
Abstract
The occurrence and development of a wide range of preeclampsia (PE), especially early-onset preeclampsia (EOPE), is closely associated with the immune system. The objective of this research is to utilize machine learning techniques to discover key immune biomarkers and evaluate their predictive potential. We sourced mRNA expression profiles from the GSE60438 + GSE75010 dataset in the Gene Expression Omnibus (GEO) and retrieved immune-related genes from the ImmPort database. Subsequently, we selected immune genes associated with EOPE and late-onset preeclampsia (LOPE) for differential expression analysis. We then carried out Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses on different immune-related genes (DIRGs). Protein‒protein interaction (PPI) networks were employed to investigate the relationships among various DIRGs. Using the least absolute shrinkage and selection operator (LASSO) and multiple support vector machine recursive feature elimination (mSVM-RFE) analyses, we identified candidate biomarkers for EOPE. Receiver operating characteristic (ROC) curves were used to assess the diagnostic capability of the candidate genes, and a nomogram was constructed to evaluate the performance of the predictive models. To further validate our findings, we analyzed additional GEO datasets (GSE22526 + GSE74341 + GSE190639*) and performed immunohistochemistry (IHC) and quantitative real-time PCR (qRT-PCR) on placental tissue to confirm the expression levels and diagnostic values of key genes. Eventually, we utilized the CIBERSORT algorithm to analyze the compositional patterns of the infiltration of 22 immune cell types in EOPE. A total of 17 differentially expressed genes (DEGs) and 7 DIRGs (HLA-DPA1, FPR1, CGB5, LYZ, LEP, PROK2, and SERPINA3) were discovered through a comparison between LOPE and EOPE. Upon conducting GO analyses, it was determined that DIRGs showed significant enrichment in positive regulation of T cell, lymphocyte, and mononuclear cell proliferation. The KEGG enrichment analysis predominantly demonstrated associations with Immune disease, Endocrine and metabolic disease, and Cardiovascular disease. We identified HLA-DPA1, a major histocompatibility complex (MHC) class II gene involved in antigen presentation and immune regulation, as a potential diagnostic biomarker for EOPE, with an area under the curve (AUC) of 0.758. Its downregulation in EOPE suggests a potential role in impaired maternal–fetal immune tolerance. Clinical sample analysis revealed that decreased expression levels of HLA-DPA1 were associated with EOPE. Moreover, immune microenvironment analysis indicated that the expression of HLA-DPA1 exhibited a negative correlation with regulatory T cells and Dendritic cells activated, a positive correlation with macrophages M1 and Mast cells resting. Immunity is a key factor in the pathogenesis of placenta in EOPE. HLA-DPA1 can be identified as a key immune gene associated with immune cells, and these findings provide novel perspectives for the diagnosis and pathogenesis of EOPE
Similar content being viewed by others
Introduction
PE is a multisystem disease characterized by newly developed hypertension after 20 weeks of pregnancy and involvement of multiple organ functions, such as the cardiovascular system, coagulation system, liver, and kidney1. It is a progressive and unpredictable severe complication of pregnancy and one of the important causes of maternal mortality. According to the onset time, PE is generally classified into two major subtypes: EOPE (< 34 weeks) and LOPE (≥ 34 weeks). An increasing amount of evidence suggests that EOPE and LOPE are two distinct diseases2. EOPE is considered to be more closely related to placental pathology compared with LOPE3, with more severe clinical symptoms, a higher risk of developing multiple organ dysfunction, and a greater likelihood of adverse pregnancy outcomes such as preterm birth, fetal distress in utero, stillbirth, and neonatal asphyxia4. In the face of the current absence of pre-symptomatic screening for EOPE, the discovery of novel diagnostic biomarkers is of utmost urgency. Identifying these biomarkers would enable the early detection of individuals at risk of developing EOPE. Additionally, the identification of therapeutic targets is equally crucial, as it holds the key to enhancing the prognosis for both fetuses and mothers affected by this pregnancy-related disorder.
EOPE is a severe pregnancy complication whose pathogenesis is closely associated with placental developmental abnormalities and maternal immune system dysregulation. The core pathological features of EOPE are defects in placental formation, particularly insufficient trophoblast invasion and failure of spiral artery remodeling5. Such abnormal placental development leads to inadequate uteroplacental perfusion, triggering placental ischemia and oxidative stress6. Patients with EOPE exhibit a pronounced polarization of the Th1-type immune response. This Th1 dominance can disrupt the maternal immune tolerance towards the fetus. The research findings are as follows: an upregulation of Th1-related cytokines (such as IFN-γ) in the placental tissue7. The abnormal expression of immune checkpoint molecules (such as PD-1 and LAG-3) may be involved in the dysregulation of immune tolerance8.NK cell subsets (such as CD8 + NK cells) in a Th1-dominant environment may attempt to restore immune balance through compensatory mechanisms9. CD200, an immunomodulatory molecule, may be involved in the development of EOPE by inhibiting the activation of classical macrophages10. However, the causal relationship has not been clearly defined.
Thus, this research aimed to use bioinformatics methods to discover new DIRGs in EOPE samples, with the intention of uncovering diagnostic immune biomarkers. Subsequently, we validated these identified DIRGs by analyzing placental samples from both EOPE and LOPE groups. Moreover, we explored the possible associations between these novel DIRGs and immune cells, aiming to stimulate further studies on the pathological mechanisms of EOPE.
Methods and materials
Collecting and processing microarray data
Relevant raw LOPE and EOPE datasets, namely GSE60438, GSE75010, GSE22526, GSE74341, and GSE190639, were retrieved from the GEO database (available at https://www.ncbi.nlm.nih.gov/gds). While the initial two datasets served as the training group, the subsequent three were consolidated into a single dataset, which was then employed as the test group. Comprising 52 LOPE samples and 88 EOPE samples, the training dataset stands in contrast to the test dataset, which is composed of 21 LOPE samples alongside 28 EOPE samples, as detailed in Table 1. Guided by their respective probe annotation files, each dataset’s probes were mapped to corresponding gene symbols (Supplementary Table S1). In instances where multiple probes corresponded to the same gene, the gene’s expression level was represented by the average of these probes’ expression values. To integrate the two and three datasets into a unified metadata cohort while eliminating batch effects, respectively. We utilized the “SVA” package within the R software. Background correction and normalization of raw data were performed using the R limma package (accessible at http://www.bioconductor.org/), with DEGs defined as those exhibiting a |log twofold change (FC)|> 0.585 and an adjusted p-value < 0.05. The merged dataset facilitated both the identification and validation of the functions associated with significant DIRGs. Immune-related genes (IRGs) were obtained by querying the ImmPort database (https://www.immport.org/shared/) as outlined in Supplementary Table S2. Following this, DIRGs were determined by intersecting IRGs with DEGs specific to EOPE.
It is acknowledged that the original inclusion and exclusion criteria may have varied across the independent studies from which the datasets were sourced. However, all studies shared the core diagnostic criterion of defining EOPE as onset at < 34 weeks and LOPE as onset at ≥ 34 weeks. To mitigate the impact of technical and procedural heterogeneity, the ComBat algorithm from the ‘SVA’ package was applied to correct for batch effects. The subsequent successful validation of findings in an independent test cohort further supports the robustness of the results across different sample populations.
Functional characterization and pathway enrichment analysis for DIRGs
Enrichment analyses for GO and KEGG11 pathways, which were performed on the genes using the R packages “clusterProfiler”, “enrichplot”, "org.Hs.eg.db", and “DOSE” to uncover enriched GO terms in three categories (cellular components, biological processes, molecular functions) and KEGG pathways, yielded results that were visualized with the assistance of R’s “ggplot2” package, and a p-value of < 0.05 was used as the criterion to determine whether enrichment was significant.
Development and computational analysis of PPI assemblies
Using the STRING database (available at https://string-db.org/)12, we searched for a protein–protein interaction (PPI) network by inputting 7 DIRGs into the “multiple proteins” module and selecting “Homo sapiens” as the organism. After deriving gene symbols from protein identifiers, any PPIs without corresponding gene names were excluded from the analysis. Once this preprocessing was complete, Cytoscape 3.10.0 was employed to construct the PPI network, with the cytoHubba plugin facilitating the identification of hub genes.
A model for predicting EOPE diagnosis was constructed using DIRGs
We leveraged the LASSO algorithm in conjunction with the mSVM-RFE algorithm to enable the identification of these biomarkers: specifically, LASSO—an overfitting-prevention tool in regression analysis that focuses on variable selection—was run via the “glmnet” R package13, and the mSVM-RFE algorithm, which utilizes resampling in every iteration to solidify feature rankings and isolates the most relevant features by eliminating feature vectors produced by SVM through supervised machine learning techniques14, was deployed using the “e1071” package. Given that mSVM-RFE carries a smaller overfitting risk than SVM-RFE, the two algorithms were combined to filter overlapping genes, which were then validated in the training set. Furthermore, the “pROC” R package was used to generate ROC curves, and the diagnostic performance of the biomarkers for distinguishing EOPE from LOPE samples was evaluated based on the AUC, a metric whose validity was also confirmed in the test cohort.
PCA & nomogram models: establishment, validation, and eope diagnostic utility
By leveraging eigenvalue decomposition to reduce the dimensionality of gene expression data and quantifying the statistical significance of between-group differences based on grouped confidence ellipses, principal component analysis (PCA) was utilized—along with the R packages “limma” and “ggplot2”—to further confirm the diagnostic capability of DIRGs for EOPE. Meanwhile, the R packages “rms” and “rmda” were used to build a nomogram model for EOPE diagnostic prediction, where each factor is assigned a “points” value, and the sum of these individual scores is termed “total points”; subsequently, calibration curves were created to assess how well the nomogram model performs in prediction.
Immune cell evaluation in EOPE and biomarker validation
The CIBERSORT algorithm (v1.03) (http://cibersort.stanford.edu/) was used to quantify how many infiltrating immune cells were present in EOPE gene expression profiles, and to estimate the putative immune cell abundance, we downloaded the LM22 gene signature matrix from the CIBERSORT webpage15—a matrix whose correlation and visualization were then conducted via the R package “corrplot”. Analysis was performed with perm = 1000 permutations for significance assessment and quantile normalization (QN) enabled. Furthermore, after visualizing the immune cell differences between LOPE and EOPE groups with the R package “boxplot”, spearman’s correlation analysis was employed to examine the links between the screened diagnostic biomarkers and infiltrating immune cell levels, with the results of this examination visualized using the R package “ggplot2”.
Patients and tissue specimens
Ethical approval
This study was approved by the Research Ethics Committee of The Second Affiliated Hospital of Fujian Medical University(Ethical Approval Number: 2024-315). All methods were carried out in accordance with relevant guidelines and regulations. This includes obtaining written informed consent from all participants prior to enrollment, and ensuring all procedures involving human participants were performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
The period from December 2020 to May 2024 saw the acquisition of 33 paraffin-embedded LOPE samples and 18 EOPE samples from the Second Affiliated Hospital of Fujian Medical University (Fujian, China), an endeavor that had received prior authorization from the institution’s Research Ethics Committee before the initiation of the study.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from placental tissues collected immediately after normal vaginal delivery or cesarean section, using TRIzol reagent (Beyotime Biotechnology, China). cDNA was then synthesized in accordance with the manufacturer’s protocol (TaKaRa, Japan). GAPDH was used as the endogenous control gene, and relative mRNA expression levels of HLA-DPA1 was computed via the 2 − ΔΔCT method. All qRT-PCR reactions were conducted in triplicate, with three independent experimental repeats for each sample. The primer sequences utilized are listed below:
HLA-DPA1
Forward: 5′- ATGTTCCATATCAGAGCTGTGATCT-3′,
Reverse: 5′- CCTGTTGGTCTATGCGTCTGTAC -3′.
GAPDH
Forward: 5′-GTCTCCTCTGACTTCAACAGCG-3′,
Reverse: 5′-ACCACCCTGTTGCTGTAGCCAA-3′.
Immunohistochemistry (IHC)
Following the methodologies described previously, we carried out the IHC staining process16. The intensity ratio of staining in specimens was categorized using an IHC staining method with an anti-HLA-DPA1 antibody (bsm-60290R, 1:100), with scoring determined by two parameters: the percentage of positive cells among all tissue cells and the staining intensity of positive cells. For the cell proportion score (A), a score of 1 was given when positive cells were < 1/3 of the total, 2 when between 1/3 and 2/3, and 3 when ≥ 2/3; for the staining intensity score (B), negative staining scored 0, pale yellow scored 1, brownish yellow scored 2, and tan scored 3. Total scores, computed as A multiplied by B, were then used to divide slide samples into low-expression (total score < 6) and high-expression (total score ≥ 6) cohorts. Two board-certified Obstetrics and Gynecology (Ob/Gyn) pathologists, who were blinded to the sample groups, independently evaluated all IHC slides. The staining intensity and the percentage of positive cells were assessed separately by each pathologist to calculate a score. In cases of initial scoring discrepancies (defined as a difference in total score of > 30%), the slides were jointly re-examined by both pathologists using a multi-headed microscope. Through this consensus meeting, a final agreed-upon score was established for each sample.
Statistical analysis
Statistical analyses were primarily performed using R software (v.4.3.3). Specifically, the construction of the nomogram and calibration curve (Fig. 5E, F) was conducted under R version 4.4.0 using the rms package. We utilized the Mann–Whitney U test to compare different groups and the Chi-square test for 2 × 2 contingency table comparisons, with the analytical suite including LASSO regression, SVM-RFE algorithm, ROC analysis, Spearman’s correlation, and unpaired t-test, and statistical significance defined as p < 0.05.
Results
Study workflow
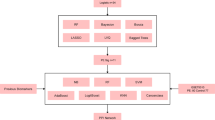
As illustrated in Fig. 1, the study’s analytical procedure involved retrieving microarray data from the GEO database, mapping microarray probes to gene symbols in each dataset using probe annotation files, and intersecting DEGs with IRGs to generate DIRGs, which were then subjected to enrichment analysis via the GO and KEGG databases. Candidate overlapping genes were further filtered through PPI networks and two machine learning algorithms (LASSO and SVM-RFE), with the predictive performance of the resulting biomarkers validated using PCA and ROC curves, a process that included additional verification in the test group (GSE22526 + GSE74341 + GSE190639). Meanwhile, the Cibersort algorithm was used to determine the compositional patterns of 22 immune cells in EOPE, correlations between these immune cells and diagnostic biomarkers were analyzed, and IHC staining was performed on paraffin-embedded specimens meeting inclusion criteria to confirm the study’s findings.

A diagram illustrating the workflow that was followed throughout the course of this research, which maps out the sequential steps and operational logic of the study from data collection to result analysis, serves as a visual representation of the research process. This figure was created with Microsoft PowerPoint (Version: office 2021).
The identification of DIRGs associated with EOPE
Through analysis of two datasets (GSE60438 + GSE75010), 17 DEGs were detected between 88 EOPE and 52 LOPE in this study, with filtering parameters of adj.P.Val < 0.05 and ||og2 fold change (FC)|> 0.585 (Fig. 2A, Supplementary Table S3)—a cohort consisting of 13 genes with significant downregulation (SELL, S100A12, S100A8, HBD, FCN1, HLA-DPA1, AMICA1, CD48, MNDA, FPR1, PROK2, LYZ, and RGS18) and the other 4 genes with marked upregulation (HTRA4, LEP, SERPINA3, and CGB5) (Fig. 2B). Subsequent intersection of these DEGs with IRGs enabled the identification of 7 EOPE-specific DIRGs (HLA-DPA1, FPR1, CGB5, LYZ, LEP, PROK2, and SERPINA3) (Fig. 2C), among which HLA-DPA1, FPR1, LYZ, and PROK2 exhibited reduced expression, and the other three DIRGs showed significant upregulation in EOPE cases.

Identification and Functional Analysis of DIRGs. (A) Heatmaps are used to visualize the expression levels of 17 DEGs between EOPE and LOPE retrieved from the GEO database, where the genes are labeled by their respective names in the row annotations—whereas the column annotations, which correspond to sample IDs, are not shown in the plots—and a color gradient spanning from red to blue is employed to signify the transition of expression levels from high to low within the heatmaps. This heatmap was generated using the pheatmap package (version 1.0.13; https://cran.r-project.org/web/packages/pheatmap/) in R software (v.4.3.3; https://www.r-project.org/). (B) Volcano plots are utilized to illustrate the 17 DEGs identified between EOPE and LOPE; in these plots, red dots are designated to indicate genes with upregulated expression, green dots to denote genes with downregulated expression, and black dots to represent genes that do not exhibit any differential expression patterns. This volcano plot was generated using the ggplot2 package (version 4.0.1; https://cran.r-project.org/web/packages/ggplot2/) in R. (C) The 7 DIRGs targeted in this study are derived from the intersection of DEGs obtained from the GSE75010 + GSE60438 dataset and IRGs that were downloaded from the ImmPort database, a process that ensures the selected DIRGs simultaneously possess differential expression characteristics and immune relevance. The Venn diagram was created using the VennDiagram package (version 1.7.3; https://cran.r-project.org/web/packages/VennDiagram/) in R. (D) A circle plot is generated to present the GO enrichment analysis results for the 7 DIRGs, providing a visual summary of the BP, CC, and MF associated with these genes. This GO circle plot was generated using R packages circlize (version 0.4.17; https://cran.r-project.org/web/packages/circlize/) and ComplexHeatmap (version 2.18.0; https://bioconductor.org/packages/ComplexHeatmap). (E) A bar graph is constructed to display the GO enrichment analysis results for the 7 DIRGs, offering a quantitative comparison of the enrichment degrees across different GO terms linked to the target genes. This bar plot was generated using the ggplot2 package in R. (F) KEGG pathway annotation is performed for the 7 DIRGs, aiming to clarify the signaling pathways and metabolic cascades in which these genes are involved. The figure presents the pathways that were significantly enriched (with p < 0.05). On the x-axis, the Gene Ratio is displayed, which is calculated as the number of genes enriched in a given pathway divided by the total number of background genes. Additionally, these pathways are sorted in descending or ascending order based on their statistical significance to reflect the strength of enrichment. This KEGG enrichment plot was generated using the ggplot2 package in R.
GO and KEGG-based functional enrichment analyses
Employing R’s “ClusterProfiler” package for functional enrichment analysis to further investigate the functions and enriched pathways of these 7 DIRGs, we found that their biological processes (BP) were predominantly dedicated to positive regulation of T cell proliferation and lymphocyte proliferation, with their cellular components (CCs) primarily located in MHC class II protein complex and MHC protein complex, and their molecular functions (MFs) mainly associated with MHC class II receptor activity, peptide hormone receptor binding, and immune receptor activity (P < 0.05, Fig. 2D, E, Supplemental S1). In addition, KEGG enrichment analysis revealed the 7 DIRGs’ primary participation in Th1 and Th2 cell differentiation, Antigen processing and presentation, and Th17 cell differentiation (P < 0.05, Fig. 2F, Supplemental S2)—results that underscore a strong association between EOPE and immunity..
Hub gene network construction based on PPI analysis
To build a PPI network, we input 7 DIRGs into the “multiple proteins” module of the STRING database (https://string-db.org/), selected Homo sapiens as the target species, and removed disconnected nodes—resulting in a PPI network that retained 7 interconnected DIRGs (Fig. 3A). Following this, the cytoHubba plugin in Cytoscape software was employed for cluster analysis of the network genes, and 7 hub nodes ranked by the MCC algorithm were identified and categorized (Fig. 3B); concurrently, expression level analyses revealed that CGB5, SERPINA3, and LEP were upregulated, whereas HLA-DPA1, FPR1, LYZ, and PROK2 showed downregulation (Fig. 3C, D).

Association Between DIRGs and Hub Genes. (A) A PPI network of the 7 DIRGs was constructed using the STRING database (https://string-db.org/) , where each node corresponds to a protein (i.e., the product of a target gene) and is colored according to its functional clustering —specifically, proteins that perform similar biological roles are assigned the same color. (B) The top 7 hub genes were identified through the MCC algorithm, which was implemented using the cytoHubba plugin within the Cytoscape software (version 3.10.0; https://cytoscape.org/). In the resulting network visualization, the intensity of the color assigned to each node is directly correlated with its MCC score: warmer colors (e.g., red) indicate higher centrality scores, thereby signifying that the corresponding gene plays a more critical role in maintaining the structure and function of the PPI network. (C) A heatmap was generated to display the expression patterns of the top 7 hub DIRGs, with the comparison focused on samples from the EOPE group and the LOPE group. The color gradient in the heatmap is based on row Z-score normalized expression values: blue hues represent low expression levels (corresponding to negative Z-scores), while red hues indicate high expression levels (corresponding to positive Z-scores). Each row in the heatmap represents an individual hub DIRG (with gene names labeled on the right-hand side), and each column corresponds to a single sample; notably, the sample groups (LOPE vs. EOPE) are clearly annotated at the top of the heatmap to facilitate group-wise comparison. This heatmap was generated using the pheatmap package (version 1.0.13; https://cran.r-project.org/web/packages/pheatmap/). (D) A volcano plot was constructed to present the expression differences of the top 7 hub DIRGs, providing a visual summary of the statistical significance and magnitude of expression changes for each of these genes across the compared groups. This volcano plot was generated using the ggplot2 (version 4.1.1) and ggrepel (version 0.9.6; https://cran.r-project.org/web/packages/ggrepel/) packages in R.
Construction of a prediction model for EOPE
To accurately pinpoint key diagnostic biomarkers for EOPE, the Lasso algorithm (Fig. 4A, B) and SVM-RFE algorithm (Fig. 4C, D) were employed to screen for potential diagnostic biomarkers—with a notable observation that the DIRGs identified by these two algorithms were fully consistent, leading to the final selection of 3 candidate genes (LEP, PROK2, and HLA-DPA1), as illustrated in Fig. 4E.

Development of a Predictive Model for EOPE. (A)The LASSO regression coefficient profiles of the 6 DIRGs depict the changing trajectory of each DIRG through a curve. This plot was generated using the glmnet package (version 4.1.8; https://cran.r-project.org/web/packages/glmnet/) in R. (B)The LASSO Cox regression model was used to plot partial likelihood deviance versus log (l). This cross-validation plot was generated using the glmnet package in R. (C) The curve of the total within sum of squared error curve under the corresponding cluster number k reached the “elbow point” when k = 3. This plot was generated as part of the SVM-RFE algorithm, using a custom mSVM-RFE script based on the implementation of the e1071 package (version 1.7.16) in R. (D) The curve representing average silhouette width for the corresponding cluster number k attained its peak at k = 3. This plot was generated as part of the SVM-RFE algorithm implementation in R. (E)The Venn diagram illustrates the three diagnostic markers shared by the LASSO and SVM-RFE algorithms. This diagram was generated using the VennDiagram package (version 1.7.3; https://cran.r-project.org/web/packages/VennDiagram/) in R.
In-depth characterization of the three key DIRGs
Figure 5A depicts the chromosomal positions of the 3 DIRGs, and principal component analysis (PCA) revealed that these 3 candidate genes had strong discriminatory capacity between EOPE and LOPE controls—results that suggest their critical importance in EOPE diagnosis (Fig. 5B). Expression levels of the 3 DIRGs differed significantly between the two groups, with HLA-DPA1 and PROK2 exhibiting downregulation and LEP exhibiting upregulation in EOPE (Fig. 5C), while ROC analyses performed to assess the predictive accuracy of potential DIRGs showed that only HLA-DPA1 had AUC values exceeding 0.7, indicating good diagnostic discriminatory value for EOPE (Fig. 5D). We then constructed a prognostic nomogram for EOPE (Fig. 5E, F) to enable risk score-based discrimination between EOPE and LOPE controls;

Supplementary Analysis of Three Key DIRGs. (A) The chromosomal locations of the three key DIRGs were mapped to illustrate their specific positions on respective chromosomes, providing a genomic context for further exploration of their potential regulatory relationships. This circos plot was generated using the circlize package (version 0.4.17; https://cran.r-project.org/web/packages/circlize/) in R. (B) A PCA plot was generated to visualize the distribution of samples, with this visualization based on the expression profiles of the three key DIRGs; in the plot, samples are color-coded according to their group affiliation (blue for the LOPE group and red for the EOPE group), and dashed ellipses are used to enclose samples from each group, representing the 95% confidence intervals for group-wise sample clustering. Meanwhile, the x-axis and y-axis correspond to the first two principal components (PC1 and PC2), respectively, and the percentage of total variance explained by each principal component is indicated in parentheses adjacent to the axis labels, allowing for assessment of the dimensionality reduction effect. This plot was generated using the ggplot2 package (version 4.0.1) in R, with principal components computed via the prcomp function. (C) The train group datasets, which integrate two individual datasets (GSE75010 + GSE60438), were used to present the comparative expression levels of three critical DIRGs between the EOPE group and the LOPE group, ensuring that the observed expression differences are supported by a combined, larger sample size. This box plot was generated using the ggpubr (version 0.6.2; https://cran.r-project.org/web/packages/ggpubr/) and ggplot2 packages in R. (D) ROC curves were constructed to validate the predictive efficacy of the three critical DIRGs for EOPE within the train group, with the AUC serving as a quantitative metric to evaluate how well each gene can distinguish between EOPE and LOPE samples. The ROC analysis and curves were generated using the pROC package (version 1.18.5; https://cran.r-project.org/web/packages/pROC/) in R. (E) A nomogram was developed to predict the risk of EOPE, and this predictive tool is based on the expression levels of three DIRGs, specifically including HLA-DPA1, PROK2, and LEP. For each of these three genes, a specific number of points is assigned based on the gene’s expression level in a given sample; the total number of points for an individual sample is then calculated by summing the points from all three genes, and this total point value is further converted to the corresponding risk of developing EOPE, enabling intuitive risk assessment. This nomogram was generated using the rms package (version 8.0.0; https://cran.r-project.org/web/packages/rms/) in R software (v.4.4.0; https://www.r-project.org/). (F) A calibration curve was generated for the nomogram to assess the agreement between the predicted risk of EOPE (displayed on the x-axis) and the actually observed risk (displayed on the y-axis). The diagonal line in the plot represents the ideal prediction scenario where predicted risk perfectly matches observed risk, while the solid line (labeled “Apparent”) shows the model’s performance before bias correction and the dashed line (labeled “Bias-corrected”) illustrates the performance after bias correction via bootstrapping with 1000 repetitions, allowing for evaluation of the model’s calibration accuracy and potential overfitting. This calibration plot was generated using the rms package in R.
HLA-DPA1 is associated with the distribution of immune cells
Employing the CIBERSORT algorithm to quantify the relative abundances of 22 immune cell types in both EOPE and LOPE samples—to deepen understanding of the association between immune cell infiltration and EOPE (Fig. 6A)—we compared immune cell infiltration patterns between EOPE samples and LOPE controls, which revealed significantly higher levels of NK cell activated, T cell regulatory, and T cell CD8 in the EOPE group (Fig. 6B). Employing the additional GEO datasets (GSE22526 + GSE74341 + GSE190639) to validate the expression levels of 3 DIRGs—to select more reliable and accurate DIRGs—we found that EOPE tissues showed significantly lower expression of HLA-DPA1 (P < 0.05, Fig. 6C). Further investigations into the relationship between key diagnostic biomarkers and distinct infiltrating immune cells showed that HLA-DPA1 expression correlated positively with Macrophages M1 and Mast cells resting T cells, alongside negatively with T cells regulatory and Dendritic cells activated (Fig. 6D)—observations that support the close link between HLA-DPA1 and immune activity and highlight its potential role in regulating immune cell function in EOPE. We can observe via hematoxylin–eosin (HE) staining of placental tissues from EOPE and LOPE cases that the placental pathological features of EOPE present as distal villous hypoplasia and intermembranous and intervillous fibrin deposition (Fig. 6E). To further validate these findings, we examined the expression levels of HLA-DPA1 via qRT-PCR, demonstrating that the low expression was linked to EOPE (Fig. 6F; p = 0.0103). Additionally, we observed the expression of HLA-DPA1 in the cytoplasm of syncytiotrophoblasts (SCT). IHC was used to assess HLA-DPA1 expression in EOPE and LOPE, revealing a significant correlation between low HLA-DPA1 expression and EOPE (Fig. 6G, P = 0.025), These findings verifying HLA-DPA1’s strong diagnostic capacity for distinguishing between these two PE types.

Immune Cell Infiltration: Distribution, Visualization, and Correlation Analysis. (A) A bar plot was constructed to illustrate the proportional distribution of 22 immune cell subtypes, with this visualization comparing samples from EOPE tissues and LOPE control pregnancies, thereby providing a clear overview of how immune cell composition varies between the two groups. This stacked bar plot was generated using base R graphics functions, with data derived from the CIBERSORT algorithm (v1.03). (B) Analyses were performed to assess differences in immune cell infiltration between the EOPE and the LOPE control pregnancy group, with the results presented to highlight which specific immune cell subtypes exhibit statistically significant differences in their infiltration levels across the two study cohorts. This box plot was generated using the ggpubr package (version 0.6.2; https://cran.r-project.org/web/packages/ggpubr/) in R. (C) The relative expression level of HLA-DPA1 compared between EOPE and LOPE samples, with this comparative analysis drawing on data from three independent datasets (GSE22526, GSE74341, and GSE190639) to ensure the robustness of the observed expression patterns. This box plot was generated using the ggpubr and ggplot2 (version 4.0.1) packages in R. (D) An investigation was conducted to explore the correlation between the expression levels of HLA-DPA1 and the infiltration levels of various immune cell subtypes in EOPE tissues, with the resulting correlation coefficients used to quantify the strength and direction of these associations. This correlation heatmap was generated using the linkET package (version 0.1.0; https://github.com/Hy4m/linkET) and ggplot2 in R. (E) Pathological features of placental tissues from EOPE and LOPE by HE staining. (F) qRT-PCR of HLA-DPA1 expression in EOPE placental tissue when compared with LOPE specimens. (G) When EOPE tissues were compared with LOPE specimens (with sample sizes of 18 for EOPE and 33 for LOPE), significantly lower expression levels of HLA-DPA1 was observed in the EOPE group; representative images of IHC staining for gene are provided at magnifications of ×40 and ×200, illustrating both high and low expression patterns across the 18 LOPE patients and 33 LOPE patients included in the analysis.
Discussion
Late-onset preeclampsia (LOPE) constitutes the majority (80–95%) of preeclampsia (PE) cases worldwide, whereas early-onset preeclampsia (EOPE), though less common, is associated with more severe maternal and neonatal morbidity and mortality[17; 18]. This clinical distinction has driven research to elucidate the distinct pathogenesis of EOPE, which is characterized by profound placental dysfunction and dysregulation of the maternal immune system at the maternal–fetal interface17,18,19. Consequently, the identification of immune-associated biomarkers is crucial not only for early diagnosis but also for understanding the molecular mechanisms underlying EOPE. Collectively, our study not only rigorously validates HLA-DPA1 as a core diagnostic biomarker for EOPE but also delivers key advancements in understanding the placental immunopathology of this severe condition. Specifically, our work provides: (1) novel histological evidence implicating the SCT in the immune pathology of PE; (2) a refined, multi-gene diagnostic signature with enhanced translational potential; and (3) crucial insights linking this signature to a distinct dysregulated immune microenvironment, thereby offering a new diagnostic and immunological model for EOPE subtyping.
Although early-onset and late-onset preeclampsia exhibit distinct clinical phenotypes and pathological underpinnings, our analysis of their placental transcriptomes identified a relatively modest set of 17 high-confidence DEGs. This number might be lower than some expectations, which can be attributed to several factors. Firstly, we applied stringent statistical thresholds (adjusted p-value < 0.05 and |log2FC|> 0.585) to minimize false positives, a approach that prioritizes reliability over the quantity of findings. Secondly, while EOPE and LOPE are considered distinct entities, they share the common clinical endpoint of preeclampsia and exhibit overlapping placental pathology. Thus, the DEGs we identified likely represent the core set of intrinsic placental transcriptional alterations that most robustly differentiate the two subtypes. Finally, the primary aim of our study was not to catalog all possible differences but to identify a concise and robust set of biomarkers with high diagnostic potential. Reassuringly, these 17 DEGs, particularly the DIRGs derived from them, demonstrated excellent diagnostic performance in subsequent machine learning models and independent validation cohorts, underscoring their biological and clinical relevance beyond mere quantity.
Dysregulation of both innate and adaptive immune cells is implicated in EOPE pathogenesis. Key alterations include an expansion of cytotoxic CD8⁺T cells and a shift in NK cells, characterized by a peripheral decrease but a cytotoxic subset increase at the maternal–fetal interface, promoting local inflammation20,21,22,23,24. Similarly, monocytopenia, particularly of the classical CD14⁺⁺CD16⁻ subset, is linked to placental hypoxia and severe systemic inflammation24,25,26. However, the role of Tregs remains complex, with studies reporting contradictory changes in their number and function21,27,28,29. This existing literature provides a context for our findings, which corroborate the increase in CD8⁺ T cells and NK cells, and the decrease in monocytes, while highlighting the need to further resolve the discrepancies regarding Tregs in EOPE (Fig. 7).

Differences in HLA-DPA1 biomarkers and Placental Immune Microenvironment between EOPE and LOPE. This figure was created with Microsoft PowerPoint (Version: office 2021).
GO/KEGG enrichment analysis revealed that positive regulation of T cell, Th1, and Th2 cell differentiation is involved in EOPE. PE is widely recognized to be associated with the failure of maternal immune tolerance mechanisms, particularly the imbalance in T cell subset differentiation. In normal pregnancy, Th1 cells (pro-inflammatory) and Th2 cells (anti-inflammatory) need to be balanced to maintain fetal tolerance. However, this balance is disrupted in PE, often manifested as excessive activation and enhanced differentiation (i.e., positive regulation) of Th1 cells. According to the research, the preponderance of Th1 over Th2 cells is a core feature of the disease, suggesting that Th1 cell differentiation may be positively regulated7. In EOPE, this imbalance is more pronounced and involves abnormalities in multiple signaling pathways, leading to dysregulated Th1/Th2 differentiation30. It has been shown that interleukin-11 (IL-11) is elevated in the serum of pregnancies that subsequently develop EOPE31. Although the underlying mechanism has not been fully elucidated, as an inflammatory factor, IL-11 may positively regulate the differentiation of Th1 or Th2 cells through indirect mechanisms, such as enhancing T cell activation pathways (e.g., NF-κB or JAK/STAT). This may contribute to the unique immune phenotype of EOPE31. The alterations in normal immune adaptations in EOPE involve dynamic dysregulation of T cell differentiation, in which the positive regulation of Th1 and Th2 cells may contribute to the exacerbation of systemic inflammation32.
Based on the 7 DIRGs that exhibited the most significant differences between the EOPE and the LOPE, the LASSO regression and mSVM-RFE models were employed to screen for candidate genes, ultimately identifying 3 genes: HLA-DPA1, PROK2, and LEP. Subsequent validation using the test set confirmed the association of HLA-DPA1 with EOPE. HLA-DPA1 is a class II gene of the major histocompatibility complex (MHC), which is responsible for antigen presentation. Researchers have reported more than 40 novel HLA-DPA1 alleles (e.g., 01:03:01:24-27, 02:01:01:14-19), which are mainly generated through single-nucleotide substitutions33,34. Variations are mostly concentrated in specific codons of exons 2–4. Variations in the 01:03:01 subtype are commonly found at codon 85 (exon 3)35, codon 120 (exon 3)36, and codon 190 (exon 4)37. Some variations may lead to the loss of allele function (e.g., the null allele 01:29N) or changes in antigen-binding properties38,39. Abnormal expression of HLA-DPA1 is directly associated with disorders of immune-inflammatory responses. In patients with idiopathic pulmonary arterial hypertension (IPAH), HLA-DPA1 is significantly downregulated in non-classical monocytes (NCMs), and the differentially expressed genes are enriched in immune and inflammatory response pathways, indicating that the loss of its expression exacerbates disease progression40. The placental tissue of patients with EOPE exhibits abnormalities in the innate immune system, such as altered gene expression of TLR1/TLR4, and a reduction in mast cells and M2 macrophages. These findings suggest that HLA molecules may be involved in local immune imbalance24. Studies have shown that specific HLA epitope mismatches between the mother and fetus (particularly those involving HLA-A, -B, -C, -DQB1, and -DRB1) may induce anti-fetal HLA antibodies, thereby promoting the development of gestational hypertension and preeclampsia41,42. Although direct data on HLA-DPA1 mismatches have not been explicitly reported in the literature, as a class II HLA molecule, it may be involved in a similar mechanism. Further research on the specific mismatches of HLA-DPA1 and the regulatory mechanisms of its expression in trophoblast cells will help deepen the understanding of the immune pathogenesis of EOPE.
We utilized the CIBERSORT algorithm and found that the proportions of CD8 + T cells, Tregs, and NK cells in EOPE were significantly higher than those in LOPE samples. In contrast, the counts of neutrophils and monocytes in EOPE samples were significantly lower when compared to those in LOPE samples. In our study, HLA-DPA1 was negatively correlated with Tregs and activated dendritic cells. No studies have directly explored the association between the HLA-DPA1 gene and Tregs. Only this particular study has reported the identification and characterization of HLA-DPA1 allelic variants in type 1 diabetes, with no extension to mechanisms at the cellular level43. HLA-DPA1 encompasses alleles harboring nonsense mutations (null mutations), synonymous mutations, or nonsynonymous mutations. These variations may result in loss of function or functional alterations, thereby indirectly affecting the antigen-presenting capacity of dendritic cells38. By analyzing three HLA-DPA1 expression subgroups (negative expression group, low expression group, and high expression group), the study observed significant differences in intercellular communication, molecular mechanisms, and metabolic activities. These differences are enriched in inflammation-related pathways, such as interferon signaling and antigen presentation processes, which may drive the differentiation of macrophages toward a pro-inflammatory phenotype40. One study has indicated that certain immune states (e.g., monocyte differentiation) may "contribute to a proinflammatory M1-like phenotype". This implies that changes in HLA-DPA1 expression or related molecular mechanisms may be involved in the polarization of macrophages toward the M1 phenotype, thereby enhancing the inflammatory response44. These findings suggest that the HLA-DPA1 gene may be closely related to the immune system and the pathogenesis of EOPE.
The translational implications of our work extend beyond the confirmation of a single biomarker. First, the construction of a multi-gene diagnostic nomogram based on HLA-DPA1, PROK2, and LEP moves the field towards a more robust and clinically applicable tool for distinguishing EOPE from LOPE, which is crucial for risk stratification and management. Second, the strong association between low HLA-DPA1 expression and a specific immune contexture (e.g., negative correlation with Tregs) indicates that measuring placental HLA-DPA1 levels could serve as a stratification biomarker, identifying an EOPE subtype driven by this particular immune dysfunction. Consequently, this insight directs future therapeutic exploration towards immune-modulatory strategies tailored to correct this specific imbalance. In conclusion, by integrating bioinformatics, machine learning, and histological validation, we have not only identified the diagnostic immune biomarkers we set out to find but have also embedded them within a broader pathophysiological framework, providing a refined model for understanding and tackling the heterogeneity of EOPE.
Our study has several limitations. First, the clinical cohort size was limited and the nomogram model requires further validation in larger, multi-center populations. Second, while our IHC findings of HLA-DPA1 localization in syncytiotrophoblast are consistent with our transcriptomic data and are supported by reports of aberrant MHC class II (HLA-DR) association with syncytiotrophoblast in PE45, they rely on a single monoclonal antibody. Orthogonal validation using an independent antibody targeting a different epitope would be a valuable future step to conclusively confirm protein specificity and exclude cross-reactivity. Nevertheless, the convergence of evidence from bioinformatics screening, independent mRNA validation, and biologically plausible IHC staining—a methodology also successfully employed to study HLA-DPA1 in other immune-mediated diseases46—strengthens the overall association of reduced HLA-DPA1 with EOPE. Third, the use of bulk RNA-sequencing data obscures the cellular origin of the HLA-DPA1 signal, a question best addressed by future single-cell studies. Finally, and most importantly, our work identifies a strong association but lacks direct functional evidence for the role of HLA-DPA1 in EOPE pathogenesis.
Conclusion
In conclusion, by integrating bioinformatics analysis with clinical validation, we have delineated a distinct immune-related molecular signature for EOPE. Our findings underscore the pivotal role of HLA-DPA1 downregulation in shaping a pro-inflammatory placental microenvironment, characterized by an altered balance of immune cells such as decreased Tregs and increased Macrophages M1, which we propose contributes significantly to the severe pathogenesis of EOPE. The proposed model, summarized in Fig. 7, not only provides a visual synthesis of our findings but also offers a foundational framework for future research into the immune mechanisms of EOPE and the development of targeted diagnostic strategies.
Data availability
The datasets generated and/or analyzed during the current study are available in the following public repositories: The mRNA expression profiles for EOPE and LOPE (GSE60438, GSE75010, GSE22526, GSE74341, GSE190639) are available in the NCBI Gene Expression Omnibus (GEO) repository, accessible via the following accession numbers: GSE60438(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60438), GSE75010(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75010), GSE22526(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22526), GSE74341(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74341).
References
Dimitriadis, E. et al. Pre-eclampsia. Nat. Rev. 9(1), 8 (2023).
Raymond, D. & Peterson, E. A critical review of early-onset and late-onset preeclampsia. Obstet. Gynecol. Surv. 66(8), 497–506 (2011).
Long, W. et al. Distinct expression profiles of lncRNAs between early-onset preeclampsia and preterm controls. Clin Chim. Acta Int. J. Clin. Chem. 463, 193–199 (2016).
Burton, G. J. et al. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 366, l2381 (2019).
Mahan, V. L. Heme oxygenase/carbon monoxide system affects the placenta and preeclampsia. Med. Gas Res. 15(2), 276–287 (2025).
Santana-Garrido, Á. et al. Oxidative and inflammatory imbalance in placenta and kidney of sFlt1-induced early-onset preeclampsia rat model. Antioxidants 11, 8 (2022).
Peng, X. et al. A comprehensive review of the roles of T-cell immunity in preeclampsia. Front. Immunol. 16, 1476123 (2025).
Szereday, L. et al. Immunological profiling of CD8(+) and CD8(-) NK Cell subpopulations and immune checkpoint alterations in early-onset preeclampsia and healthy pregnancy. Int. J. Mol. Sci. 25, 15 (2024).
Meggyes, M. et al. CD8 and CD4 Positive NKT subpopulations and immune-checkpoint pathways in early-onset preeclampsia and healthy pregnancy. Int. J. Mol. Sci. 24, 2 (2023).
Tsubokura, H. et al. Significance of placental CD200 expression in patients with preeclampsia: Comparison between early- and late-onset patients. Mol. Med Rep. 27, 1 (2023).
Chen, S. et al. Association of the LEP gene with immune infiltration as a diagnostic biomarker in preeclampsia. Front. Mol. Biosci. 10, 1209144 (2023).
von Mering, C. et al. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31(1), 258–261 (2003).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33(1), 1–22 (2010).
Zhou, X. & Tuck, D. P. MSVM-RFE: extensions of SVM-RFE for multiclass gene selection on DNA microarray data. Bioinformatics 23(9), 1106–1114 (2007).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12(5), 453–457 (2015).
Chen, H. et al. Association between FOXM1 and hedgehog signaling pathway in human cervical carcinoma by tissue microarray analysis. Oncol. Lett. 12(4), 2664–2673 (2016).
Huppertz, B. The Critical Role of Abnormal Trophoblast Development in the Etiology of Preeclampsia. Curr. Pharm. Biotechnol. 19(10), 771–780 (2018).
Gupta, A. K. et al. Analysis of plasma elastase levels in early and late onset preeclampsia. Arch. Gynecol. Obstet. 273(4), 239–242 (2006).
Aneman, I. et al. Mechanisms of key innate immune cells in early- and late-onset preeclampsia. Front. Immunol. 2020, 11 (1864).
Robillard, P. Y. et al. Progress in the understanding of the pathophysiology of immunologic maladaptation related to early-onset preeclampsia and metabolic syndrome related to late-onset preeclampsia. Am. J. Obstet. Gynecol. 226(2s), S867-s875 (2022).
Zhou, W. et al. Characterizing immune variation and diagnostic indicators of preeclampsia by single-cell RNA sequencing and machine learning. Commun. Biol. 7(1), 32 (2024).
Cornelius, D. C. et al. Inflammatory mediators: A causal link to hypertension during preeclampsia. Br. J. Pharmacol. 176(12), 1914–1921 (2019).
Elfarra, J. et al. Natural killer cells mediate pathophysiology in response to reduced uterine perfusion pressure. Clin. Sci. 131(23), 2753–2762 (2017).
Broekhuizen, M. et al. The placental innate immune system is altered in early-onset preeclampsia, but not in late-onset preeclampsia. Front. Immunol. 12, 780043 (2021).
Moore, J. K. et al. Patients with the worst outcomes after paracetamol (acetaminophen)-induced liver failure have an early monocytopenia. Aliment. Pharmacol. Ther. 45(3), 443–454 (2017).
Li, G. et al. Hypoxic stress disrupts HGF/Met signaling in human trophoblasts: implications for the pathogenesis of preeclampsia. J. Biomed. Sci. 29(1), 8 (2022).
Green, S. et al. Regulatory T cells in pregnancy adverse outcomes: A systematic review and meta-analysis. Front. Immunol. 12, 737862 (2021).
Piccinni, M. P. et al. Cytokines, hormones and cellular regulatory mechanisms favoring successful reproduction. Front. Immunol. 12, 717808 (2021).
Tsuda, S. et al. CD4(+) T cell heterogeneity in gestational age and preeclampsia using single-cell RNA sequencing. Front. Immunol. 15, 1401738 (2024).
Geldenhuys, J. et al. Disruption in the regulation of immune responses in the placental subtype of preeclampsia. Front. Immunol. 9, 1659 (2018).
Menkhorst, E. et al. IL11 activates the placental inflammasome to drive preeclampsia. Front. Immunol. 14, 1175926 (2023).
Han, X. et al. Differential dynamics of the maternal immune system in healthy pregnancy and preeclampsia. Front. Immunol. 10, 1305 (2019).
Vianna, R., Lopes, G. & Porto, L. C. Two novel HLA-DPA1*01:03:01 variants identified in Brazilian individuals: DPA1*01:03:01:38 and DPA1*01:03:01:39. Hla 96(6), 755–756 (2020).
Vianna, R. et al. The discovery of two HLA-DPA1*02:01:01 variants, found in Brazilian individuals. Hla 96(4), 555–556 (2020).
Blandin, L. et al. Characterization of the novel HLA-DPA1*01:03:34 allele by sequencing-based typing. Hla 99(3), 227–228 (2022).
Cargou, M. et al. Characterization of the novel HLA-DPA1*01:159 allele by sequencing-based typing. Hla 102(5), 647–648 (2023).
Cargou, M. et al. Characterization of the novel HLA-DPA1*01:150 allele by sequencing-based typing. Hla 102(4), 543–545 (2023).
Hu, Q. et al. Discovery of three novel HLA-DPA1 alleles, HLA-DPA1*01:147N, 01:03:47, and 02:106 using next-generation sequencing. Hla 102(3), 385–387 (2023).
Keppen, C. et al. Description of the novel null allele, HLA-DPA1*01:29N in a Lotha individual from Nagaland. North-East India. Hla 95(1), 82–83 (2020).
Chen, M. et al. Integrating single-cell RNA-seq, bulk RNA-seq, and Mendelian randomization to elucidate the role of HLA-DPA1 expression levels and non-classical monocytes in the pathogenesis of idiopathic pulmonary arterial hypertension. Int. J. Biol. Macromol. 319(Pt 3), 145284 (2025).
Stefańska, K. et al. High maternal-fetal HLA eplet compatibility is associated with severe manifestation of preeclampsia. Front. Immunol. 14, 1272021 (2023).
Nilsson, L. L. et al. Distribution of HLA-G extended haplotypes and one HLA-E polymorphism in a large-scale study of mother-child dyads with and without severe preeclampsia and eclampsia. Hla 88(4), 172–186 (2016).
Xia, Y. et al. The association of HLA-DP loci with autoimmune diabetes in Chinese. Diab. Res. Clin. Pract. 173, 108582 (2021).
Anderson-Baucum, E. et al. Deoxyhypusine synthase promotes a pro-inflammatory macrophage phenotype. Cell Metab. 33(9), 1883-1893.e7 (2021).
Tersigni, C. et al. HLA-DR is aberrantly expressed at feto-maternal interface in pre-eclampsia. J. Reprod. Immunol. 129, 48–52 (2018).
Huang, F. et al. Exploration of the combined role of immune checkpoints and immune cells in the diagnosis and treatment of ankylosing spondylitis: A preliminary study immune checkpoints in ankylosing spondylitis. Arth. Res. Ther. 26(1), 115 (2024).
Acknowledgements
The authors would like to express their gratitude to the GEO database, which has made available data related to EOPE and LOPE, and to the Import database, as both databases have provided valuable data on IRGs—a contribution that has laid a critical foundation for the completion of this study.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Joint funds for the Fujian Provincial Health Technology Project (2024GGA044), the innovation of science and technology, Fujian province (Grant number: 2024Y9412), the innovation of science and technology, Fujian province (Grant number: 2023Y9234), and the Second Affiliated Hospital of Fujian Medical University Doctoral Miaopu Project (BS202401).
Author information
Authors and Affiliations
Contributions
Z.W.: Writing–review and editing, writing–original draft, visualization, validation, formal analysis, data curation, conceptualization, funding acquisition. Y.X.: Writing–review and editing, writing–original draft, visualization, validation, formal analysis, data curation, conceptualization. W.C. and Z.Z.: Writing–review and editing, formal analysis. L.H. and L.S.: Writing–review and editing, validation, investigation. Y.W. and B.C.: Writing–review and editing, visualization. C.Y. and Y.K.: Conceptualization, writing review and editing, project administration, supervision, funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All human-related studies obtained ethical approval from the Second Affiliated Hospital of Fujian Medical University, and they were implemented in strict compliance with both local legislative regulations and the specific requirements of the hosting institution—two prerequisites that ensure the study’s adherence to ethical and legal standards. Furthermore, each participant involved in the research voluntarily provided written informed consent before their enrollment, a step that is fundamental to upholding the principles of participant autonomy and ethical research practice.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Z., Xie, Y., Chen, W. et al. HLA-DPA1 as a diagnostic biomarker differentiating early- and late-onset preeclampsia. Sci Rep 16, 8206 (2026). https://doi.org/10.1038/s41598-026-39050-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-39050-0
