
Abstract
Radiotherapy is an essential treatment for non-small cell lung cancer (NSCLC), but its effectiveness is often reduced by radioresistance. miR-320a has been shown to improve radiosensitivity in NSCLC, but the molecular mechanisms are not well understood. This study investigates how miR-320a regulates RAD51 to affect the radiosensitivity of NSCLC. miR-320a expression in NSCLC tissues and cell lines was evaluated using The Cancer Genome Atlas (TCGA) data and qRT-PCR. RAD51 was predicted as a miR-320a target using bioinformatic tools (TargetScan, miRDB, miRTarBase) and validated by dual-luciferase reporter assays. Functional experiments were conducted to examine the effects of miR-320a and RAD51 manipulation on NSCLC cell responses to varying radiation doses. Ferroptosis was examined by measuring lipid reactive oxygen species (ROS) and GPX4 expression levels. miR-320a expression was markedly reduced in NSCLC tissues and cell lines relative to normal controls and showed a positive association with clinical radiosensitivity. Functional experiments demonstrated that miR-320a overexpression increased radiosensitivity by inhibiting post-irradiation cell proliferation, colony formation, and migration. RAD51 was validated as a direct post-transcriptional target of miR-320a. Mechanistically, RAD51 expression inversely correlated with miR-320a (R = −0.16, P < 0.001) and was associated with reduced radiosensitivity both in vitro and in patient samples. Importantly, RAD51 knockdown reversed the radioresistance induced by miR-320a inhibition. Further analyses revealed that RAD51 positively regulated GPX4 expression, thereby suppressing ferroptosis. Inhibition of RAD51 or restoration of miR-320a led to enhanced lipid peroxidation, as evidenced by increased lipid ROS accumulation and reduced GPX4 expression, ultimately sensitizing NSCLC cells to radiotherapy. Our results indicate that miR-320a promotes NSCLC radiosensitivity through a negative regulation of RAD51. The miR-320a/RAD51/GPX4 axis may be used as a key pathway in regulating NSCLC radiosensitivity.
Similar content being viewed by others


Introduction
Non-small cell lung cancer (NSCLC) accounts for about 85% of all lung cancer diagnoses, making it the most common histological type1. The lack of distinct early clinical manifestations often results in diagnoses at advanced stages2. Radiotherapy is applicable to NSCLC across all disease stages, with nearly 50% of patients receiving this modality3. However, the development of radiotherapy resistance frequently leads to disease progression or metastasis4, and the underlying mechanisms contributing to such resistance remain poorly understood. Therefore, elucidating the mechanisms of radioresistance and identifying effective therapeutic targets are of great clinical importance.
MicroRNAs (miRNAs), a major class of non-coding RNAs, are critical post-transcriptional regulators involved in both physiological homeostasis and tumor initiation and progression5,6. Owing to their stability and detectability in body fluids, miRNAs have emerged as promising biomarkers for cancer prognosis and therapeutic decision-making7,8,9. Numerous studies have reported that miRNAs participate in key signaling pathways and DNA damage repair mechanisms, implicating them in cellular radiosensitivity10,11,12. Previous studies have indicated that microRNAs, such as miR-192, miR-221, and miR-223, modulate cell cycle progression and contribute to resistance against radiotherapy and chemotherapy by targeting p2713. In this context, miR-320a has attracted increasing attention as a potential radiosensitizing miRNA that negatively regulates radiation resistance in cancer cells14. According to Xu et al., miR-320a reduces radiotherapy resistance in NSCLC by suppressing PTEN methylation mediated by HIF-1α15. Interestingly, Wang et al. demonstrated that ZEB1 promotes the expression of RAD51AP1 by repressing miR-320a, thereby facilitating NSCLC progression16. However, whether miR-320a directly modulates core DNA repair effectors and thereby influences radiosensitivity remains unclear.
RAD51 is a central effector of the homologous recombination repair pathway and plays a pivotal role in maintaining genomic stability following ionizing radiation-induced DNA double-strand breaks. Elevated expression of RAD51 has been observed in lung, prostate, and breast cancers, among others17,18, and has long been recognized as a contributor to radiotherapy resistance19. For instance, research by Wu et al. demonstrated that suberoylanilide hydroxamic acid (SAHA) boosts radiosensitivity in pancreatic cancer cells by suppressing RAD51 expression20. In NSCLC, Piotto et al. reported that upregulation of hsa-miR-96-5p enhances radiation sensitivity by inhibiting RAD5121. Importantly, recent studies have suggested that defects in homologous recombination repair can influence the cellular response to radiation not only by increasing unrepaired DNA damage but also by altering susceptibility to regulated cell death pathways22.
Ferroptosis is a distinct form of regulated cell death driven by iron-dependent lipid peroxidation. Accumulating evidence indicates that radiotherapy can trigger ferroptotic cell death in cancer cells. As a central suppressor of ferroptosis, glutathione peroxidase 4 (GPX4) preserves membrane integrity by detoxifying lipid hydroperoxides23. Notably, emerging studies suggest that DNA repair proficiency may modulate cellular susceptibility to radiation-induced ferroptosis24; however, the underlying molecular mechanisms remain poorly defined. In this context, whether RAD51 participates in GPX4-mediated ferroptosis regulation during radiotherapy remains to be elucidated.
These observations led us to hypothesize that a miR-320a/RAD51/GPX4 axis may be involved in regulating radioresistance in NSCLC. In this study, we observed that high RAD51 expression correlates with reduced radiosensitivity in NSCLC cells, while miR-320a exerts an inhibitory effect on RAD51. From these results, we suggest that miR-320a could be a radiosensitizing agent that influences NSCLC’s reaction to radiation by regulating RAD51 and pathways related to ferroptosis.
Materials and methods
Clinical data and sample collection
Clinical characteristics and transcriptomic (RNA-seq) data of NSCLC patients were retrieved from The Cancer Genome Atlas (TCGA) via the UCSC Xena browser (http://xena.ucsc.edu/welcome-to-ucsc-xena/). Transcriptomic data were normalized using the transcripts per million (TPM) method. Following approval from the Ethics Committee of Affiliated Zhongshan Hospital of Dalian University (PJ-KY2025-184–1), we collected 60 tumor tissue specimens from patients diagnosed with NSCLC, between January 2016 and December 2021 (Supplementary Table 1). Patients who had not received any anti-tumor treatment within one month before radiotherapy were included, and those with contraindications to radiotherapy were excluded. Treatment response was evaluated according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Radiological assessments were performed using contrast-enhanced CT at approximately 5–9 weeks after completion of radiotherapy. Imaging assessments were conducted by two senior oncologists with extensive experience in tumor response evaluation, following standardized institutional protocols. Patients achieving complete response (CR) or partial response (PR) were classified as responders, whereas those with stable disease (SD) or progressive disease (PD) were classified as non-responders. All fresh tissue samples were immediately snap-frozen in liquid nitrogen and preserved at −80 °C for subsequent analysis.
Cell lines and cell culture
Human NSCLC cell lines (A549, H1299, and H1975) and the normal human bronchial epithelial cell line (BEAS-2B) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). These cell lines were maintained in RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin, and incubated at 37 °C in a humidified environment with 5% CO₂.
Quantitative real-time PCR
Total RNA was isolated from cultured cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA concentration and purity were determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Germany). From 1 µg of total RNA, complementary DNA (cDNA) was generated using the SweScript All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (Servicebio, China), following the manufacturer’s protocol. Quantitative PCR was performed using 2 × SYBR Green RT-PCR Master Mix (None ROX; Servicebio, China) on a real-time PCR system (Heal Force, China). The CT method used 2-ΔΔCT to calculate the relative expression levels of miR-320a and RAD51.
Western blot analysis
Cells were lysed using RIPA lysis buffer supplemented with protease and phosphatase inhibitors to extract total protein. Equal amounts of protein were separated by 10% SDS-PAGE (Servicebio, China) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). 5% non-fat dry milk (Solarbio, China) diluted in TBST was used to block membranes for 1 h at ambient temperature. Membranes were then incubated with primary antibodies at 4 °C overnight. After rinsing with TBST, the membranes were processed for chemiluminescence detection using a CLINX imaging system. Protein bands were visualized, and their intensities were quantified using ImageJ software for gel image analysis. RAD51 antibody (rabbit polyclonal; ABclonal, A6268) was diluted 1:1000 in TBST, whereas the GPX4 antibody (mouse monoclonal; Proteintech, 67763-1-Ig) was diluted 1:2000.
Plasmids transfection
MiR-320a mimics, miR-320a inhibitor, and their corresponding controls were purchased from RiboBio, pLenti. miRNA mimic was compared with a corresponding mimic negative control with a non-targeting sequence (UUGUACUACACAAAAGUACUG). For miRNA inhibition experiments, an inhibitor negative control with a non-targeting sequence (CAGUACUUUUGUGUAGUACAA) was used. The transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The cells were collected for analysis 24 h after transfection.
Immunohistochemical staining
Tissue sections were treated with minocycline-ethylenediaminetetraacetic acid to remove paraffin and rehydrated through a series of graded ethanol solutions. Endogenous peroxidase activity was blocked by immersing sections in a 0.3% methanol-peroxidase solution for 30 min. For antigen retrieval, sections were heated in citric acid buffer using a microwave oven at 100 °C for 15 min. After washing three times with phosphate-buffered saline (PBS) (Thermo Scientific, Germany), sections were incubated with primary antibodies at 4 °C overnight. Staining was performed using 3,3'-diaminobenzidine, followed by counterstaining with hematoxylin. The Aipathwell system (Servicebio, China) was employed to quantify the proportion of immunoreactive cells and assess staining intensity.
Dual luciferase reporter assay
The HEK-293FT cells were cultured in a 24 well plate (1 × 105 cells/well) and transfected using Lipofectamine 2000 (Invitrogen) with RAD51-3UTR-wt or RAD51-3UTR-mt and hsa-miR-320a-3pmimics or mimics NC, respectively. After 24 h, luciferase activity was detected using the double luciferase assay kit (Beyotime, China) and standardized to Renilla Luciferase activity.
Wound-healing assay
Cells were seeded in 6-well plates (1 × 105 cells/well) and cultured overnight to form a confluent monolayer. Cells were then treated with mitomycin C (30 µM) for 2 h, followed by irradiation or other indicated treatments. Twenty-four hours after irradiation, a uniform linear wound was created using a sterile 200 µL pipette tip. Wound closure was monitored and photographed at 0 and 48 h after scratching under a light microscope. The wound area was quantified using ImageJ software.
Colony formation assay
The cells were cultured in a 6-well plate (1000 cells per well), and then irradiated with 0, 2, 4, 6, and 8 Gy of X-rays and incubated in a culture incubator for 14 days. The cells were fixed in polymethanal (paraformaldehyde) and stained with crystal violet for counting. Cell survival rates in response to the different irradiation doses were evaluated using the results of 0 Gy irradiation as a control.
Cell counting kit-8 assay
Irradiation was carried out using an Elekta Synergy linear accelerator with 6 MV X-rays in an isocentric setup. The source-to-axis distance (SAD) was 100 cm, and the gantry was positioned at 0°. The dose rate was 512 cGy/min. Dose calibration was performed by medical physicists using a standard ionization chamber in accordance with institutional quality assurance procedures based on the AAPM TG-51 protocol. The cells were cultured in a 96 well plate (2 × 10^3 cells/well), and then irradiated with 0, 2, 4, 6, and 8 Gy X-rays. Adding 10 μl CCK-8 solution to each well and incubate for 4 h Absorbance was measured at 450 nm using an enzyme-linked immunosorbent assay (ELISA) reader, and the data was recorded.
Flow cytometric detection of lipid ROS
Cells were resuspended in 500 μL of C11-BODIPY 581/591 staining solution (final concentration: 5 μM) and incubated at 37 °C in the dark for 30 min, with gentle mixing every 5–10 min. After staining, the cells underwent two PBS washes, were resuspended in PBS, and promptly analyzed by flow cytometry via the FL1-A channel.
Statistical analysis
Statistical analyses were conducted with R (v4.0.3) and GraphPad Prism (v9). Gene expression and survival analyses were conducted based on data retrieved from TCGA. Potential targets of miR-320a were identified through integration of predictions from TargetScan, miRDB, and miRTarBase. The prognostic relevance of gene expression levels was evaluated using Kaplan–Meier survival analysis. The clinical sample size was calculated to ensure a statistical power of at least 80%. All in vitro experiments were performed with independent biological replicates, and each biological replicate consisted of multiple technical measurements. For data meeting assumptions of normality and homoscedasticity, parametric tests were applied; otherwise, appropriate nonparametric tests were used. Comparisons between two groups were performed using Student’s t-test. Multiple-group comparisons were conducted using one-way ANOVA, followed by Tukey’s post hoc test. Results are presented as mean ± standard error of the mean (SEM). A p-value < 0.05 was considered statistically significant. The following notation was applied to indicate levels of significance: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Results
miR-320a is linked to the radiosensitivity of non-small cell lung cancer
The regulatory role of miR-320a in NSCLC radiosensitivity was evaluated using clinical specimens and in vitro models. In NSCLC tumor tissues, miR-320a levels were significantly lower compared to the surrounding non-cancerous lung tissues (Fig. 1A). This pattern was further confirmed in multiple NSCLC cell lines (A549, H1299, H1975), which exhibited substantially lower miR-320a expression than nonmalignant bronchial epithelial cells (BEAS-2B) (Fig. 1B). Survival analysis suggested a potential link between low miR-320a expression and poor prognosis in NSCLC patients (Fig. 1C), while functional experiments provided more conclusive evidence of its role. In A549 cells, inhibition of miR-320a enhanced clonogenic survival following irradiation and accelerated wound closure in scratch assays, reflecting increased cell proliferation, migration, and radioresistance (Figs. 1D-F; Supplementary Fig. 1).

Low miR-320a expression reduces radiosensitivity in NSCLC cells. (A) Comparison of mRNA levels between NSCLC tissues (n = 502) and adjacent normal tissues (n = 52). (B) Relative expression of miR-320a in NSCLC cell lines compared to normal bronchial epithelial BEAS-2B cells (n = 3). (C) Kaplan–Meier survival analysis revealed a correlation between miR-320a expression and overall survival in NSCLC patients undergoing radiotherapy (n = 277). (D) miR-320a expression following treatment with a specific inhibitor (n = 3). (E) Clonogenic survival of cells with miR-320a inhibition subjected to escalating doses of radiation (0, 2, 4, 6, 8 Gy). (F) Wound healing assay evaluating migration capacity of miR-320a-inhibited cells at 48 h following irradiation at varying doses (0, 2, 4, 6, 8 Gy). Data are presented as mean ± SEM. Differences between the two groups were assessed using Student’s t-test (A, D). * p < 0.05.
Conversely, enforced overexpression of miR-320a in H1299 cells suppressed colony-forming ability and delayed wound closure after irradiation, indicating reduced cell viability and impaired repair capacity (Figs. 2A-C; Supplementary Fig. 2). These phenotypic changes suggest that elevated miR-320a expression sensitizes tumor cells to radiation. According to Fig. 2D, the levels of miR-320a were notably higher in the CR + PR group than in the stable or SD + PD group, as indicated by the clinical response correlation analysis. Collectively, these data establish miR-320a as a positive regulator of radiosensitivity in NSCLC, potentially mediated through suppression of migratory and reparative capacities, warranting further molecular elucidation.

High miR-320a expression enhances radiosensitivity. (A) Induction of miR-320a expression following treatment with miR-320a mimics (n = 3). (B) Clonogenic survival of miR-320a-overexpressing cells after irradiation (0, 2, 4, 6, 8 Gy). (C) Migration capacity of miR-320a-overexpressing cells assessed by wound healing assay at 48 h after irradiation (0, 2, 4, 6, 8 Gy). (D) Comparison of miR-320a levels between NSCLC patients exhibiting SD + PD and those achieving CR + PR; n = 60 NSCLC patients. Data are presented as mean ± SEM. Differences between the two groups were assessed using Student’s t-test (A, D). *** p < 0.001.
RAD51 is a target of miR-320a
Integrated bioinformatic analyses using miRDB, miRTarBase, and TargetScan consistently predicted RAD51 as a direct target of miR-320a (Fig. 3A). Correlation analysis revealed an inverse relationship between miR-320a expression and RAD51 mRNA levels in NSCLC samples (R = −0.16, P < 0.001; Fig. 3B). Functional validation in A549 cells demonstrated that inhibition of miR-320a led to a marked increase in both RAD51 mRNA and protein expression (Figs. 3C and D). Conversely, overexpression of miR-320a in H1299 cells did not significantly alter RAD51 mRNA levels (Fig. 3E), but substantially reduced RAD51 protein expression (Fig. 3F), suggesting that miR-320a regulates RAD51 primarily at the post-transcriptional level.

miR-320a negatively regulates RAD51. (A) Prediction of miR-320a target genes using miRDB, miRTarBase, and TargetScan. (B) Correlation analysis between miR-320a and RAD51 mRNA expression (n = 473). (C, D) Changes in RAD51 mRNA and protein levels after miR-320a inhibition (n = 3). (E, F) Changes in RAD51 mRNA and protein levels after miR-320a overexpression (n = 3). (G) Luciferase activity changes following transfection with miR-320a mimics (n = 3). Data are presented as mean ± SEM. Differences between the two groups were assessed using Student’s t-test (B, E, G).
To validate the direct interaction between miR-320a and RAD51, a dual-luciferase reporter assay was conducted. Co-transfection of miR-320a mimics with reporter constructs containing either the wild-type RAD51 3’UTR led to a significant decrease in luciferase activity. This suggests that miR-320a suppresses RAD51 expression through binding to its 3’UTR (Fig. 3G). Collectively, these results confirm RAD51 as a genuine post-transcriptional target of miR-320a.
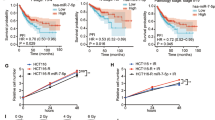
RAD51 is associated with radiotherapy resistance
Analysis of TCGA lung adenocarcinoma (LUAD) datasets revealed that RAD51 mRNA expression is markedly elevated in tumor tissues relative to adjacent normal lung tissues (Fig. 4A). This upregulation was also confirmed across multiple LUAD-derived cell lines (Fig. 4B). Survival analysis demonstrated that patients with higher RAD51 expression exhibit poorer overall survival compared to those with lower expression, suggesting its potential prognostic value (Fig. 4C). To assess the role of RAD51 in radiosensitivity, the NSCLC cell lines H1299 and H1975, which have different levels of RAD51 expression, were exposed to increasing doses of radiation (0, 2, 4, 6, and 8 Gy). Colony formation assays showed that cell proliferation declined with increasing radiation dose in both cell lines; however, no statistically significant difference in clonogenic survival was observed between the two (Figs. 4D-F). In contrast, scratch wound healing and CCK-8 assays revealed that H1299 cells displayed greater migratory and proliferative capacity than H1975 cells under irradiation, indicating a higher resistance to radiotherapy (Figs. 4G-K). Clinically, RAD51 expression was significantly lower in NSCLC patients achieving CR + PR to radiotherapy compared to those with SD + PD (Fig. 4L), further supporting the association between elevated RAD51 levels and radiotherapy resistance. Collectively, these findings suggest that RAD51 contributes to reduced radiosensitivity in NSCLC by promoting cellular survival and migration following irradiation.

RAD51 promotes radioresistance in NSCLC. (A) Comparison of RAD51 mRNA expression between NSCLC tumor tissues and adjacent normal tissues. (B) RAD51 mRNA and protein levels in BEAS-2B, A549, H1299, and H1975 cell lines (n = 3). (C) Kaplan–Meier analysis showing the association between RAD51 expression and overall survival in NSCLC patients. (D–F) Clonogenic survival of H1299 and H1975 cells exposed to increasing doses of radiation (0, 2, 4, 6, and 8 Gy). (G–I) Wound healing assay evaluating migration capacity of H1299 and H1975 cells at 48 h after irradiation. (J, K) Cell viability analysis using CCK-8 assay in irradiated H1299 and H1975 cells. (L) RAD51 protein expression in SD + PD vs. CR + PR patient subgroups. n = 60 NSCLC patients; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
RAD51 reverses miR-320a-mediated radiosensitivity
To clarify the interplay between miR-320a and RAD51 in modulating radiosensitivity, we conducted loss- and gain-of-function experiments in A549 cells. RAD51 knockdown via shRNA significantly reduced its mRNA and protein levels compared to controls (Fig. 5A). Inhibition of miR-320a markedly increased RAD51 expression, an effect that was partially reversed by RAD51 silencing (Fig. 5B). Functionally, suppression of miR-320a enhanced clonogenic survival across a radiation dose range of 0–8 Gy, indicating reduced radiosensitivity. This effect was significantly attenuated upon RAD51 knockdown, which restored cellular sensitivity to irradiation (Fig. 5C). Similarly, scratch wound healing assays demonstrated accelerated wound closure following miR-320a inhibition, consistent with enhanced migratory capacity and radioresistance; RAD51 silencing mitigated this effect (Fig. 5D). These data collectively indicate that RAD51 functions downstream of miR-320a, mediating its regulatory impact on NSCLC radiosensitivity. Specifically, RAD51 overexpression counteracts the radiosensitizing effects of miR-320a, emphasizing its pivotal role in resistance to radiotherapy.

RAD51 reverses miR-320a–mediated radiosensitivity in NSCLC cells (A) RAD51 mRNA and protein expression following shRNA-mediated knockdown, detected by qRT-PCR and Western blot (n = 3). (B) RAD51 expression in cells treated with miR-320a inhibitor alone or in combination with RAD51 knockdown (n = 3). (C) Clonogenic survival following miR-320a inhibition with or without RAD51 silencing. (D) Migration ability assessed by wound healing assay at 48 h after irradiation in cells with miR-320a inhibition, with or without RAD51 knockdown, at varying doses (0, 2, 4, 6, 8 Gy). Data are presented as mean ± SEM. P-values were calculated using either Student’s t-test (A) or one-way ANOVA (B).
RAD51 regulates GPX4 to modulate radiosensitivity
To elucidate the mechanism by which RAD51 influences radiosensitivity in NSCLC, we employed both pharmacological inhibition and gene overexpression strategies. H1299 cells were treated with the RAD51 inhibitor B02, while H1975 cells were transfected to overexpress RAD51. We first examined GPX4 expression across graded radiation doses (0, 2, 4, 6, and 8 Gy) under different RAD51 conditions. In both cell lines, GPX4 expression varied with increasing radiation dose. However, the patterns of change differed between RAD51-inhibited and RAD51-overexpressing cells (Fig. 6A). These observations indicate that irradiation alters GPX4 expression, but the dose-escalation experiment alone does not establish whether this effect is directly mediated by radiation or secondary to differences in RAD51 status. To specifically evaluate RAD51-dependent regulation, we focused on 4 Gy and compared GPX4 expression under controlled RAD51 manipulation. At this fixed radiation dose, pharmacologic inhibition of RAD51 significantly reduced GPX4 expression relative to control cells, whereas RAD51 overexpression increased GPX4 levels (Figs. 6B and C). Because radiation exposure was held constant in these experiments, the observed differences in GPX4 expression can be attributed to RAD51 status. Functional assays further supported this regulatory relationship. Under 4 Gy irradiation, GPX4 overexpression enhanced cell viability, whereas GPX4 knockdown reduced viability (Supplementary Fig. 3). Together, these findings indicate that RAD51 positively regulates GPX4 expression under irradiation conditions and thereby contributes to radioresistance in NSCLC cells.

RAD51 reduces radiosensitivity by upregulating GPX4 expression. (A) GPX4 protein levels in NSCLC cells following exposure to varying doses of irradiation (0, 2, 4, 6, 8 Gy). (B, C) Western blot analysis of GPX4 expression after RAD51 inhibition (via B02) or overexpression; **** p < 0.0001.
miR-320a modulates ferroptosis via regulation of RAD51
To further explore the molecular mechanism through which miR-320a regulates RAD51 to influence radiosensitivity, we investigated its role in ferroptosis modulation. Flow cytometric analysis revealed that miR-320a inhibition significantly reduced lipid ROS levels relative to the negative control, suggesting a decrease in ferroptosis (Fig. 7A and B). However, simultaneous silencing of RAD51 in miR-320a-inhibited cells restored lipid ROS levels, reversing the effect of miR-320a inhibition (Fig. 7A and B). Western blot analysis corroborated these findings, showing that GPX4 expression increased in A549 cells after miR-320a inhibition but decreased when RAD51 was knocked down (Fig. 7C). In TCGA-LUAD clinical samples, miR-320a showed a significant negative correlation with GPX4 (R = −0.16, P < 0.001; Supplementary Fig. 4). These results suggest that miR-320a downregulates RAD51, resulting in decreased GPX4 expression, which subsequently enhances ferroptosis and alters the radiosensitivity of NSCLC (Fig. 8).

miR-320a promotes ferroptosis by negatively regulating RAD51. (A, B) Lipid ROS levels detected by flow cytometry in cells treated with miR-320a inhibitor alone or in combination with RAD51 knockdown (n = 3). (C) GPX4 protein expression in cells treated with miR-320a inhibitor with or without RAD51 silencing. Data are presented as mean ± SEM. P-values were calculated using one-way ANOVA (B). ** p < 0.01, *** p < 0.001.

Schematic diagram illustrating the regulatory role of miR-320a in radiosensitivity.
Discussion
Radiotherapy is a cornerstone in the treatment of NSCLC; however, radioresistance frequently leads to disease progression or metastasis4. Elucidating the mechanisms underlying radiotherapy resistance and identifying potential therapeutic targets are therefore crucial for improving treatment outcomes and prolonging patient survival.
Previous studies have suggested that miR-320a may serve as a promising biomarker and therapeutic target in various malignancies. Aberrant expression of miR-320a has been documented in malignancies such as colorectal25, breast26, gastric27, and prostate cancers28, where it typically exerts tumor-suppressive effects by inhibiting cell proliferation and migration. In NSCLC, downregulation of miR-320a has also been observed29. Supporting these observations, our analysis of TCGA datasets revealed a marked reduction in miR-320a levels in tumor samples relative to adjacent normal lung tissues.
Malachowska et al. observed that serum concentrations of multiple miRNAs, including miR-320a, are altered in response to irradiation, showing an increase after radiation treatment30. In line with this, Zheng et al. observed that miR-320a expression in cancer cells rises in a manner dependent on radiation dose and exposure time14. Together, these findings suggest a role for miR-320a in modulating radiosensitivity. Consistently, our results revealed higher miR-320a levels in NSCLC patients who were radiosensitive compared to those exhibiting radioresistance, reinforcing its potential function in promoting radiosensitivity.
Nevertheless, the molecular mechanisms by which miR-320a regulates radiosensitivity have not been fully elucidated. Through bioinformatic analysis, we predicted a potential regulatory interaction between miR-320a and RAD51. Although the observed correlation coefficient was modest (R = −0.16), such weak correlations are not unexpected in bulk tumor datasets characterized by substantial inter-patient heterogeneity and multi-factorial regulation. Therefore, the biological relevance of this association should not be interpreted based on correlation strength alone. Instead, it is supported by consistent functional in vitro data, which demonstrate that modulation of miR-320a leads to reproducible changes in RAD51, in line with the proposed mechanistic framework. The convergence of correlative clinical data and functional experimental evidence strengthens the biological plausibility of our findings. Based on this, we focused our mechanistic investigations on RAD51. As a central component of the homologous recombination repair pathway, RAD51 plays a pivotal role in the cellular response to DNA damage and has been associated with poor prognosis in lung cancer31. Multiple studies have indicated that miR-34a, miR-96, miR-182, and miR-4429 can all regulate DNA damage repair through RAD51, and some have even been implicated in modulating cellular radiosensitivity21,32,33,34. Building on these findings, we further validated RAD51 as a direct target of miR-320a using dual-luciferase reporter assays. Functionally, we demonstrated that RAD51 overexpression diminishes the radiosensitizing effects of miR-320a, while RAD51 knockdown reverses the radioresistant phenotype induced by miR-320a inhibition. These data firmly establish RAD51 as a key downstream effector of miR-320a in regulating NSCLC cell response to radiation. However, these studies did not further elucidate how these miRNAs regulate radiosensitivity at the mechanistic level.
Interestingly, recent evidence suggests that activation of the RAD51-mediated ferroptotic signaling axis drives intracellular ROS accumulation, thereby overcoming radioresistance and enhancing cellular radiosensitivity35. We also observed that RAD51 may influence radiosensitivity through regulation of GPX4. GPX4 functions by reducing lipid peroxides and thereby protecting cells from iron-dependent oxidative damage36,37. Increasing evidence indicates that ferroptosis is crucial for inhibiting tumors38,39. Emerging evidence indicates that GPX4 inhibition induces ferroptotic vulnerability, thereby reversing radioresistance40. Notably, integration of radiotherapy with ferroptosis-targeted approaches exerts synergistic effects on tumor radiosensitization and tumor growth inhibition41,42. In our study, RAD51-overexpressing NSCLC cells exhibited elevated GPX4 expression, while RAD51 inhibition led to decreased GPX4 levels. Additionally, we discovered that GPX4 expression is altered by radiation in a manner dependent on the dose, notably within the 0–4 Gy range. Evidence suggests that GPX4 is particularly responsive to low-dose radiation, whereas once the irradiation dose exceeds a critical threshold, cellular antioxidant and damage-repair processes may become saturated, thereby limiting further transcriptional modulation43,44,45. These findings indicate that RAD51 may contribute to radioresistance by upregulating GPX4 and attenuating lipid peroxidation. Further mechanistic analyses revealed that miR-320a promotes ferroptosis by targeting RAD51 and thereby reducing GPX4 expression. Inhibition of miR-320a decreased lipid ROS accumulation and increased GPX4 levels, consistent with suppressed ferroptosis. These effects were reversed upon RAD51 silencing, reinforcing the regulatory axis involving miR-320a, RAD51, and GPX4 in ferroptosis and radiosensitivity. Unlike miR-34a, miR-96, miR-182, and miR-442921,32,33,34, which mainly act on canonical homologous recombination repair components, miR-320a uniquely bridges the DNA repair machinery with lipid peroxidation defense, thereby providing a distinct mechanistic pathway that complements existing miRNA-mediated regulatory models.
Of course, the present study is mainly based on mechanistic investigations conducted in vitro, and thus lacks in vivo confirmation, validation in radiation-exposed human tissues, and support from a larger patient cohort. Consequently, the proposed mechanisms may not fully recapitulate the complexity of the tumor microenvironment in vivo. We also acknowledge that although GPX4 appears to participate in the miR-320a/RAD51 regulatory axis based on expression trends, its functional role has not been fully validated through GPX4 knockdown or overexpression. GPX4 should therefore be considered a putative mediator rather than a definitively established mechanistic driver. Despite these limitations, our findings suggest that miR-320a and RAD51 may serve as potential predictive biomarkers for radiosensitivity.
Conclusion
In conclusion, this study suggests the involvement of a miR-320a/RAD51/GPX4 regulatory axis in modulating radiosensitivity in NSCLC. Our data indicate that miR-320a may enhance radiosensitivity by suppressing RAD51, accompanied by reduced GPX4 expression and increased ferroptosis. However, these observations are primarily based on in vitro evidence, and further in vivo studies as well as functional validation of GPX4 are required to confirm the biological relevance and therapeutic implications of this pathway in radiotherapy resistance.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Gridelli, C. et al. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 1, 15009 (2015).
Ferro, A. et al. The multidisciplinary approach in stage III non-small cell lung cancer over ten years: From radiation therapy optimisation to innovative systemic treatments. Cancers (Basel) https://doi.org/10.3390/cancers14225700 (2022).
Kong, F. M., Zhao, J., Wang, J. & Faivre-Finn, C. Radiation dose effect in locally advanced non-small cell lung cancer. J. Thorac. Dis. 6, 336–347 (2014).
Sitthideatphaiboon, P. et al. STK11/LKB1 mutations in NSCLC are associated with KEAP1/NRF2-dependent radiotherapy resistance targetable by glutaminase inhibition. Clin. Cancer Res. 27, 1720–1733 (2021).
Goodall, G. J. & Wickramasinghe, V. O. RNA in cancer. Nat. Rev. Cancer 21, 22–36 (2021).
Slack, F. J. & Chinnaiyan, A. M. The role of non-coding RNAs in oncology. Cell 179, 1033–1055 (2019).
Zen, K. & Zhang, C. Y. Circulating microRNAs: A novel class of biomarkers to diagnose and monitor human cancers. Med. Res. Rev. 32, 326–348 (2012).
Weber, J. A. et al. The microRNA spectrum in 12 body fluids. Clin. Chem. 56, 1733–1741 (2010).
Wang, K. et al. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 38, 7248–7259 (2010).
Wang, Y. et al. MicroRNA regulation of ionizing radiation-induced premature senescence. Int. J. Radiat. Oncol. Biol. Phys. 81, 839–848 (2011).
Hu, H. & Gatti, R. A. MicroRNAs: New players in the DNA damage response. J. Mol. Cell Biol. 3, 151–158 (2011).
Wang, J. et al. MiR-320b/RAD21 axis affects hepatocellular carcinoma radiosensitivity to ionizing radiation treatment through DNA damage repair signaling. Cancer Sci. 112, 575–588 (2021).
Arghiani, N. & Shah, K. Modulating microRNAs in cancer: Next-generation therapies. Cancer Biol. Med. 19, 289–304 (2021).
Hu, Z. et al. Transcriptional activation of miR-320a by ATF2, ELK1 and YY1 induces cancer cell apoptosis under ionizing radiation conditions. Int. J. Oncol. 53, 1691–1702 (2018).
Xu, L. M. et al. Overcoming of radioresistance in non-small cell lung cancer by microRNA-320a through HIF1alpha-suppression mediated methylation of PTEN. Front. Cell Dev. Biol. 8, 553733 (2020).
Wang, H. et al. ZEB1 induces non-small cell lung cancer development by targeting microRNA-320a to increase the expression of RAD51AP1. Exp. Cell Res. 405, 112687 (2021).
Hine, C. M., Seluanov, A. & Gorbunova, V. Use of the Rad51 promoter for targeted anti-cancer therapy. Proc. Natl. Acad. Sci. U. S. A. 105, 20810–20815 (2008).
Budke, B., Lv, W., Kozikowski, A. P. & Connell, P. P. Recent developments using small molecules to target RAD51: How to best modulate RAD51 for anticancer therapy?. ChemMedChem 11, 2468–2473 (2016).
Zhao, Y. & Chen, S. Targeting DNA double-strand break (DSB) repair to counteract tumor radio-resistance. Curr. Drug Targets 20, 891–902 (2019).
Wu, Z. et al. The effects of SAHA on radiosensitivity in pancreatic cancer cells by inducing apoptosis and targeting RAD51. Biomed. Pharmacother. 89, 705–710 (2017).
Piotto, C., Biscontin, A., Millino, C. & Mognato, M. Functional validation of miRNAs targeting genes of DNA double-strand break repair to radiosensitize non-small lung cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 1861, 1102–1118 (2018).
Szmyd, R. et al. Homologous recombination promotes non-immunogenic mitotic cell death upon DNA damage. Nat. Cell Biol. 27, 59–72 (2025).
Yang, W. S. & Stockwell, B. R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 26, 165–176 (2016).
Jiang, K. et al. STC2 activates PRMT5 to induce radioresistance through DNA damage repair and ferroptosis pathways in esophageal squamous cell carcinoma. Redox Biol. 60, 102626 (2023).
Zhang, W. et al. miR-320a/SP1 negative reciprocal interaction contributes to cell growth and invasion in colorectal cancer. Cancer Cell Int. 21, 175 (2021).
Zhang, C. et al. LINC00460 Facilitates Cell Proliferation and Inhibits Ferroptosis in Breast Cancer Through the miR-320a/MAL2 Axis. Technol Cancer Res Treat 22, 15330338231164360 (2023).
Ge, X. et al. miR-320a modulates cell growth and chemosensitivity via regulating ADAM10 in gastric cancer. Mol. Med. Rep. 16, 9664–9670 (2017).
Bozgeyik, E. et al. miR-320a promotes p53-dependent apoptosis of prostate cancer cells by negatively regulating TP73-AS1 invitro. Biochem. Biophys. Res. Commun. 619, 130–136 (2022).
Wang, J. et al. MicroRNA-320a is downregulated in non-small cell lung cancer and suppresses tumor cell growth and invasion by directly targeting insulin-like growth factor 1 receptor. Oncol. Lett. 13, 3247–3252 (2017).
Malachowska, B. et al. Circulating microRNAs as biomarkers of radiation exposure: A systematic review and meta-analysis. Int. J. Radiat. Oncol. Biol. Phys. 106, 390–402 (2020).
Pataer, A. et al. Major pathologic response and RAD51 predict survival in lung cancer patients receiving neoadjuvant chemotherapy. Cancer Med. 7, 2405–2414 (2018).
Cortez, M. A. et al. In vivo delivery of miR-34a sensitizes lung tumors to radiation through RAD51 regulation. Mol. Ther. Nucleic Acids 4, e270 (2015).
Lai, T. H. et al. HDAC inhibition induces microRNA-182, which targets RAD51 and impairs HR repair to sensitize cells to Sapacitabine in acute myelogenous leukemia. Clin. Cancer Res. 22, 3537–3549 (2016).
Sun, H., Fan, G., Deng, C. & Wu, L. MiR-4429 sensitized cervical cancer cells to irradiation by targeting RAD51. J. Cell. Physiol. 235, 185–193 (2020).
Ye, D. M. et al. Histone H3 acetylation reverses radioresistance in breast cancers through BRD3-mediated inhibition of RAD51 and inducing ferroptosis. Free Radic. Biol. Med. 241, 760–772 (2025).
Liang, D. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 186(2748–2764), e2722 (2023).
Lei, G., Zhuang, L. & Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381–396 (2022).
Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 20, 1181–1192 (2018).
Mao, C. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021).
Aishajiang, R. et al. Engineering ferroptosis radiosensitizer for SPARC-targeted degradation: A strategy to reverse radioresistant non-small cell lung cancer. Biomaterials 326, 123675 (2026).
Liu, S. et al. Tubastatin A potently inhibits GPX4 activity to potentiate cancer radiotherapy through boosting ferroptosis. Redox Biol. 62, 102677 (2023).
Chen, Y. et al. GSTM3 enhances radiosensitivity of nasopharyngeal carcinoma by promoting radiation-induced ferroptosis through USP14/FASN axis and GPX4. Br. J. Cancer 130, 755–768 (2024).
Yin, J. et al. Identification of ferroptosis biomarker in AHH-1 lymphocytes associated with low dose radiation. Health Phys. 120, 541–551 (2021).
Shuryak, I. & Brenner, D. J. Review of quantitative mechanistic models of radiation-induced non-targeted effects (Nte). Radiat. Prot. Dosimetry 192, 236–252 (2020).
Zheng, D. et al. Mathematical modeling in radiotherapy for cancer: A comprehensive narrative review. Radiat. Oncol. 20, 49 (2025).
Funding
This study received no funding.
Author information
Authors and Affiliations
Contributions
RW, ZC, and CZ were responsible for the study’s conception and design. JL, ZW, RW and XQ recruited patients. CZ, XR and SL collected clinical information. JL, CZ, FG and ZW analyzed and reviewed clinical data. JL and XR performed experiments. FG, CZ and ZC conducted data analysis. JL and CZ wrote original draft preparation. JL, XQ, and XR interpreted the findings and contributed to manuscript revision and editing. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Human ethics approval (PJ-KY2025-184-1) was obtained from Affiliated Zhongshan Hospital of Dalian University. All participants, or their legally authorized representatives, provided informed consent prior to enrollment. The study was conducted in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lv, J., Zhang, C., Ren, X. et al. miR-320a enhances radiosensitivity in non-small cell lung cancer by targeting RAD51 and modulating ferroptosis via GPX4. Sci Rep 16, 11397 (2026). https://doi.org/10.1038/s41598-026-41692-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-41692-z
