
Abstract
Endometriosis is a disease characterized by fibrosis and adhesions. There are still no treatment methods targeting these conditions. One reason for this is the lack of useful animal models to investigate the mechanisms of fibrosis and adhesion in endometriosis. We previously proposed that Homeobox C8 (HOXC8) acts as an upstream regulator involved in endometriosis development, promoting fibrosis by activating transforming growth factor β (TGFB) signaling in vitro. This study was undertaken to establish a xenograft mouse model for endometriosis-associated fibrosis using HOXC8-overexpressing cells. Stable lines of immortalized human endometrial stromal cells overexpressing HOXC8 (HOXC8-imESCs) and mock control lines (Control-imESCs) were established and evaluated for cellular functions in vitro. These cell lines were then transplanted under the renal capsule of severely immunodeficient mice, and the xenografts were harvested five weeks after transplantation. In vitro, cell migration, invasion, and fibrotic capacity were enhanced in HOXC8-imESCs compared to Control-imESCs. In the xenografts, HOXC8-imESCs exhibited significant collagen fiber production compared to Control-imESCs. Additionally, immunohistochemistry of the xenografts detected phosphorylated SMAD2/3 exclusively in HOXC8-imESCs, indicating that HOXC8 overexpression activates the TGFB/SMAD pathway aberrantly in vivo. In this study, a new animal model for endometriosis-associated fibrosis was established using the HOXC8-imESC xenograft model system.
Similar content being viewed by others


Introduction
Endometriosis is a common estrogen-dependent gynecological disorder affecting approximately 10% of reproductive-age women. The primary pathological features of endometriosis are fibrosis and adhesions, which cause dysmenorrhea, chronic pelvic pain, and infertility, thereby reducing women’s quality of life1. Nonsteroidal anti-inflammatory drugs (NSAIDs) and other analgesics are used to treat dysmenorrhea and chronic pelvic pain, but these approaches are symptomatic and not curative2. Low-dose estrogen-progestin combination drugs (LEP), progestins, and gonadotropin-releasing hormone (GnRH) analogs can temporarily relieve symptoms by suppressing estrogen action or secretion. However, these drugs are also not curative and cannot be used with fertility treatments3,4,5,6. Therefore, there is a growing need to develop therapeutic strategies targeting the primary pathological features of endometriosis-associated fibrosis and adhesions. However, such treatments have not yet been established. One of the reasons is the lack of necessary animal models to investigate the mechanisms of fibrosis and adhesion in endometriosis.
Numerous studies have shown that the characteristics of endometriosis, including cell proliferation, adhesion, and fibrosis, are associated with the aberrant activation of transforming growth factor β (TGFB) signaling7,8,9,10,11,12,13,14,15. Additionally, increased local estradiol production and aberrant estrogen receptor expression have also been reported to contribute to lesion development15. In our previous study, transcriptome analysis of endometrial stromal cells isolated from endometriosis revealed aberrant expression of approximately 800 genes13. We suggested that these genes disrupt various cellular functions and signaling pathways, thereby contributing to endometriosis development. However, the mechanisms underlying this pathological process remain largely unknown.
In recent years, the role of upstream regulators (URs) in disease pathogenesis has become more widely recognized16,17,18. Mutations or aberrant expression of URs have been reported to cause widespread changes in the expression of their downstream target genes, thereby contributing to disease pathogenesis19,20,21,22,23. To identify URs involved in the onset and development of endometriosis, we performed an integrated analysis combining transcriptome data and gene regulatory network information from stromal cells of endometriotic and normal endometrium, along with in silico simulations of disease treatment using mathematical modeling. Through this approach, Homeobox C8 (HOXC8), which is highly expressed in endometriosis, was identified as a candidate of the URs24. HOXC8 has been reported to be dysregulated in breast and lung cancers, promoting tumor invasion and metastasis by activating the TGFB signaling pathway25,26. To investigate the role of HOXC8 in endometriosis, we established primary endometrial stromal cells overexpressing HOXC8 (HOXC8-euESCs) and a mock control cell line (Control-euESCs) and analyzed their cellular functions in vitro. Proliferation, migration, and fibrotic activities were enhanced in HOXC8-euESCs compared to Control-euESCs24. Signaling pathway and Gene Ontology (GO) analyses based on transcriptome data revealed that the TGFB signaling pathway and enrichment of GO terms related to cell proliferation, migration, and fibrosis were more strongly activated in HOXC8-euESCs compared to Control-euESCs24. In addition, HOXC8-euESCs exhibited increased SMAD2/3 phosphorylation, a marker of TGFB pathway activation24. Their fibrotic activity was suppressed by treatment with a TGFB signaling inhibitor24. These in vitro findings suggested that HOXC8 promotes fibrotic processes through TGFB signaling and is thus one of the URs in endometriosis.
In the present study, we evaluated the role of HOXC8 in endometriosis-associated fibrosis in vivo. Immortalized endometrial stromal cell lines overexpressing HOXC8 were established and used for a xenograft model, in which human cells are transplanted into severely immunodeficient mice. These xenograft mice exhibited fibrotic lesions with SMAD2/3 phosphorylation, demonstrating that HOXC8 promotes fibrosis through the TGFB signaling pathway in vivo. Thus, this study provided a novel animal model to investigate the mechanisms of endometriosis-associated fibrosis.
Results
In vitro analysis of cellular functions in HOXC8-overexpressing immortalized endometrial stromal cell lines (HOXC8-imESCs)
To establish a xenograft model in this study, we first produced immortalized endometrial stromal cell lines stably overexpressing HOXC8 (HOXC8-imESCs) and the mock control lines (Control-imESCs). HOXC8 overexpression in the cell lines was confirmed by real-time RT-PCR and Western blotting. HOXC8 mRNA expression was significantly higher in HOXC8-imESCs than in Control-imESCs (Fig. 1A). HOXC8 protein was detected only in HOXC8-imESCs (Fig. 1B).
We next evaluated cellular functions in the established cell lines in vitro. The cell proliferation assay showed no significant difference in cell number between HOXC8-imESCs and Control-imESCs (Fig. 1C), indicating that, unlike in primary cultured cells, HOXC8 overexpression did not promote cell proliferation in the immortalized cell line. However, the HOXC8-imESCs differed from the Control-imESCs in having a significantly higher number of migrating cells (Fig. 1D), significantly higher cell invasion rate (Fig. 1E), and significantly higher wound healing rate (Fig. 1F), and in causing significantly more gel contraction (Fig. 1G). These findings indicate that HOXC8-imESCs exhibit enhanced migration, invasion, and fibrotic activities compared to Control-imESCs in vitro.

In vitro functional analysis of HOXC8-overexpressing immortalized endometrial stromal cells (HOXC8-imESCs). (A) HOXC8 mRNA expression in Control-imESCs and HOXC8-imESCs analyzed by real-time RT-PCR. Experiments were performed three times in triplicate, with GAPDH used as an internal control. The relative HOXC8 expression in HOXC8-imESCs was normalized to Control-imESCs as 1 and is presented as mean ± SD. *p < 0.05. (B) Western blot analysis of HOXC8 protein in Control-imESCs and HOXC8-imESCs. TUBB was used as an internal control. The images of HOXC8 and TUBB are cropped from different blots. The original blot images are presented in Supplementary Figure S1. (C) Cell proliferation assay in Control-imESCs and HOXC8-imESCs. Cell numbers were counted at 24, 48, 72, and 96 hours after seeding and are shown as mean ± SD. Experiments were performed three times in triplicate. (D) Cell migration assay in Control-imESCs and HOXC8-imESCs. Representative images for each cell line are shown. Scale bar = 100 μm. Migrated cells were counted in four randomly selected fields at 200× magnification and are shown as mean ± SD. Experiments were repeated three times. *p < 0.05. (E) Cell invasion assay in Control-imESCs and HOXC8-imESCs. Representative images for each cell line are shown. Scale bar = 100 μm. Invaded cells were counted in four randomly selected fields at 200× magnification. Experiments were repeated three times. Invasion rate (%) was calculated by dividing the number of invaded cells by the number of migrated cells, multiplied by 100, and shown as mean ± SD. *p < 0.05. (F) Wound healing assay in Control-imESCs and HOXC8-imESCs. Representative images at 0 and 24 hours after scratching are shown. Scale bar = 50 μm. Wound healing rate (%) was calculated as the area covered by migrating or proliferating cells at 24 hours divided by the initial wound area, multiplied by 100, and presented as mean ± SD. Experiments were performed three times in triplicate. *p < 0.05 . (G) Collagen gel contraction assay in Control-imESCs and HOXC8-imESCs. Representative images at 24 hours after collagen gel culture are shown. The contraction area was calculated by subtracting the gel area from the whole well area. The contraction rate (%) was calculated by dividing the contraction area by the whole well area, multiplied by 100, and presented as mean ± SD. Experiments were performed three times in triplicate. *p < 0.05
Expression of fibrosis-related genes in HOXC8-imESCs
Major regulators of fibrosis include collagen and matrix metalloproteinases (MMPs), as well as their endogenous inhibitors, the tissue inhibitor of metalloproteinases (TIMPs)27. To investigate the effects of HOXC8 overexpression on these regulators, the mRNA expression levels of representative genes from each family were analyzed using real-time RT-PCR in vitro (Fig. 2). COL1A1 and TIMP3 were significantly upregulated in HOXC8-imESCs compared to Control-imESCs. In contrast, no significant differences in MMP1, MMP2, or MMP14 were observed between HOXC8-imESCs and Control-imESCs. However, it should be noted that altered expression of MMP family members has been reported in endometriosis28. Although the upregulation of TIMP3 mRNA in HOXC8-imESCs appears to be inconsistent with the increased invasion abilities observed in Fig. 1E, the TIMP3 expression level is approximately 32-fold lower than that of MMP2 and MMP14. Increased expression of TIMP3 in HOXC8-imESC cells is expected to be insufficient to effectively inhibit MMP activity. This does not necessarily contradict the invasion assay results. These findings therefore suggest that HOXC8 overexpression primarily contributes to increased collagen production.

mRNA expression of fibrosis-related genes in HOXC8-imESCs. Real-time RT-PCR analysis of the fibrosis-related gene mRNA expression in Control-imESCs and HOXC8-imESCs. The analysis included the following genes: COL1A1 and COL1A2 (collagen), MMP1, MMP2, and MMP14 (matrix metalloproteinases), and TIMP1, TIMP2, and TIMP3 (tissue inhibitor of metalloproteinases). Experiments were performed three times in triplicate. GAPDH was used as an internal control. The relative expression levels in HOXC8-imESCs were normalized to Control-imESCs as 1 and are presented as mean ± SD. *p < 0.05
Establishment of a xenograft model using HOXC8-imESCs
Five weeks after transplantation, xenograft lesions were visibly identified in both whole and cross-sectional views of the kidneys in which the cell lines had been transplanted. Xenograft lesions derived from HOXC8-imESCs were easy to detect because they were thicker than those derived from Control-imESCs (Fig. 3C). Histological evaluation by hematoxylin and eosin (HE) staining showed a clear boundary between the xenograft and renal parenchyma in Control-imESCs, whereas HOXC8-imESC-derived xenografts exhibited an indistinct boundary (Fig. 3D). This suggests that inflammatory cells infiltrated the HOXC8-imESC xenografts. Indeed, immunohistochemical analysis using the pan-leukocyte marker CD45 and the macrophage marker CD68 revealed the presence of CD45- and CD68-positive cells in HOXC8-imESC xenograft lesions and mouse renal parenchyma (Fig. S3). These findings support the notion that HOXC8-imESCs may promote an inflammatory response that extends into host kidney tissue.
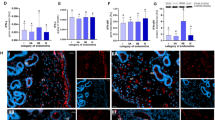
Collagen fibers stained blue with trichome staining were observed in the xenograft lesions from both cell lines (Fig. 3D). HOXC8-imESCs had much more collagen fibers than Control-imESCs (Fig. 3D). Immunohistochemistry revealed positive staining for ACTA2, a myofibroblast marker, in the xenograft lesions from both cell lines, and the proportion of ACTA2-positive cells did not differ significantly between HOXC8-imESCs (62.5% ± 2.8%) and Control-imESCs (65.1% ± 1.2%) (p = 0.398) (Figs. 3E, F). In contrast, COL1A1 was strongly detected in HOXC8-imESCs, with a positive area of 15.8% ± 2.5%, which was significantly higher than that in Control-imESCs (4.1% ± 2.8%) (p = 0.00558), indicating a marked increase in collagen production associated with HOXC8 overexpression (Figs. 3E, F). These findings suggest that HOXC8 overexpression promotes fibrosis, supporting the utility of the xenograft model using HOXC8-imESCs as a valid model for studying endometriosis-associated fibrosis. Although immunohistochemical staining was performed for MMP2, TIMP2, and COL4A2 to evaluate fibrosis more clearly, no signals were detected in the Control-imESC- or HOXC8-imESC-derived xenograft lesions (Fig. S2).
Next, to confirm that the xenograft lesions were indeed derived from the human cell lines (Control-imESCs or HOXC8-imESCs), the cells were immunostained for vimentin (VIM), a mesenchymal and ESC marker, and STEM101, a human-specific nuclear marker, and checked for HOXC8 signals. Cells positive for VIM and STEM101 were detected in xenograft lesions derived from both cell lines, clearly confirming that the lesions originated from the transplanted imESCs (Fig. 3G). HOXC8 signals were strongly detected in HOXC8-imESC xenografts but not in Control-imESC xenografts, indicating that HOXC8 overexpression was maintained in HOXC8-imESCs, even after transplantation (Fig. 3G). These findings demonstrate that both Control-imESCs and HOXC8-imESCs can be engrafted and maintained under the renal capsule in this xenograft mouse model.


Establishment of a xenograft model using HOXC8-imESCs and histological analysis. (A) Summary of the xenograft mouse model. Cell aggregates of Control-imESCs or HOXC8-imESCs (Fig. 3B) were transplanted under the renal capsules of immunodeficient NOD/Scid mice and harvested five weeks after transplantation for morphological evaluation. (B) Detection of GFP-expressing cells in cell aggregates of both cell lines. Cell viability within the aggregates, which were embedded in collagen gel for transplantation, was confirmed by GFP expression. Representative fluorescent images of Control-imESCs one day after culture are shown. Scale bar = 200 μm. (C) Whole and cross-sectional images of kidneys transplanted with Control-imESCs and HOXC8-imESCs, which were harvested five weeks after transplantation. Scale bar = 1 mm. (D) HE and trichrome stained images of xenograft lesions in Control-imESCs or HOXC8-imESCs. Scale bar = 100 μm. (E) Immunofluorescence staining of xenograft lesions for ACTA2 and COL1A1, as well as the corresponding HE and DAPI-staining. In ACTA2 and COL1A1 staining, the images on the right show magnified views of the regions outlined by dashed lines in the corresponding images on the left. Nuclei were counterstained with DAPI. Scale bar = 100 μm. (F) COL1A1-positive area and ACTA2-positive cell proportions. The collagen-positive area rate (%) was calculated by the average of the positive area ratios within the field of view at 200x magnification. The ACTA2-positive cells rate (%) was calculated as the ratio of ACTA2-positive cells to VIM-positive cells. Quantification was performed using three independent tissue sections (n = 3) for both Control-imESCs and HOXC8-imESCs xenografts. All quantified values are presented as the mean ± SD. **p < 0.01. (G) Immunofluorescence staining of xenograft lesions for VIM, human nuclear marker, and HOXC8, along with HE and DAPI staining of adjacent sections. In VIM, human nuclear marker, and HOXC8 staining, the images on the right are magnified views of the areas outlined by dashed lines in the corresponding images on the left. Nuclei were counterstained with DAPI. Scale bar = 100 μm.
Activation of TGFB/SMAD signaling pathway in HOXC8-imESCs
In the TGFB signaling pathway, SMAD2 and SMAD3 are activated via phosphorylation by TGFB receptors and translocated into the nucleus to regulate gene expression29,30. To assess the activation of the TGFB/SMAD pathway, the expression and localization of phosphorylated SMAD2 and SMAD3 (pSMAD2/3) were examined in both cultured cells and xenograft lesions (Fig. 4).
Immunocytochemistry of cultured cells revealed that pSMAD2/3 was exclusively localized to the nucleus in HOXC8-imESCs, with positive cell rates of 98.2% ± 3.17% for pSMAD2 and 100% for pSMAD3, whereas no pSMAD2/3-positive cells were observed in Control-imESCs (Figs. 4A, B). This difference in pSMAD2-positive cell rates was statistically significant (p = 0.000000725), while pSMAD3 was consistently positive in all HOXC8-imESCs and absent in Control-imESCs (Fig. 4B). Additionally, immunohistochemical analysis of xenograft lesions revealed that pSMAD2/3 were localized to the nucleus only in HOXC8-imESC xenografts (Fig. 4C). These findings demonstrate that HOXC8 overexpression can activate the TGFB/SMAD signaling pathway in vivo in endometriosis.

Detection of phosphorylated SMAD2 and SMAD3 in culture and the xenograft lesions in HOXC8-imESCs. (A) Immunocytochemistry for SMAD2, SMAD3, and their phosphorylated forms (pSMAD2 and pSMAD3) in monolayer cultures of Control-imESCs and HOXC8-imESCs. Nuclei were counterstained with DAPI. Scale bar = 20 μm. (B) pSMAD2- and pSMAD3-positive cell proportions. The positive cell rate (%) was calculated relative to the number of DAPI-positive nuclei and presented as the mean ± SD. Quantification was performed in three independent experiments (n = 3). ***p < 0.001. (C) Immunofluorescence staining with a pan-phospho-SMAD2/3 (pSMAD2/3) in xenograft lesions in Control-imESCs and HOXC8-imESCs, along with corresponding HE and DAPI-staining. The images on the right of the pSMAD2/3 staining show magnified views of the regions outlined by dashed lines in the corresponding images on the left. Nuclei were counterstained with DAPI. Scale bar = 20 μm.
Discussion
Using an in vivo xenograft model, this study demonstrated that HOXC8 plays a role in fibrosis, a key pathological feature of endometriosis. Previously reported animal models of endometriosis commonly involved transplanting minced mouse endometrial tissue into the peritoneal cavity of immunodeficient mice31,32. However, in these models, there is a possibility that non-endometrial stromal cells, such as myometrial cells, influence lesion formation. Alternatively, the regenerated tissue at adhesion sites may simply reflect normal endometrial tissue. Therefore, a model that more accurately recapitulates the pathophysiology of the disease is needed. This study showed that transplanting HOXC8-imESCs under the renal capsule of severely immunodeficient mice created fibrotic lesions. This fibrosis was likely driven by activation of the TGFB/SMAD signaling pathway. These results support establishing a new animal model that better reflects the fibrotic pathology of endometriosis.
HOXC8 is a transcription factor belonging to the HOX gene family, which plays crucial roles in embryonic development and morphogenesis33,34. HOXC8 overexpression has been observed in various cancers, including breast, prostate, and pancreatic cancers25,35,36. In breast and lung cancers, HOXC8 contributes to invasion and metastasis by activating the TGFB pathway25,26. Additionally, HOXC8 has been reported to be involved in liver fibrosis in a mouse model, where its expression is increased, and suppression of HOXC8 leads to reduced fibrosis37. The present results suggest that HOXC8 is involved in the pathogenesis of endometriosis, particularly by promoting fibrosis.
In this study, both Control-imESCs and HOXC8-imESCs expressed the myofibroblast marker ACTA2 in the xenograft lesions. However, only the HOXC8-imESCs accumulated large amounts of collagen. In general, tissue damage activates fibroblasts, which then differentiate into myofibroblasts. Persistent stimuli, such as chronic inflammation, lead to excessive collagen accumulation and fibrosis progression38. TGFB1 is a major profibrotic factor that promotes collagen production through myofibroblast activation39. Based on these reports and our xenograft results, excessive collagen production in endometriosis appears to require not only the differentiation of fibroblasts into myofibroblasts but also activation of the TGFB/SMAD signaling pathway in the differentiated myofibroblasts via HOXC8. Thus, our model system may be a valuable tool for further investigations of the mechanisms of fibrosis in endometriosis. However, one limitation of this study is that it demonstrates only a correlation based on immunostaining results, and further mechanistic studies are needed to establish causality.
We previously found that treatment of HOXC8-euESCs with an ALK5 inhibitor, which blocks the TGFB signaling pathway, significantly reduced collagen gel contraction24. These results imply that ALK5 inhibitors could be promising therapeutic agents for treating fibrosis associated with endometriosis. Furthermore, our model system may also contribute to the development of future targeted therapies using agents such as ALK5 inhibitors.
In this study, HE staining of kidneys transplanted with HOXC8-imESCs revealed clusters of small cells in the renal parenchyma. We performed immunostaining using markers for macrophages (CD68) and leukocytes (CD45) since NOD/Scid mice lack almost all T and B cells. Cells positive for these markers were detected in the xenograft lesions and renal parenchyma only in mice transplanted with HOXC8-imESCs, indicating inflammatory cell infiltration in the recipient mice. Fibrosis in endometriosis is closely associated with inflammation. Macrophages play a key role in driving fibrotic progression by activating fibroblasts40,41,42,43. In addition, IL-8 expression is upregulated in endometriotic tissues44. In monkeys with spontaneous endometriosis, administration of an anti-IL-8 antibody has been shown to reduce fibrosis and adhesion formation45. A limitation of this study was that we were unable to confirm the presence of cytokines. Another limitation was that the cells transplanted in the intra-abdominal space did not engraft, resulting in the formation of subrenal capsule lesions only.
In this study, we established an animal model of endometriosis-associated fibrosis using a xenograft model system with immortalized endometrial stromal cells overexpressing HOXC8, an upstream regulator associated with endometriosis. This animal model is expected to be a useful tool for investigating the mechanisms of fibrosis in endometriosis and for developing therapeutic strategies to suppress fibrosis.
Methods
Ethical statement
This study was approved by the Institutional Review Board of Yamaguchi University Graduate School of Medicine (No. H47-026). All animal procedures were approved by the Animal Experimentation Ethics Committee of Yamaguchi University Graduate School of Medicine, performed according to the relevant guidelines and regulations, and confirmed compliance with ARRIVE guidelines.
Establishment of HOXC8-overexpressing human immortalized endometrial stromal cell lines (HOXC8-imESCs)
An expression vector co-expressing HOXC8 and GFP was constructed as previously reported19,24. The HOXC8 coding sequence and the IRES-AcGFP sequence were amplified by PCR using the pMX_HOXC8 vector24 and the pLVSIN-CMV_hyg_IRES-AcGFP vector19 as templates, respectively, with primer sets listed in Table 1. PCR amplification was performed using PrimeSTAR GXL DNA polymerase (Takara, Kyoto, Japan) under the following conditions: 98 °C for 10 s, 60 °C for 15 s, and 68 °C for 2.5 min, for 35 cycles. The amplified HOXC8 and IRES-AcGFP sequences were cloned into the multiple cloning site of the pMXs retroviral expression vector (Cell Biolabs Inc., San Diego, CA) using the In-Fusion HD Cloning Kit (Takara). As a mock control, an expression vector containing only the IRES-AcGFP sequence was also constructed.
To produce the retrovirus, the HOXC8-overexpressing or the mock control vector was co-transfected with the packaging plasmids into the HEK293FT packaging cell line using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Retroviral supernatants were collected 48 h after transfection and concentrated using PEG-it Virus Precipitation Solution (System Biosciences, Palo Alto, CA). Human immortalized endometrial stromal cells (imESCs)46 were cultured in 12-well plates at a density of 6 × 10⁴ cells per well. After 24 h, the cells were incubated for 48 h with concentrated retrovirus added to the culture medium containing 5 µl/well of polybrene (Sigma-Aldrich, St. Louis, MO). The cells transduced with either the HOXC8-overexpressing or mock control vector were subjected to drug selection with 10 µg/ml hygromycin for one month, resulting in the establishment of stable HOXC8-overexpressing cell lines (HOXC8-imESCs) and corresponding mock control lines (Control-imESCs).
Cell culture
HOXC8-imESCs, Control-imESCs, and parental cell line imESCs were maintained by 10 cm collagen-coated culture dishes (Corning Life Science, Tewksbury, MA) in DMEM/F12 medium (Wako, Osaka, Japan), supplemented with 10% FBS (Gibco, Rockville, MD), 2 mM L-glutamine (Gibco), and a 1% antibiotic-antimycotic solution (Wako), at 37 °C in a humidified atmosphere of 5% CO₂ and 95% air. The medium was exchanged every other day.
Quantitative RT-PCR
Total RNA was extracted from HOXC8-imESCs and Control-imESCs using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. cDNA was synthesized using the QuantiTect Reverse Transcription Kit (Qiagen) with random hexamers as previously described47. Real-time RT-PCR was performed in triplicate using SYBR Premix Ex Taq (Takara) and the primer sets listed in Table 1 on a CFX384 thermal cycler (Bio-Rad, Hercules, CA). Relative mRNA expressions were calculated using the ΔΔCt method with GAPDH as an internal control. Expression levels in HOXC8-imESCs were normalized to those in control-imESCs as 1 and presented as mean ± standard deviation (SD).
Western blotting
Western blotting was performed as previously reported48. HOXC8-imESCs and Control-imESCs were lysed in RIPA buffer (Wako), and total protein lysates were extracted. Ten micrograms of protein were separated by SDS-PAGE and transferred to PVDF membranes (New England Biolabs, Ipswich, MA). The membranes were blocked for 60 min, followed by incubation with either mouse anti-TUBB monoclonal antibody (Sigma-Aldrich, RRID: AB_477577) or rabbit anti-HOXC8 monoclonal antibody (Abcam, Tokyo, Japan, RRID: AB_1925078) at a 1:1000 dilution. The secondary antibodies used were anti-mouse IgG/HRP (Dako, Santa Clara, CA, RRID: AB_2636929) or anti-rabbit IgG/HRP (Dako, RRID: AB_2617141), both diluted 1:1000. The target protein signals were detected by chemiluminescence using ECL™ Prime Solution (Cytiva, Tokyo, Japan) and visualized with the ChemiDoc™ XRS Plus imaging system (Bio-Rad).
Cell proliferation assay
HOXC8-imESCs and Control-imESCs were seeded in 6-well plates at a density of 5 × 10⁴ cells per well. At 24, 48, 72, and 96 h after seeding, the cells were trypsinized to obtain single-cell suspensions, and cell numbers were measured using a Vi-CELL XR cell counter (Beckman Coulter, Tokyo, Japan). Each experiment was performed three times in triplicate. The results are presented as mean ± SD.
Cell migration and invasion assays
Cell migration and invasion assays were performed using non-Matrigel-coated and Matrigel-coated culture inserts (BioCoat Matrigel Invasion Chamber, Corning), respectively, according to the manufacturer’s instructions49. A total of 4 × 10⁵ HOXC8-imESCs or Control-imESCs suspended in serum-free medium were added to the upper chamber of the culture insert, while 750 µl of medium containing 10% FBS was placed in the lower chamber of a 24-well plate and incubated for 24 h. The cells that remained on the upper surface of the culture insert membrane were gently removed to eliminate the non-migrated/non-invaded cells. The migrated cells on the lower surface of the membrane were stained with Diff-Quick (Sysmex, Kobe, Japan), and the cells were counted in four randomly selected fields at 200× magnification. The migration assay was performed three times, and the number of cells that migrated through the non-coated insert membranes was expressed as the mean ± SD. The invasion assay was performed three times. The cell invasion rate (%) was calculated by dividing the number of cells that passed through the Matrigel-coated insert membrane by the number of migrated cells, multiplied by 100, and presented as mean ± SD.
Wound healing assay
HOXC8-imESCs and Control-imESCs were cultured in 6-well plates until confluence, and a linear scratch was made in the center of each well using a sterile 1000 µl pipette tip. Immediately after scratching and at 24 h, images of the same fields were captured under a microscope at 40× magnification, and the area of the cell-free region was measured using ImageJ software. The assay was performed in triplicate and repeated three times. The wound healing rate (%) was calculated as [(initial wound area - wound area at 24 h)/initial wound area] × 100, and shown as mean ± SD.
Collagen gel contraction assay
Collagen gel contraction was evaluated according to previously reported methods24,50. A total of 1.0 × 10⁵ HOXC8-imESCs or Control-imESCs were mixed with 100 µl of collagen gel solution [40% rat tail collagen type I (final concentration: 0.8 mg/ml; Sigma-Aldrich), 20% 5× DMEM, 10% FBS, 10% 0.44 M NaHCO₃, and 5% 0.18 N NaOH], and the mixture was plated into 96-well plates pre-coated with 10% BSA. After incubation at 37 °C for 30 min to solidify the gel, 50 µl of DMEM was added to each well and incubated for 24 h. The gels were imaged under a microscope, and their surface area was measured using ImageJ software. The assay was performed three times in triplicate. Gel contraction area was calculated by subtracting the measured gel area from the whole well area. The collagen gel contraction rate (%) was calculated by dividing the gel contraction area by the whole well area, multiplied by 100, and presented as mean ± SD.
Xenograft model
We performed a xenograft model based on the previously established protocol for uterine leiomyoma cells in our laboratory with some modifications. In brief, HOXC8-imESCs and Control-imESCs were mixed with a collagen gel solution (same composition as described above, except that the final concentration of type I collagen was 1.6 mg/ml) at a density of 2 × 10⁷ cells/ml. The mixture was incubated at 37 °C for 30 minutes to form solid cell aggregates. The cell aggregates were then maintained under humidified conditions at 37 °C with 5% CO₂ in the same culture medium used for cell culture. GFP expression, which is used as an indicator of viable cells, was observed in both cell lines in the aggregates. Figure 3B shows a representative aggregate formed from Control-imESCs. Twenty-four hours later, the cell aggregates were transplanted under the renal capsules of both kidneys of ovariectomized, severely immunodeficient NOD/Scid mice (Kyudo, Tosu, Japan) at a density of 1.2 × 10⁶ cells per kidney. The NOD/Scid mice provided by Kyudo are ‘NOD/ShiJic-scid Jcl’ mice produced and supplied by CLEA Japan, Inc. The mice were anesthetized for surgery with a combination anesthetic consisting of 0.2 mg/kg of medetomidine, 2.66 mg/kg of midazolam, and 3.33 mg/kg of butorphanol. A total of 26 mice were transplanted with Control-imESC aggregates, and 30 mice were transplanted with HOXC8-imESC aggregates. Five weeks later, the transplanted mice were euthanized by cervical dislocation. The transplanted kidneys were harvested, and the xenografts were collected (Fig. 3A). The xenografts were then fixed in 4% Paraformaldehyde (PFA), embedded in paraffin, and processed for histological evaluation.
Trichrome staining
Collagen fibers were stained using a Trichrome Stain Kit (TRM-1; ScyTec Laboratories Inc., West Logan, UT) as previously reported51. In brief, 5-µm sections of paraffin-embedded tissue were deparaffinized and fixed in Bouin’s solution overnight. The sections were stained with Weigert’s iron hematoxylin for 10 min, Biebrich scarlet/acid fuchsin solution for 10 min, phosphomolybdic/phosphotungstic acid solution for 15 min, and aniline blue solution for 15 min. Finally, the sections were immersed in 1% acetic acid for 1 min.
Immunofluorescence staining
Immunocytochemistry was performed as previously reported51. In brief, HOXC8-imESCs and Control-imESCs cultured in 24-well plates were fixed with 4% PFA for 15 min, permeabilized with 0.5% Triton X-100, and blocked for 60 min using a blocking solution containing 10% fetal bovine serum and 1% bovine serum albumin in PBST. The cells were incubated overnight at 4 °C with one of the following primary antibodies: rabbit anti-SMAD2 (Cell Signaling Technology, Danvers, MA, RRID: AB_823638, 1:200 dilution), rabbit anti-SMAD3 (Cell Signaling Technology, RRID: AB_2193182, 1:200 dilution), rabbit anti-phospho-SMAD2 (Abcam, RRID: AB_2732791, 1:100 dilution), or rabbit anti-phospho-SMAD3 (Cell Signaling Technology, RRID: AB_2193207, 1:100 dilution). Then, the cells were incubated for 60 min in the dark with Alexa Fluor 594-conjugated anti-rabbit IgG (Abcam, RRID: AB_2734147) diluted 1:1000 as the secondary antibody. Nuclei were counterstained with DAPI (500 ng/ml, Nacalai, Kyoto, Japan).
Immunohistochemical staining was also performed as previously reported51. In brief, 5-µm paraffin-embedded tissue sections were deparaffinized, fixed with 4% PFA for 5 min, and permeabilized with 0.5% Triton X-100. After blocking with a blocking solution for 60 min, the following primary antibodies were used: mouse anti-ACTA2 (Abcam, RRID: AB_2313736; 1:500 dilution), rabbit anti-COL1A1 (Abcam, RRID: AB_2861258; 1:500 dilution), rabbit anti-VIM (Abcam, RRID: AB_10562134; 1:1000 dilution), mouse specific for human Ku80 nuclear marker (Takara Bio, Shiga, Japan, RRID: AB_2895096; 1:500 dilution), rabbit anti-HOXC8 (Proteintech, Rosemont, IL, RRID: AB_2878140; 1:100 dilution), rabbit anti-MMP2 (Proteintech, RRID: AB_2250823; 1:200 dilution), rabbit anti-TIMP2 (Proteintech, RRID: AB_ 2287495; 1:200 dilution), rabbit anti-COL4A2 (Abcam, RRID: AB_10976330; 1:500 dilution), or rabbit anti-phospho-SMAD2/3 (Abcam, RRID: AB_882596; 1:100 dilution), and incubated overnight at 4 °C. As secondary antibodies, Alexa Fluor 594-conjugated anti-mouse IgG (Abcam, RRID: AB_2732073) or Alexa Fluor 594-conjugated anti-rabbit IgG were used at a 1:1000 dilution and incubated for 60 min in the dark. Additionally, to perform double immunofluorescence staining, mouse anti-VIM (Abcam, RRID: AB_306907; 1:500 dilution) and rabbit anti-CD45 (Abcam, RRID: AB_442810; 1:100 dilution) or rabbit anti-CD68 (Abcam, RRID: AB_10975465; 1:100 dilution) were used as primary antibodies, followed by Alexa Fluor 488-conjugated anti-mouse IgG (Abcam, RRID: AB_2576208) and Alexa Fluor 594-conjugated anti-rabbit IgG as secondary antibodies. After incubation, the sections were treated with a Vector TrueVIEW™ Autofluorescence Quenching Kit (Vector Laboratories Inc., Newark, CA) to reduce tissue autofluorescence. Nuclei were counterstained with DAPI.
Quantification
The positive area of COL1A1 and the percentage of ACTA2-positive cells were quantified from immunohistochemical images of xenograft lesions using ImageJ software (version 1.53t, National Institutes of Health, USA) as previously reported51. The collagen-positive area was calculated by the average of the positive area ratios within the field of view at 200x magnification. The percentage of ACTA2-positive cells was calculated as the ratio of ACTA2-positive cells to VIM-positive cells, which served as a stromal cell marker.
The number of pSMAD2- and pSMAD3-positive cells in the immunocytochemistry of cultured cells was measured individually, and the percentage of positive cells was calculated relative to the number of DAPI-positive nuclei.
Quantification was performed using three independent tissue sections (n = 3) for both Control-imESCs and HOXC8-imESCs xenografts in immunohistochemical analyses (COL1A1 and ACTA2). For immunocytochemical analyses (pSMAD2 and pSMAD3), quantification was performed in three independent experiments (n = 3). All quantified values are presented as the mean ± SD.
Statistical analysis
Statistical significance was analyzed using Student’s t-test. P-values less than 0.05 were considered statistically significant. All statistical analyses were performed using the R software program (version 3.6.1).
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Rogers, P. A. W. et al. Priorities for endometriosis research: Recommendations from an international consensus workshop. Reprod. Sci. 16, 335–346 (2009).
Brown, J. et al. Nonsteroidal anti-inflammatory drugs for pain in women with endometriosis. Cochrane Database Syst. Rev. 23, CD004753 (2017).
Saunders, P. T. K. & Horne, A. W. Endometriosis: Etiology, pathobiology, and therapeutic prospects. Cell 184, 2807–2824 (2021).
Chapron, C. et al. Rethinking mechanisms, diagnosis and management of endometriosis. Nat. Rev. Endocrinol. 15, 666–682 (2019).
Zondervan, K. T. et al. Endometriosis. Nat. Rev. Dis. Primers 4, 10. https://doi.org/10.1038/s41572-018-0008-5 (2018).
Becker, C. M. et al. ESHRE guideline: Endometriosis. Hum. Reprod. Open 2022, hoac009. https://doi.org/10.1093/hropen/hoac009 (2022).
Barjaste, N. et al. Genome-wide DNA methylation profiling in ectopic and eutopic of endometrial tissues. J. Assist. Reprod. Genet. 36, 1743–1752 (2019).
Bulun, S. E. et al. Molecular biology of endometriosis: From aromatase to genomic abnormalities. Semin. Reprod. Med. 33, 220–224 (2015).
Dyson, M. T. et al. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet 10, e1004158. https://doi.org/10.1371/journal.pgen.1004158 (2014).
Yotova, I. et al. Epigenetic alterations affecting transcription factors and signaling pathways in stromal cells of endometriosis. PLoS One 12, e0170859. https://doi.org/10.1371/journal.pone.0170859 (2017).
Naqvi, H., Ilagan, Y., Krikun, G. & Taylor, H. S. Altered genome-wide methylation in endometriosis. Reprod. Sci. 21, 1237–1243 (2014).
Khan, M. A., Sengupta, J., Mittal, S. & Ghosh, D. Genome-wide expressions in autologous eutopic and ectopic endometrium of fertile women with endometriosis. Reprod. Biol. Endocrinol. 10, 84. https://doi.org/10.1186/1477-7827-10-84 (2012).
Yamagata, Y. et al. Genome-wide DNA methylation profiling in cultured eutopic and ectopic endometrial stromal cells. PLoS One 9, e83612. https://doi.org/10.1371/journal.pone.0083612 (2014).
Yamagata, Y. et al. Retinoic acid has the potential to suppress endometriosis development. J. Ovarian Res. 8, 49. https://doi.org/10.1186/s13048-015-0179-6 (2015).
Maekawa, R. et al. Aberrant DNA methylation suppresses expression of estrogen receptor 1 (ESR1) in ovarian endometrioma. J. Ovarian Res. 12, 14. https://doi.org/10.1186/s13048-019-0489-1 (2019).
Vierbuchen, T. et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 (2010).
Ieda, M. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 (2010).
Szabo, E. et al. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 468, 521–526 (2010).
Sato, S. et al. SATB2 and NGR1: Potential upstream regulatory factors in uterine leiomyomas. J. Assist. Reprod. Genet. 36, 2385–2397 (2019).
Althubaiti, S. et al. Ontology-based prediction of cancer driver genes. Sci. Rep. 9, 17405. https://doi.org/10.1038/s41598-019-53454-1 (2019).
Liu, S. H. et al. DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res. 48, D863–D870 (2020).
Ma, M. et al. Identify cancer driver genes through shared mendelian disease pathogenic variants and cancer somatic mutations. Pac. Symp. Biocomput. 22, 473–484 (2017).
Song, J., Peng, W., Wang, F. & Wang, J. Identifying driver genes involving gene dysregulated expression, tissue-specific expression and gene-gene network. BMC Med. Genomics 12(Suppl 7), 168. https://doi.org/10.1186/s12920-019-0619-z (2019).
Mihara, Y. et al. An integrated genomic approach identifies HOXC8 as an upstream regulator in ovarian endometrioma. J. Clin. Endocrinl Metab. 105, 1–16 (2020).
Gong, C. et al. Upregulation of MGP by HOXC8 promotes the proliferation, migration, and EMT processes of triple-negative breast cancer. Mol. Carcinog. 58, 1863–1875 (2019).
Liu, H. et al. HOXC8 promotes proliferation and migration through transcriptional up-regulation of TGFβ1 in non-small cell lung cancer. Oncogenesis 7(1), s41389. https://doi.org/10.1038/s41389-017-0016-4 (2018).
Duarte, S., Baber, J., Fujii, T. & Coito, A. J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 44–46, 147–156 (2015).
Ke, J., Ye, J., Li, M. & Zhu, Z. The role of matrix metalloproteinases in endometriosis: A potential target. Biomolecules 11, 1739. https://doi.org/10.3390/biom11111739 (2021).
Hu, H. H. New insights into TGFB/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 292, 76–83 (2018).
Piersma, B., Bank, R. A. & Boersema, M. Signaling in fibrosis: TGFB, WNT, and YAP/TAZ converge. Front. Med. 2, 59. https://doi.org/10.3389/fmed.2015.00059 (2015).
Burns, K. A. et al. Role of estrogen receptor signaling required for endometriosis-like lesion establishment in a mouse model. Endocrinology 153, 3960–3971 (2012).
Hirata, T. et al. Development of an experimental model of endometriosis using mice that ubiquitously express green fluorescent protein. Hum. Reprod. 20, 2092–2096 (2005).
Lei, H., Wang, H., Juan, A. H. & Ruddle, F. H. The identification of Hoxc8 target genes. Proc. Natl. Acad. Sci. U. S. A. 102, 2420–2424 (2005).
Lei, H., Juan, A. H., Kim, M. S. & Ruddle, F. H. Identification of a Hoxc8- regulated transcriptional network in mouse embryo fibroblast cells. Proc. Natl. Acad. Sci. U. S. A. 103, 10305–10309 (2006).
Axlund, S. D., Lambert, J. R. & Nordeen, S. K. HOXC8 inhibits androgen receptor signaling in human prostate cancer cells by inhibiting SRC-3 recruitment to direct androgen target genes. Mol. Cancer Res. 8, 1643–1655 (2010).
Adwan, H. et al. Expression of HOXC8 is inversely related to the progression and metastasis of pancreatic ductal adenocarcinoma. Br. J. Cancer 105, 288–295 (2011).
Zhang, N., Guo, F. & Song, Y. HOXC8/TGFB1 positive feedback loop promotes liver fibrosis and hepatic stellate cell activation via activating Smad2/Smad3 signaling. Biochem. Biophys. Res. Commun. 662, 39–46 (2023).
Klingberg, F., Hinz, B. & White, E. S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 229, 298–309 (2013).
Kendall, R. T. & Feghali-Bostwick, C. A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 5, 123. https://doi.org/10.3389/fphar.2014.00123 (2014).
Garcia Garcia, J. M. et al. Endometriosis: Cellular and molecular mechanisms leading to fibrosis. Reprod. Sci. 30, 1453–1461 (2022).
Vissers, G. et al. The role of fibrosis in endometriosis: A systematic review. Hum. Reprod. Update 30, 706–750 (2024).
Muraoka, A. et al. Fusobacterium infection facilitates the development of endometriosis through the phenotypic transition of endometrial fibroblasts. Sci. Transl. Med. 15, eadd1531. https://doi.org/10.1126/scitranslmed.add1531 (2023).
Stratopoulou, C. A. et al. Impact of peritoneal macrophage depletion on endometriotic lesion development in a mouse model. J. Endometr. Uterine Disord. 10, 100113. https://doi.org/10.1016/j.jeud.2025.100113 (2025).
Konno, R. et al. Role of immunoreactions and mast cells in pathogenesis of human endometriosis–morphologic study and gene expression analysis. Hum. Cell 3, 141–149 (2003).
Nishimoto-Kakiuchi, A. et al. A long-acting anti–IL-8 antibody improves inflammation and fibrosis in endometriosis. Sci. Transl. Med. 15, eabq5858. https://doi.org/10.1126/scitranslmed.abq5858 (2023).
Tamura, K. et al. Involvement of stathmin in proliferation and differentiation of immortalized human endometrial stromal cells. J. Reprod. Dev. 53, 525–533 (2007).
Maekawa, R. et al. Genome-wide DNA methylation analysis reveals a potential mechanism for the pathogenesis and development of uterine leiomyomas. PLoS One 8, e66632. https://doi.org/10.1371/journal.pone.0066632 (2013).
Kajimura, T. et al. Overexpression of carbonyl reductase 1 inhibits malignant behaviors and epithelial mesenchymal transition by suppressing TGFB signaling in uterine leiomyosarcoma cells. Oncol. Lett. 18, 1503–1512 (2019).
Hayashi-Okada, M. et al. Identification of long noncoding RNAs downregulated specifically in ovarian high-grade serous carcinoma. Reprod. Med. Biol. 23, e12572. https://doi.org/10.1002/rmb2.12572 (2024).
Yuge, A. et al. Collagen gel contractility is enhanced in human endometriotic stromal cells: A possible mechanism underlying the pathogenesis of endometriosis-associated fibrosis. Hum. Reprod. 22, 938–944 (2007).
Sakai, T. et al. Establishment of a 3D spheroid culture system to evaluate the responsiveness of uterine leiomyoma cells to female hormones. Reprod. Med. Biol. 24, e12627. https://doi.org/10.1002/rmb2.12627 (2025).
Acknowledgements
This study has been funded by JSPS KAKENHI Grants (24K12533 to Sato S, 25K20119 to Tamehisa T, 24K12579 to Tamura I, and 23K27734 to Sugino N) for Scientific Research from the Ministry of Education, Science, and Culture, Japan.
Author information
Authors and Affiliations
Contributions
H.T-K. was responsible for the conception and design of the study, data collection, analysis, interpretation, and manuscript drafting. S.S. contributed to the conception and design of the study, data analysis and interpretation, and manuscript revision. T.T. participated in the study design, experiments, and data analysis. T.Y., A.S., and T.F. were involved in the experiments and data analysis. I.T. and R.M. contributed to the analysis and interpretation of the data. N.S. supervised the entire study, provided overall guidance, and revised the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Takasaki-Kawasaki, H., Sato, S., Tamehisa, T. et al. Establishment of a xenograft model of endometriosis-associated fibrosis using human immortalized endometrial stromal cells overexpressing HOXC8. Sci Rep 16, 11318 (2026). https://doi.org/10.1038/s41598-026-41956-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-41956-8
