
Abstract
Appendiceal Adenocarcinoma (AA) is a rare gastrointestinal cancer with no FDA-approved targeted therapies. Here, we retrospectively compare BRAF-mutant AA and colorectal cancer (CRC). BRAF mutation is rare in AA (3%). Unlike CRC, BRAFV600E AA is not associated with poor prognosis, female sex, microsatellite instability, mucinous histology, or poor differentiation. In both cancers, BRAFV600E but not atypical BRAF mutations are mutually exclusive with other Ras-activating mutations. BRAFV600E + EGFR inhibition shows efficacy in BRAFV600E AA (disease control rate = 80%, median progression-free survival = 7.1 months).
Similar content being viewed by others

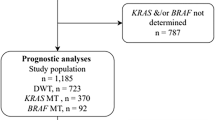

Appendiceal adenocarcinoma (AA) is a rare cancer, with an age-adjusted incidence rate of 1.3 (per 100,000)1 and also an orphan disease with no FDA-approved therapies. The best standard-of-care therapy remains cytoreductive surgery with hyperthermic intraperitoneal chemotherapy (CRS + HIPEC). However many patients are not good surgical candidates. Traditionally AA has been thought to respond poorly to systemic chemotherapy1, but more recent studies have suggested activity of targeted, immune, and intraperitoneally directed therapies2,3,4. Here we sought to characterize BRAF-mutant AA for the first time and evaluate the efficacy of BRAF inhibition specifically in AA.
Despite the appendix’s anatomic proximity to the colon, major clinical, demographic, and molecular differences exist between AA and colorectal cancer (CRC). AA frequently metastasizes to the peritoneal cavity and rarely exhibits hematogenous or lymphatic spread; in contrast, the two most common sites of CRC metastasis are the liver and the lungs5,6,7. Additionally, compared to CRC, the AA genomic landscape is markedly different with rare APC mutation and less frequent microsatellite instability (MSI) in AA8,9. Despite these clear clinical and molecular differences, and prospective data suggesting that low-grade AA does not respond to 5-FU-based chemotherapy1, current National Comprehensive Cancer Network guidelines for colon cancer still suggest similar chemotherapy treatment for AA10.
BRAF is a notable proto-oncogene in many cancers, including melanoma and CRC. BRAF is part of the MAPK pathway and is located downstream of RAS to activate MEK which subsequently activates ERK. This in turn turns on proliferation and survival signals11. BRAF variants are separated into three classes based on how the mutation affects BRAF function. Class I mutations are V600-mutations which can signal as monomers independent of upstream RAS signaling. Non-V600-mutations, or atypical BRAF (aBRAF) mutations, are divided into two classes. Class II mutations signal as constitutively active, RAS-independent homodimers, while Class III mutations have low kinase activity and signal as RAS-dependent heterodimers with CRAF12,13,14,15. Differences exist within each BRAF class, with some suggesting further subdivisions to capture these differences16. Each unique variant also has unique clinical and molecular features, highlighting the complexity of BRAF signaling in cancer17,18.
BRAF-mutant CRC has been thoroughly characterized including response to BRAFV600E inhibition. Although BRAFV600E inhibitor monotherapy is not effective in BRAFV600E CRC due to EGFR feedback activation, combination therapy with the BRAFV600E inhibitor encorafenib and the EGFR inhibitor cetuximab have been shown to be efficacious in BRAFV600E CRC19,20. Yet, to date, no analysis has been performed on BRAF-mutant AA. Here, we perform a comprehensive retrospective analysis of BRAF-mutant AA patients treated at MD Anderson Cancer Center (MDACC) from 2003 to 2023. A total of 856 patients with AA were tested for BRAF mutation identifying 26 (3.0%) with BRAF mutation; of 7256 patients with CRC 617 (8.5%) were found to have BRAF mutation (Supplementary Fig. 1, Supplementary Table 1). These proportions are comparable to other analyses of the molecular landscape of AA9,21. Age, sex, race/ethnicity, tumor grade, histological features, microsatellite status, and mutation profiles were also collected (Supplementary Table 1).
Several differences between the AA and CRC cohort were identified. First, BRAF mutation was significantly more frequent in CRC compared to AA (Fig. 1A, B; 8.50% vs. 3.04%, p < 0.0001). BRAFV471I and BRAFT241M were the two unclassified BRAF mutations in AA (Supplementary Table 2). Although unclassified, BRAFV471I is located in the P-Loop of the kinase domain of BRAF and therefore may be functionally significant22. BRAFT241M is also unclassified but not in the kinase domain and is therefore most likely a passenger mutation. Previous studies found that relative to BRAFWT, BRAFV600E-CRC is associated with female sex, mucinous histology, high microsatellite instability (MSI-H), and poor differentiation23,24. The MDACC CRC cohort recapitulated these differences between BRAFWT and BRAFV600E tumors, but interestingly none of these comparisons were significant in BRAFV600E AA (Fig. 1C–F. Supplementary Fig. 2). Notably, none of the BRAFV600E AA tumors were MSI-H while 130/366 (35.5%) of BRAFV600E mutant CRC were MSI-H (Fig. 1D), suggesting that unlike in CRC, microsatellite instability in AA is not associated with BRAFV600E hypermethylation activity25. Interestingly, all of the class II and III BRAF mutations we identified were in moderately or poorly differentiated tumors.

A Variant proportions in BRAF mutant AA and CRC sorted by BRAF mutation class. B Proportions of BRAF mutation in AA and CRC split by BRAF mutation class. C Comparison of patient sex in BRAFWT (gray) vs. BRAFV600E (blue) vs. aBRAF (skyblue) AA and CRC. D Comparison of proportion of mucinous tumors (mucinous adenocarcinoma and mucinous features) in BRAFWT (gray) vs. BRAFV600E (blue) vs. aBRAF (skyblue) AA and CRC. E Comparison of high-level microsatellite instability in BRAFWT (gray) vs. BRAFV600E (blue) vs. aBRAF (skyblue) AA and CRC. F Comparison of poor differentiation (grade 3+) in BRAFWT (gray) vs. BRAFV600E (blue) vs. aBRAF (skyblue) AA and CRC. P-value by Pearson’s Chi-Squared. *, <0.05; **, <0.01; ***, <0.001; ****, <0.0001. No comparison shown within cancers = not significant.
Next, we explored the mutation profiles of both cohorts (Supplementary Data 1, 2). Because BRAFV600E is a strong activator of the Ras/MAPK pathway in CRC BRAFV600E is known to be mutually exclusive with activating mutations of KRAS or other Ras/MAPK genes26,27. This was confirmed in our MDACC cohort, with KRAS mutation significantly more frequent in BRAFWt and aBRAF tumors relative to BRAFV600E (50.1% Wt vs. 0.9% V600E, p < 0.0001; 28.4% aBRAF vs. 0.9% V600E, p < 0.0001; Fig. 2C). KRAS mutation was significantly more frequent in BRAFWt CRC relative to aBRAF CRC (p < 0.0001). Despite sharing none of the earlier histological or demographic features with BRAFV600E CRC, BRAFV600E AA showed significantly less frequent KRAS mutation relative to BRAFWT and aBRAF (45.2% WT vs. 0% V600E, p < 0.0001; 55.6% aBRAF vs. 0% V600E, p = 0.005; Fig. 2A–C).

A Oncoprint plot of the mutation profile of BRAFV600E AA. Bar charts display relative proportion of tumors with mutations in a given gene (red), wildtype (black), or were untested (light gray). B Oncoprint plot of the mutation profile of aBRAF AA. C Comparison of KRAS comutation in BRAFWT (gray) vs. BRAFV600E (blue) vs. aBRAF (skyblue) AA and CRC. D Comparison of TP53 comutation in BRAFWT (gray) vs. BRAFV600E (blue) vs. aBRAF (skyblue) AA and CRC. E Comparison of GNAS comutation in BRAFWT (gray) vs. BRAFV600E (blue) vs. aBRAF (skyblue) AA and CRC. P value by Pearson’s Chi-Squared. *, <0.05; **, <0.01; ***, <0.001; ****, <0.0001. No comparison shown within cancers = not significant.
Like KRAS, TP53 and GNAS are commonly mutated in AA9. While there was not a significant association of TP53 mutation with frequency of BRAF mutation in our AA cohort, BRAFV600E CRC showed significantly less TP53 mutation compared to BRAFWT CRC (56.3% vs. 68.3%, p < 0.0001; Fig. 2D) similar to prior report28. GNAS mutation was not associated with BRAF mutation in either AA or CRC (Fig. 2E).
Previous studies of BRAFV600E CRC have found that BRAFV600E is associated with poor prognosis23. The MDACC CRC cohort confirmed this finding (median OS = 63 with 95% Confidence Interval of [61–66] mos. WT vs. 41 [35–49] mos. BRAFV600E, Hazard Ratio [HR] = 1.4 and p < 0.001, Fig. 3A). While Class II, Class III, and unclassified BRAF mutant CRC did not have a significantly different OS compared to BRAFWT, Class III had a trend toward worse OS (Median OS: 47 [31-Not Reached (NR)], HR = 1.4, p = 0.052). Previous studies have also shown that microsatellite stable (MSS) BRAFV600E CRC is associated with particularly poor prognosis23. The results from the MDACC CRC cohort support these earlier findings: MSS BRAFV600E CRC showed the worst median OS (28 [23–42] mos., HR = 2.0 and p < 0.001 relative to BRAFWT MSS CRC) and microsatellite instability-high (MSI-H) BRAFWT patients showed the best median OS (215 [201-NR] mos., HR = 0.35 and p < 0.001 relative to BRAFWT MSS CRC) (Fig. 3B). In contrast, BRAFV600E mutation did not significantly impact the median OS in our AA cohort (92 [78-105] mos. BRAFWT vs. 110 [75-NR] mos. BRAFV600E, HR = 1.1 with p = 0.88; Fig. 2c). The same was true of aBRAF (41 [31.6-Not Reached], HR = 1, p = 0.95). These relationships did not change when aBRAF was split into Class II, Class III, and unclassified BRAF mutations (Supplementary Fig. 3).

A Kaplan-Meier plot of CRC split into groups based on BRAF mutation status. B Kaplan-Meier plot of CRC split into groups based on both BRAF mutation status and microsatellite status. C Kaplan-Meier plot of AA split into groups based on BRAF mutation status. P-value and hazard ratios (HR) by Cox Proportional Hazard Model. Overall survival (OS) by Kaplan-Meier method. 95% Confidence Intervals are shown in parentheses. NR not reached.
Finally, treatment history for patients with BRAFV600E AA was manually collected to study the effectiveness of BRAFV600E inhibition (Supplementary Fig. 4). Most BRAFV600E AA patients were surgically treated before initiation of chemotherapy (16/17, 94.1%). First-line chemotherapy was always a 5-fluorouracil (5-FU)-based-treatment. There were three categories of 5-FU-based treatments: 5-FU (n = 4 individual treatment regimens; 50% 5-FU, 50% 5-FU+Bevacizumab [Bev]), FOLFOX (Folinic Acid+5-FU+oxaliplatin; n = 14; 57.1% FOLFOX, 35.7% FOLFOX+Bevacizumab, 7.1% XELOX/CAPEOX), and FOLFIRI (Folinic Acid+5-FU+and irinotecan; n = 13; 23.1% FOLFIRI, 7.7% FOLFIRI + EGFR Inhibitor, 69.2% FOLFIRI+Bevacizumab).
After progressing on 5-FU-based treatments, 9/17 patients received at least one regimen of monotherapy or combination therapy with a BRAFV600E inhibitor for a total of 10 regimens. Half of the regimens were 2nd line while the rest ranged from 3rd line to 5th line (Fig. 4a). Pt BRAF 20 was treated with 2 separate BRAFV600E inhibitor regimens: one before surgery and one after recurrence. Pt BRAF 16 was also treated with 2 regimens but passed away 4 days after starting the second regimen. The second regimen was not included in this analysis. Patients were initially treated with BRAFV600E + EGFR inhibition (7/10, 70%); BRAFV600E + EGFR inhibition+irinotecan (2/10, 20%); or BRAFV600E + RTK inhibitors (1/10, 10%) (Supplementary Table 3). 1/7 patients with BRAFV600E + EGFR inhibition were reduced to only BRAF inhibition due to drug toxicity. Half of patients with initial BRAFV600E + EGFR+Irinotecan were transitioned to only BRAFV600E + EGFR due to drug toxicity.

A Distribution of line of therapy for BRAFV600E inhibition treatment regimens. B Time-on-treatment ratio (TOTr) of the best performing prior line of 5-FU-based treatment versus BRAFV600E inhibition. Horizontal line = 1.3, arrow = still on treatment at last follow-up. C Best radiographic response (PR partial response, SD stable disease, PD progressive disease). D Longitudinal tumor marker information for Pt BRAF 15 showing CA 19-9 (U/mL, blue), CA 125 (U/mL, red), and CEA (ng/mL, gray). Shaded regions represent periods where patients were treated with the labeled chemotherapy. Dashed lines are the upper limit of the reference range for each tumor marker.
First, we compared time-on-treatment with BRAFV600E inhibition compared to the longest time-on-treatment for any prior 5-FU-based treatment regimens. Despite this conservative approach, 7/9 patients showed a longer time-on-treatment with BRAFV600E inhibition (Fig. 4b). 6/9 passed the accepted cutoff time-on-treatment ratio (TOTr) of 1.329,30. Moreover, 3/9 patients being treated with BRAFV600E inhibition had stable disease at the last follow-up before December 2023 (Fig. 4b).
Next, we checked the radiographic best response for a given treatment regimen. The radiographic disease control rate (partial response or stable disease) was 80% with a median PFS of 217 days (7.1 mos.) (Fig. 4c, Supplementary Fig. 5). The observed PFS in BRAFV600E AA treated with BRAFV600E inhibitor ± EGFR inhibitor is longer than previously reported in a prospective trial with BRAFV600E CRC19. Pt BRAF 8 had progression in the lungs at first follow-up; however, they continued with BRAFV600E inhibition because they had stable disease in the abdomen (Supplementary Fig. 4).
Finally, we checked for biochemical response in all patients treated with BRAFV600E inhibition. CEA, CA19-9, and CA125 are commonly used in the management of gastrointestinal cancers and have been associated with overall survival in AA31. One patient who received BRAFV600E + EGFR inhibitors (Pt BRAF 15) had longitudinal TMs collected; all 3 TMs were stable or decreased during the BRAFV600E inhibitor treatment period (Fig. 4d). During three prior lines of 5-FU-based treatments, both CA19-9 and CEA decreased; however, it was only with BRAFV600E inhibition that all three tumor markers were brought within normal ranges (Fig. 4d). There was also a 14.8% decrease in CA19-9 and a 60.7% decrease in CEA during BRAFV600E inhibition. The biochemical response together with the radiographic findings points to the efficacy of BRAFV600E inhibition in patients with BRAFV600E AA.
To our knowledge, this study is the first analysis of BRAF-mutant appendiceal adenocarcinoma and the first evidence supporting a precision oncology approach in AA. We find marked differences between CRC and AA in BRAF mutation frequency. Furthermore, unlike in CRC, BRAFV600E in AA is not associated with female sex, poor differentiation, microsatellite instability, and poor prognosis. Critically, we show an 80% disease control rate for BRAFV600E inhibition±EGFR inhibition in patients with BRAFV600E AA with median PFS of 7.1 mo; 7 of 9 patients had a longer TOTr with BRAFV600E inhibition±EGFR inhibition relative to the best-performing prior 5-FU-based treatment. These real-world results lead us to support using systemic BRAFV600E + EGFR inhibitors in the treatment of BRAFV600E AA. Recent preclinical research has suggested that unlike BRAFV600E CRC, BRAFV600E AA responds to BRAFV600E inhibitor monotherapy without feedback activation of EGFR. As a result, addition of EGFR inhibition may not be necessary in BRAFV600E AA although we are not aware of any appendiceal cancer patients treated long-term with BRAF inhibitor monotherapy32. With regards to class II and class III BRAF mutations, there is evidence that tumors with class II BRAF mutations in other cancers respond to dual BRAFV600E + MEK inhibition16,33. Additionally, class III BRAF mutant CRC has been reported to respond to EGFR inhibition without BRAF inhibition34. A new generation of pan-RAF inhibitors which inhibit MEK1/2 activation from all of the RAF genes (ARAF, BRAF, CRAF) are in clinical development which have shown activity against both class II and III BRAF mutations in pre-clinical models11,35. In conclusion, given our real-world findings, functional data in preclinical models, and the molecular differences between CRC and AA, it is clear that BRAFV600E AA must be studied and potentially treated as a unique entity distinct from BRAFV600E CRC.
Methods
Cohort generation
The MD Anderson adapted version of the Palantir-Foundry software system was used to perform an automated query of the MD Anderson GI Medical Oncology database to identify patients with AA treated between 2004 and Dec 2023. Eligible patients had a pathologic diagnosis of AA, mucinous adenocarcinoma, signet ring adenocarcinoma, or goblet cell adenocarcinoma along with a history of BRAF mutational profiling in the MD Anderson Molecular Diagnostic Laboratory (MDL), a CLIA-certified diagnostic laboratory. A cohort of patients with CRC was similarly created (Supplementary Fig. 1). This study was approved by the University of Texas MD Anderson Cancer Center Institutional Review Board (protocol 09-0373). A waiver of informed consent was granted in accordance with US federal regulation 45 CFR § 46.116 (Common Rule) and the study’s minimal risk to patients. This study was conducted in accordance with the ethical guidelines stated in the Declaration of Helsinki. We followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for cohort studies (Supplementary Table 4).
Data collection
Mutation profiles and other easily accessible information (age, sex, date of diagnosis, date of last follow-up, date of death, labs for tumor marker data, and microsatellite instability) were collected using the Palantir-Foundry software system. Patients with germline TP53 mutations (5 patients in the BRAFWT CRC) were grouped with non-germline TP53 mutations for presentation. A manual chart review was performed on BRAF-mutant AA patients to confirm the specific BRAF mutation. BRAF variant class assignments were determined by following prior literature11,36. Histological features and tumor grade were collected in a semi-automated fashion using a regular expression on pathology notes and confirmed by manual review. The MD Anderson clinical panels for microsatellite stability were used to define microsatellite status. These panels were either PCR-based or immunohistochemistry-based. We prioritized PCR results over IHC results, meaning that if a patient was tested with both IHC and PCR, the PCR results were used to define microsatellite status.
Overall survival was determined by calculating the difference between the date of diagnosis and either the date of last follow-up or the date of death. Median overall survival (OS) for each group was determined using the Kaplan-Meier method; p-value by the Cox Proportional Hazard model.
Treatment information was collected via manual chart review. In several cases, the notes only included the month of treatment initiation and cessation. In these cases, the 1st was assigned as the day of initiation. For analysis, 5-FU and its prodrug capecitabine were grouped.
Treatment response determination
Radiographic best response for BRAFV600E treatment were manually determined using radiologist interpretations of abdominal or thoracic CT scans with or without contrast, abdominal MRI scans with or without contrast, and FDG PET/CT scans. When data were available, the RECISTv1.1 criteria37 were used to determine the best radiographic response to treatment. Due to inadequate information in patient notes, the modified peritoneal RECIST criteria (mpRECIST) could not be used (3).
Biochemical response for treatment was determined using tumor marker information. Normal Tumor marker (TM) levels and biological response were defined as previously described31,38.
Statistics
Baseline characteristics of the study population were summarized by frequency, and differences were compared using the chi-square test. Time-to-event endpoints were visualized using Kaplan-Meier curves, and differences in OS were compared using Cox’s proportional hazards model.
All statistics were performed using R (v4.3.2) in RStudio (2023.09.01).
Data availability
All data except those containing PHI are included in the manuscript and supplementary data. Aggregated patient data are included in supplemental tables. Anonymized data are available for non-commercial use from the corresponding author upon request pending data usage agreement and/or IRB-approved collaboration.
Code availability
The code used to analyze the data and generate figures in R is deposited in Zenodo: https://doi.org/10.5281/zenodo.13769394.
References
Shen, J. P. et al. Efficacy of systemic chemotherapy in patients with low-grade mucinous appendiceal adenocarcinoma: a randomized crossover trial. JAMA Netw. Open 6, e2316161 (2023).
More, A. et al. Oncogene addiction to GNAS in GNASR201 mutant tumors. Oncogene 41, 4159–4168 (2022).
Hornstein, N. J. et al. Efficacy and safety of atezolizumab and bevacizumab in appendiceal adenocarcinoma. Cancer Res. Commun. 4, 1363–1368 (2024).
Ito, I. et al. Intraperitoneal paclitaxel is a safe and effective therapeutic strategy for treating mucinous appendiceal adenocarcinoma. Cancer Res. 83, 3184–3191. https://doi.org/10.1158/0008-5472.CAN-23-0013 (2023).
Riihimäki, M., Hemminki, A., Sundquist, J. & Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci Rep. 6, 29765 (2016).
Sugarbaker, P. H. Pseudomyxoma peritonei. A cancer whose biology is characterized by a redistribution phenomenon. Ann. Surg. 219, 109–111 (1994).
Pattalachinti, V. K. et al. Peritoneal microenvironment promotes appendiceal adenocarcinoma growth: a multi-omics approach using patient-derived xenografts. Mol. Cancer Res. 22, 329–336. https://doi.org/10.1158/1541-7786.MCR-23-0749 (2024).
Taggart, M. W. et al. High-level microsatellite instability in appendiceal carcinomas. Am. J. Surg. Pathol. 37, 1192–1200 (2013).
Ang, C. S.-P. et al. Genomic landscape of appendiceal neoplasms. JCO Precis. Oncol. 1–18 https://doi.org/10.1200/PO.17.00302 (2018).
Benson, A. B. et al. Colon Cancer, Version 3.2024, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 22, e240029 (2024).
Hanrahan, A. J., Chen, Z., Rosen, N. & Solit, D. B. BRAF—a tumour-agnostic drug target with lineage-specific dependencies. Nat. Rev. Clin. Oncol. 21, 224–247 (2024).
Yao, Z. et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238 (2017).
Yao, Z. et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 28, 370–383 (2015).
Schirripa, M. et al. Class 1, 2, and 3 BRAF-mutated metastatic colorectal cancer: a detailed clinical, pathologic, and molecular characterization. Clin. Cancer Res. 25, 3954–3961 (2019).
Sahin, I. H. & Klostergaard, J. BRAF mutations as actionable targets: a paradigm shift in the management of colorectal cancer and novel avenues. JCO Oncol. Pract. 17, 723–730. https://doi.org/10.1200/OP.21.00160 (2021).
Dankner, M. et al. Dual MAPK inhibition is an effective therapeutic strategy for a subset of class II BRAF mutant melanomas. Clin. Cancer Res. 24, 6483–6494 (2018).
Noeparast, A. et al. Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib. Oncotarget 8, 60094 (2016).
Zengarini, C. et al. BRAF V600K vs. BRAF V600E: a comparison of clinical and dermoscopic characteristics and response to immunotherapies and targeted therapies. Clin. Exp. Dermatol. 47, 1131 (2022).
Kopetz, S. et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–mutated colorectal cancer. N. Engl. J. Med. 381, 1632–1643 (2019).
Prahallad, A. et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103 (2012).
Foote, M. B. et al. Molecular classification of appendiceal adenocarcinoma. JCO 41, 1553–1564 (2023).
Schreck, K. C. et al. Integrated molecular and clinical analysis of BRAF-mutant glioma in adults. npj Precis. Onc. 7, 1–11 (2023).
Tran, B. et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 117, 4623–4632 (2011).
Clarke, C. N. & Kopetz, E. S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 6, 660–667 (2015).
Fang, M., Ou, J., Hutchinson, L. & Green, M. R. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol. Cell 55, 904–915 (2014).
Rajagopalan, H. et al. RAF/RAS oncogenes and mismatch-repair status. Nature 418, 934–934 (2002).
Zhao, Y. et al. Assessment of RAS dependency for BRAF alterations using cancer genomic databases. JAMA Netw. Open 4, e2035479 (2021).
Lee, C. S. et al. Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci. Rep. 11, 8154 (2021).
Von Hoff, D. D. et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. JCO 28, 4877–4883 (2010).
Mock, A. et al. Community-driven development of a modified progression-free survival ratio for precision oncology. ESMO Open 4, e000583 (2019).
Yousef, A. et al. Serum tumor markers and outcomes in patients with appendiceal adenocarcinoma. JAMA Netw. Open 7, e240260 (2024).
Martínez-Quintanilla, J. et al. Precision oncology and systemic targeted therapy in Pseudomyxoma Peritonei. Clin. Cancer Res. 30, 4082–4099. https://doi.org/10.1158/1078-0432.CCR-23-4072 (2024).
Pradervand, S. et al. Real-world occurrence, therapy, and outcome of patients with class 2 or 3 BRAF compared with class 1 BRAF-mutated cancers. ESMO Real World Data Digit. Oncol. 6, 100075 (2024).
Yaeger, R. et al. Response to anti-EGFR therapy in patients with BRAF non-V600–mutant metastatic colorectal cancer. Clin. Cancer Res. 25, 7089–7097 (2019).
Yaeger, R. et al. A next-generation BRAF inhibitor overcomes resistance to BRAF inhibition in patients with BRAF-mutant cancers using pharmacokinetics-informed dose escalation. Cancer Discov. 14, 1599–1611 (2024).
Owsley, J. et al. Prevalence of class I–III BRAF mutations among 114,662 cancer patients in a large genomic database. Exp. Biol. Med. 246, 31–39 (2021).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Dansby, J. et al. Taxane-Based Chemotherapy Is Effective in Metastatic Appendiceal Adenocarcinoma. The Oncologist 28, e1303–e1305 (2023).
Acknowledgements
This work was supported by the Col. Daniel Connelly Memorial Fund, the Andrew Sabin Family Fellowship Award, the National Cancer Institute (K22 CA234406 to J.P.S.), and the Cancer Center Support Grant (P30 CA016672), the Cancer Prevention & Research Institute of Texas (RR180035 to J.P.S., J.P.S. is a CPRIT Scholar in Cancer Research), and a Conquer Cancer Career Development Award (CDA-7604125121 to J.P.S). Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the American Society of Clinical Oncology® or Conquer Cancer.
Author information
Authors and Affiliations
Contributions
V.K.P. conceived, designed, and wrote the paper. E.H. collected clinical annotations and treatment information. M.Y. and A.Y. collected clinical annotations. S.C. created figures. M.O., C.M.P., V.K.M., B.K., R.W.H., and K.R. provided critical review and provided patient data. C.M.C. aided during the revision process. J.P.S. wrote the paper and conceived, designed, and supervised the study and provided patient data.
Corresponding author
Ethics declarations
Competing interests
J.P.S.—Scientific Advisory Board for Engine Biosciences, NaDeNo Nanoscience; Research funding from BostonGene, Celsius Therapeutics. All other authors declare no COI. M.O.—Consultant for Roche, BMS, Medimmune, Merck, Amgen, Takeda, Janssen, Pfizer, Array, Gritstone, 3DMed, Nouscom, Atreca, Bayer; Research funding from Takeda, Roche, Lilly, Merck, Medimmune, BMS, Nouscom, Phanes.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pattalachinti, V.K., Haque, E., Yousef, M. et al. BRAF mutant appendiceal adenocarcinoma differs from colorectal cancer but responds to BRAF-targeted therapy. npj Precis. Onc. 9, 38 (2025). https://doi.org/10.1038/s41698-025-00821-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00821-z
