion Solvent Evaporation method. The optimized formulation exhibited a monodisperse structure (136.37 nm, PDI: 0.122), a high encapsulation efficiency (69.22 ± 4.84%), and a sustained drug release profile governed by Fickian and non-Fickian mechanisms. Stability studies confirmed the need for protective strategies in acidic environments, and the addition of trehalose effectively enhanced nanoparticle stability. DSC, FT-IR, and ¹H-NMR analyses confirmed the successful encapsulation of donepezil hydrochloride, ensuring structural integrity. Biological evaluations demonstrated that I-DNP exhibited strong anti-AChE and anti-BuChE activity, confirming its potential in Alzheimer’s disease therapy. Additionally, CAM assays revealed anti-angiogenic properties, suggesting a broader therapeutic potential. Future research will focus on long-term stability testing, blood-brain barrier (BBB) penetration, the incorporation of alternative carrier systems, and in vivo Alzheimer’s models to further validate the formulation’s clinical potential. These investigations are crucial for optimizing bioavailability, targeting efficiency, and overall therapeutic effectiveness, paving the way for the translation of the I-DNP formulation into clinical applications.
Data availability
All results obtained during this study are included in the article, and a supplementary file containing NMR analyses is provided to the presented version.
References
Korczyn, A. D. & Grinberg, L. T. Is alzheimer disease a disease? Nat. Rev. Neurol. 20, 245–251 (2024).
Better, M. A. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 20, 3708–3821 (2024).
Self, W. K. & Holtzman, D. M. Emerging diagnostics and therapeutics for alzheimer disease. Nat. Med. 29, 2187–2199 (2023).
Bateman, R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804 (2012).
Vermunt, L. et al. Duration of preclinical, prodromal, and dementia stages of alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 15, 888–898 (2019).
Tzioras, M., McGeachan, R. I., Durrant, C. S. & Spires-Jones, T. L. Synaptic degeneration in alzheimer disease. Nat. Rev. Neurol. 19, 19–38 (2023).
Winslow, B. T., Onysko, M. K., Stob, C. M. & Hazlewood, K. A. Treatment of alzheimer disease. Am. Fam Physician. 83, 1403–1412 (2011).
Zhang, P. et al. In vitro and in vivo evaluation of donepezil-sustained release microparticles for the treatment of Alzheimer’s disease. Biomaterials 28, 1882–1888 (2007).
Brewster, J. T., Dell’Acqua, S., Thach, D. Q. & Sessler, J. L. Classics in chemical neuroscience: donepezil. ACS Chem. Neurosci. 10, 155–167 (2018).
Liu, T. et al. A donepezil/cyclodextrin complexation orodispersible film: Effect of cyclodextrin on taste-masking based on dynamic process and in vivo drug absorption. Asian J. Pharm. Sci. 14, 183–192 (2019).
Tripathi, S. K. et al. Donepezil loaded PLGA Nanoparticles, from modified nano-precipitation, an advanced drug delivery system to treat Alzheimer disease. J. Phys.: Conf. Seri. 1849 12001 (2021).
Nakashima, K., Itoh, K., Kono, M., Nakashima, M. N. & Wada, M. Determination of donepezil hydrochloride in human and rat plasma, blood and brain microdialysates by HPLC with a short C30 column. J. Pharm. Biomed. Anal. 41, 201–206 (2006).
Zhang, Q. et al. Multi targeted therapy for Alzheimer’s disease by guanidinium-modified calixarene and cyclodextrin co-assembly loaded with insulin. ACS Nano 18, 33032–33041 (2024).
Karati, D. et al. Glycogen synthase kinase 3 (GSK3) Inhibition: A potential therapeutic strategy for Alzheimer’s disease. Naunyn Schmiedebergs Arch. Pharmacol. 1–24 (2024).
Meur, S. & Karati, D. Fyn kinase in Alzheimer’s disease: Unraveling molecular mechanisms and therapeutic implications. Mol. Neurobiol. 1–18 (2024).
Luo, H. et al. Apelin-13 suppresses neuroinflammation against cognitive deficit in a streptozotocin-induced rat model of Alzheimer’s disease through activation of BDNF-TrkB signaling pathway. Front. Pharmacol. 10, 395 (2019).
Li, H. et al. Untargeted metabolomics analysis of the hippocampus and cerebral cortex identified the neuroprotective mechanisms of Bushen Tiansui formula in an aβ25-35-induced rat model of Alzheimer’s disease. Front. Pharmacol. 13, 990307 (2022).
Wang, H. et al. A strategy of monitoring acetylcholinesterase and screening of natural inhibitors from uncaria for Alzheimer’s disease therapy based on near-infrared fluorescence probe. Sens. Actuators B Chem. 424, 136895 (2025).
Solak, B., Soyer, P., Tilki, E. K. & Öztürk, A. A. Evaluation of antimicrobial properties and antioxidant activity of rosmarinic acid-loaded resomer RG 502 H-based nanoparticles against tert-butyl hydroperoxide-induced oxidative stress: A detailed formulation, characterization and efficacy determination study. J. Drug Deliv Sci. Technol. 106721 (2025).
Öztürk, A. A. & Kıyan, H. T. Treatment of oxidative stress-induced pain and inflammation with dexketoprofen trometamol loaded different molecular weight chitosan nanoparticles: Formulation, characterization and anti-inflammatory activity by using in vivo HET-CAM assay. Microvasc Res. 128, 103961 (2020).
Öztürk, A. A., Yenilmez, E. & Özarda, M. G. Clarithromycin-loaded Poly (lactic-co-glycolic acid) (PLGA) nanoparticles for oral administration: Effect of polymer molecularweight and surface modification with chitosan on formulation, nanoparticle characterization and antibacterial effects. Polym. (Basel) 11, (2019).
Öztürk, A. A., Banderas, L. M., Otero, C., Yenilmez, M. D., Yazan, Y. & E. & New approach to hypertension treatment: Carvediol-Loaded PLGA nanoparticles, preparation, in vitro characterization and gastrointestinal stability. Lat Am. J. Pharm. 37, 1730–1771 (2018).
Kaval, B., Şen, F. D., Batmaz, K. K., Ekinci, M. & Öztürk, A. A. Latest research about active pharmaceutical ingredient loaded poly lactic acid-co-glycolic acid (PLGA) based drug delivery system in Türkiye. Eur. J. Life Sci. 1, 127–139 (2023).
Kaya-Tilki, E., Öztürk, A. A., Engür-Öztürk, S. & Dikmen, M. Enhanced anti-angiogenic effects of aprepitant-loaded nanoparticles in human umbilical vein endothelial cells. Sci. Rep. 14, 19837 (2024).
Alshetaili, A., Almutairy, B. K., Alshehri, S. M. & Repka, M. A. Development and characterization of sustained-released donepezil hydrochloride solid dispersions using hot melt extrusion technology. Pharmaceutics 13, 213 (2021).
Ekinci, M., Öztürk, A. A., Santos-Oliveira, R. & İlem-Özdemir, D. The use of lamivudine-loaded PLGA nanoparticles in the diagnosis of lung cancer: Preparation, characterization, radiolabeling with 99mTc and cell binding. J. Drug Deliv Sci. Technol. 69, 103139 (2022).
ÖZTÜRK, A. A. & KIRIMLIOĞLU, G. Y. Preparation and in vitro characterization of lamivudine loaded nanoparticles prepared by acid and/or ester terminated PLGA for effective oral anti-retroviral therapy. J. Res. Pharm. 23, 897–913 (2019).
Öztürk, A. A., Çinar, N. İ. & Yenilmez, E. Development of nano-sized ketoprofen lysine incorporated Eudragit ® S100 nanomedicine by double emulsion solvent evaporation and in vitro characterization. J. Pharm. Pharmacogn Res. 7, 47–58 (2019).
Baghirova, L., Kaya Tilki, E. & Öztürk, A. A. Evaluation of cell proliferation and wound healing effects of vitamin A palmitate-loaded PLGA/chitosan-coated PLGA nanoparticles: Preparation, characterization, release, and release kinetics. ACS Omega. 8, 2658–2668 (2023).
Öztürk, A. A., Gündüz, A. B. & Ozisik, O. Supervised machine learning algorithms for evaluation of solid lipid nanoparticles and particle size. Comb. Chem. High. Throughput Screen. 21, 693–699 (2018).
Öztürk, A. A., Merve Güven, U., Yenilmez, E. & Şenel, B. Effects of different derivatives of Eudragit polymer on entrapment efficiency, in vitro dissolution, release kinetics and cell viability results on extended release flurbiprofen loaded nanomedicines. Lat Am. J. Pharm. 37, 1981–1992 (2018).
Ozturk, A., Yenilmez, G. Ü. V. E. N. U., Senel, B. & E. & Effects of different derivatives of Eudragit polymer on entrapment efficiency, in vitro dissolution, release kinetics and cell viability results on extended release flurbiprofen loaded nanomedicines. Lat. Am. J. Pharm. 37, 1981–1992 (2018).
Arınmış, K. N., Kıyan, H. T. & Ozturk, A. A. Preparation, characterization, antioxidant activities, and determination of anti-Alzheimer effects of PLGA-based DDSs containing ferulic acid. ACS Omega. 9, 11321–11338 (2024).
Martín-Banderas, L. et al. Cannabinoid derivate-loaded PLGA nanocarriers for oral administration: formulation, characterization, and cytotoxicity studies. Int. J. Nanomed. 7, 5793 (2012).
Committee, I. C. H. & S. ICH Q2B validation of analytical procedures: Methodology. (European agency for the evaluation of medicinal products, International commission on harmonisation, London (CPMP/ICH/281/95), 1996).
Guideline, I. C. H. H. Validation of analytical procedures Q2 (R2) (ICH, Geneva, Switzerland, 2022).
Öztürk, A. A., Güven, U. M. & Yenilmez, E. Flurbiprofen loaded gel based topical delivery system: formulation and in vitro characterization with new developed UPLC method. ACTA Pharm. Sci. 56, 81–105 (2018).
Öztürk, A. A. & Merve Güven, U. Cefaclor monohydrate loaded microemulsion formulation for topical application: Characterization with new developed UPLC method and stability study. J. Res. Pharm. 23, 426–440 (2019).
Borman, P. & Elder, D. Q2 (R1) validation of analytical procedures. ICH Qual. Guidelines 127–166 (2017).
Çevikelli, T., Güven, U. M. & Öztürk, A. A. Metronidazole loaded novel microemulsion formulation for topical delivery and characterization with validated new UPLC method. Fabad J. Pharm. Sci. 49, 111–128 (2024).
Elmaskaya, A., Alper Öztürk, A., Büyükköroǧlu, G. & Yenilmez, E. Spray-dried ketoprofen lysine-incorporated PLGA nanoparticles; formulation, characterization, evaluation and cytotoxic profile. Indian J. Pharm. Sci. 81, (2019).
Öztürk, A., Namlı, İ. & Aygül, A. Cefaclor monohydrate-loaded colon-targeted nanoparticles for use in COVID-19 dependent coinfections and intestinal symptoms: Formulation, characterization, release kinetics, and antimicrobial activity. Assay. Drug Dev. Technol. 19, 156–175 (2021).
Güven, U. M., Yenİlmez, E. & Öztürk, A. A. & Evaluation of carvedilol-loaded Eudragit® nanoparticles. J. Res. Pharm. 24, 71–81 (2020).
Zuo, J., Gao, Y., Bou-Chacra, N. & Löbenberg, R. Evaluation of the DDSolver software applications. Biomed. Res. Int. (2014). (2014).
Zhang, Y. et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 12, 263–271 (2010).
Ismailovi, N., Kıyan, H. T. & Öztürk, A. A. A novel phytotherapy application: Preparation, characterization, Antiox-idant activities and determination of Anti-inflammatory effects by in vivo HET-CAM assay of chitosan-based DDSs containing endemic Helichry-sum pamphylicum PH Davis & Kupicha methanolic extract. Curr. Drug Deliv (2023).
Ozturk, A., Yenilmez, E., Arslan, R., Şenel, B. & Yazan, Y. Dexketoprofen Trometamol loaded solid lipid nanoparticles (SLNs): formulation, in vitro and in vivo evaluation. J. Res. Pharm. 24, (2020).
Öztürk, A. A. & Arpagaus, C. Nano spray-dried drugs for oral administration: A review. Assay. Drug Dev. Technol. 19, 412–441 (2021).
Öztürk, A. A., Aygül, A. & Şenel, B. Influence of glyceryl behenate, tripalmitin and stearic acid on the properties of clarithromycin incorporated solid lipid nanoparticles (SLNs): Formulation, characterization, antibacterial activity and cytotoxicity. J. Drug Deliv Sci. Technol. 54, 101240 (2019).
Öztürk, A. A., Yenilmez, E., Arslan, R., Şenel, B. & Yazan, Y. Dexketoprofen trometamol-loaded kollidon® SR and eudragit® RS 100 polymeric nanoparticles: Formulation and in vitro-in vivo evaluation. Lat. Am. J. Pharm. 36, (2017).
Öztürk, A. A., Yenilmez, E. & Yazan, Y. Dexketoprofen trometamol-loaded eudragit® rl 100 nanoparticle formulation, characterization and release kinetics. Acta Pharm. Sci. 57, (2019).
Öztürk, A. A., Ünver, H. & Kıyan, H. T. Antioxidant and antiangiogenic effect of homoleptic palladium (II) car-boxamide complex loaded chitosan modified PLGA nanoparticles: In vitro evaluation and in vivo chick embryo chorioallantoic membrane (CAM) assay. Lett. Drug Des. Discov. 20, 1158–1170 (2023).
Öztürk, A. A., Tilki, E. K., Güven, U. M. & Kıyan, H. T. Ketorolac tromethamine loaded nano-spray dried nanoparticles: Preparation, characterization, cell viability, COL1A1 gene simulation and determination of anti-inflammatory activity by in vivo HET-CAM assay. Curr. Drug Deliv. 20, 830–840 (2023).
Ekambaram, P. & Sathali, A. A. H. Formulation and evaluation of solid lipid nanoparticles of Ramipril. J. Young Pharmacists3, 216–220 (2011).
Şenel, B. & Öztürk, A. A. New approaches to tumor therapy with siRNA-decorated and chitosan-modified PLGA nanoparticles. Drug Dev. Ind. Pharm. 45, 1835–1848 (2019).
Zirak, M. B. & Pezeshki, A. Effect of surfactant concentration on the particle size, stability and potential zeta of beta carotene nano lipid carrier. Int. J. Curr. Microbiol. Appl. Sci. 4, 924–932 (2015).
Koppolu, B., Rahimi, M., Nattama, S., Wadajkar, A. & Nguyen, K. T. Development of multiple-layer polymeric particles for targeted and controlled drug delivery. Nanomedicine 6, 355–361 (2010).
Mainardes, R. M. & Evangelista, R. C. PLGA nanoparticles containing praziquantel: Effect of formulation variables on size distribution. Int. J. Pharm. 290, 137–144 (2005).
Kurban, B. et al. Elemi essential oil nanocapsulated drug ameliorates lung cancer via oxidative stress, apoptosis and inflammation pathway. J. Cell. Mol. Med. 27, 1887–1899 (2023).
Pinheiro, R. G. R. et al. Quercetin lipid nanoparticles functionalized with transferrin for Alzheimer’s disease. Eur. J. Pharm. Sci. 148, 105314 (2020).
Revdekar, A. & Shende, P. Block copolymers in Alzheimer’s disease therapy: A perceptive to revolutionize biomaterials. J. Controlled Release. 340, 271–281 (2021).
Madhu, S., Komala, M. & Pandian, P. Formulation development and characterization of withaferin-A loaded polymeric nanoparticles for Alzheimer’s disease. Bionanoscience 11, 559–566 (2021).
Saraiva, C. et al. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Controlled Release 235, 34–47 (2016).
Nikandish, N., Hosseinzadeh, L., Azandaryani, A. H. & Derakhshandeh, K. The role of nanoparticle in brain permeability: An in-vitro BBB model. Iran. J. Pharm. Res. 15, 403 (2016).
Lockman, P. R., Koziara, J. M., Mumper, R. J. & Allen, D. D. Nanoparticle surface charges alter blood–brain barrier integrity and permeability. J. Drug Target. 12, 635–641 (2004).
Wu, D. et al. The blood–brain barrier: Structure, regulation and drug delivery. Signal. Transduct. Target. Ther. 8, 217 (2023).
Lima, I. A. et al. Mucoadhesive chitosan-coated PLGA nanoparticles for oral delivery of ferulic acid. Artif. Cells Nanomed. Biotechnol. 46, 993–1002 (2018).
Öztürk, A. A. A. Design of Cefaclor monohydrate containing nanoparticles with extended antibacterial effect by nano-spray dryer: A Nanoenglobing study. J. Res. Pharm. 24, 1–12 (2020).
Müller, R. H., Mäder, K. & Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery–a review of the state of the art. Eur. J. Pharm. Biopharm. 50, 161–177 (2000).
Durán-Lobato, M., Martín-Banderas, L., Gonçalves, L. M. D., Fernández-Arévalo, M. & Almeida, A. J. Comparative study of chitosan-and PEG-coated lipid and PLGA nanoparticles as oral delivery systems for cannabinoids. J. Nanopart. Res. 17, 1–17 (2015).
Abdelwahed, W., Degobert, G., Stainmesse, S. & Fessi, H. Freeze-drying of nanoparticles: Formulation, process and storage considerations. Adv. Drug Deliv Rev. 58, 1688–1713 (2006).
Öztürk, A. A. et al. Dexketoprofen trometamol-loaded poly-lactic-co-glycolic acid (PLGA) nanoparticles: Preparation, in vitro characterization and cyctotoxity. Trop. J. Pharm. Res. 18, 1–11 (2019).
Rapalli, V. K. et al. UV spectrophotometric method for characterization of curcumin loaded nanostructured lipid nanocarriers in simulated conditions: Method development, in-vitro and ex-vivo applications in topical delivery. Spectrochim Acta Mol. Biomol. Spectrosc. 224, 117392 (2020).
Enggi, C. K. et al. Validation of spectrophotometric method to quantify cabotegravir in simulated vaginal fluid and porcine vaginal tissue in ex vivo permeation and retention studies from thermosensitive and mucoadhesive gels. Spectrochim Acta Mol. Biomol. Spectrosc. 267, 120600 (2022).
Migues, V. H., David, J. M., Gomes, A. F. & David, J. P. Determination of soybean isoflavone by HPLC/DAD and simple UV spectroscopic analysis: A comparative study. Food Anal. Methods. 15, 367–376 (2022).
Yenilmez, E., Öztürk, A. & Başaran, E. Preparation and in vitro, ex vivo evaluation of benzidamine hydrochlorideloaded fast dissolving oral strip formulations: Treatment of oral mucositis due to side effects of chemotherapy and radiotherapy. Lett. Drug Des. Discov. 20, (2023).
Panyam, J., Williams, D., Dash, A., Leslie-Pelecky, D. & Labhasetwar, V. Solid‐state solubility influences encapsulation and release of hydrophobic drugs from PLGA/PLA nanoparticles. J. Pharm. Sci. 93, 1804–1814 (2004).
AnjiReddy, K. & Karpagam, S. In vitro and in vivo evaluation of oral disintegrating nanofiber and thin-film contains hyperbranched chitosan/donepezil for active drug delivery. J. Polym. Environ. 29, 922–936 (2021).
Bulut, E. & Şanli, O. Optimization of release conditions of Alzheimer’s drug donepezil hydrochloride from sodium alginate/sodium carboxymethyl cellulose blend microspheres. J. Macromol. Sci. Part. B. 53, 902–917 (2014).
Raman, S., Khan, A. A. & Mahmood, S. Nose to brain delivery of selegiline loaded PLGA/lipid nanoparticles: Synthesis, characterisation and brain pharmacokinetics evaluation. J. Drug Deliv Sci. Technol. 77, 103923 (2022).
Sutthapitaksakul, L., Dass, C. R. & Sriamornsak, P. Donepezil—an updated review of challenges in dosage form design. J. Drug Deliv Sci. Technol. 63, 102549 (2021).
Dash, S., Murthy, P. N., Nath, L. & Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 67, 217–223 (2010).
Goldsmith, J. A., Randall, N. & Ross, S. D. On methods of expressing dissolution rate data. J. Pharm. Pharmacol. 30, 347–349 (1978).
Langenbucher, F. Letters to the editor: Linearization of dissolution rate curves by the Weibull distribution. J. Pharm. Pharmacol. 24, 979–981 (1972).
Öztürk, A. A., Namlı, İ., Güleç, K. & Kıyan, H. T. Diclofenac sodium loaded PLGA nanoparticles for inflammatory diseases with high anti-inflammatory properties at low dose: formulation, characterization and in vivo HET-CAM analysis. Microvasc Res. 130, 103991 (2020).
Yang, H. et al. Design of poly (lactic-co-glycolic acid)(PLGA) nanoparticles for vaginal co-delivery of Griffithsin and Dapivirine and their synergistic effect for HIV prophylaxis. Pharmaceutics 11, 184 (2019).
Singh, G., Kaur, T., Kaur, R. & Kaur, A. Recent biomedical applications and patents on biodegradable polymer-PLGA. Int. J. Pharmacol. Pharm. Sci. 1, 30–42 (2014).
Kirimlioğlu, G. Y. & Öztürk, A. A. Levocetirizine dihydrochloride-loaded Chitosan nanoparticles: formulation and in vitro evaluation. Turk. J. Pharm. Sci. 17, 27 (2020).
Mulla Saira, M. R. N. Synthesis, isolation & and characterization of donepezil open ring impurity. Int. J. Pharm. Pharm. Sci. 5, 826–829 (2013).
Copeland, R. A., Harpel, M. R. & Tummino, P. J. Targeting enzyme inhibitors in drug discovery. Expert Opin. Ther. Targets 11, 967–978 (2007).
Anand, P. & Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 36, 375–399 (2013).
Ribatti, D. et al. Chorioallantoic membrane capillary bed: A useful target for studying angiogenesis and anti-angiogenesis in vivo. Anat. Record: Offi. Publ. Am. Assoc. Anatomists 264, 317–324 (2001).
Liekens, S., De Clercq, E. & Neyts, J. Angiogenesis: Regulators and clinical applications. Biochem. Pharmacol. 61, 253–270 (2001).
Ribatti, D. The crucial role of vascular permeability factor/vascular endothelial growth factor in angiogenesis: A historical review. Br. J. Haematol. 128, 303–309 (2005).
Ribatti, D. History of Research on Tumor Angiogenesis (Springer Science & Business Media, 2008).
Ribatti, D. The chick embryo chorioallantoic membrane (CAM). A multifaceted experimental model. Mech. Dev. 141, 70–77 (2016).
Helmlinger, G., Endo, M., Ferrara, N., Hlatky, L. & Jain, R. K. Formation of endothelial cell networks. Nature 405, 139–141 (2000).
Paper, D. H., Karall, E., Kremser, M. & Krenn, L. Comparison of the antiinflammatory effects of Drosera rotundifolia and Drosera Madagascariensis in the HET-CAM assay. Phytother. Res.: Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 19, 323–326 (2005).
Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389–395 (2000).
Carmeliet, P. & Jain, R. K. Angiogenesis in cancer and other diseases. Nature 407, 249–257 (2000).
Potente, M., Gerhardt, H. & Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887 (2011).
Acknowledgements
This work has been supported by Anadolu University Scientific Research Projects Coordination under grant number 2304S027 (Project ID: 1956). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Data AvailabilityAll results obtained during this study are included in the article, and a supplementary file containing NMR analyses is provided to the presented version.Competing interestsThe authors declare no competing interests.Additional informationCorrespondence and requests for materials should be addressed to A.A.Ö.
Author information
Authors and Affiliations
Contributions
Author contributionsThe study’s conceptualization and design were led by A.A.Ö. The pharmaceutical technology-related analyses, including data collection and evaluation, were conducted by M.K. and A.A.Ö. The biological activity studies were carried out by H.T.K.; A.A.Ö. contributed to the discussion and interpretation of the results from both the pharmaceutical technology and biological activity studies. AAÖ wrote this manuscript with contributions of all co-authors.Author informationMerve Kömür (M.K.) Department of Pharmaceutical Technology, Faculty of Pharmacy, Anadolu University, Eskişehir, Türkiye.Hülya Tuba Kıyan (H.T.K) Department of Pharmacognosy, Faculty of Pharmacy, Anadolu University, Eskişehir, Türkiye.A. Alper Öztürk (A.A.Ö) *Department of Pharmaceutical Technology, Faculty of Pharmacy, Anadolu University, Eskişehir, Türkiye.*Corresponding author. aaozturk@anadolu.edu.tr.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kömür, M., Kıyan, H.T. & Öztürk, A.A. Development of donepezil hydrochloride-loaded PLGA-based nanoparticles for Alzheimer’s disease treatment. Sci Rep 15, 13184 (2025). https://doi.org/10.1038/s41598-025-95792-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-95792-3
