
Abstract
PTEN hamartoma tumor syndrome (PHTS) is associated with increased lifetime risks of breast, thyroid, kidney, endometrial, and colorectal cancers, as well as melanoma (collectively, component cancers). We sought to characterize non-component cancers (NCC) in PHTS. Of 701 research participants with PHTS, 340 (49%) had cancer, with 101 (30%) having at least one NCC. Interestingly, 71 (70%) of those with NCC had at least one other PHTS component malignancy. Patients with pathogenic PTEN variants showed higher risks for prostate cancer and soft tissue sarcomas at younger ages than the general population. A literature survey showed independent cases of NCC in PHTS, with PTEN-related molecular changes including second-hit somatic PTEN alterations in a subset of various specimens. We recommend increased awareness regarding NCC in individuals with PHTS, particularly increased risks for prostate cancer and sarcoma. Further studies are needed to define age-related penetrance and accordingly, the appropriate strategies for cancer risk management.
Similar content being viewed by others
Introduction
PTEN hamartoma tumor syndrome (PHTS) is a term encompassing individuals with disease-causing germline PTEN variants regardless of clinical diagnosis1,2. PHTS includes Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRRS), PTEN-related Proteus syndrome (PS), and PTEN-related Proteus-like syndrome1. Even prior to the discovery of PTEN as the first susceptibility gene for CS, it was recognized that the syndrome was associated with elevated lifetime risks of breast and thyroid cancers3,4. Subsequent studies following the identification of germline PTEN variants in individuals with CS led to the expansion of the spectrum of cancers associated with CS and the establishment of more accurate cancer risk assessment and surveillance strategies5,6,7.
Studies in adult patients with PHTS indicate that germline PTEN variants are associated with increased lifetime risks of breast, thyroid, endometrial, kidney, and colon cancers, as well as melanoma (collectively referred to as PHTS component cancers)6,8,9,10. However, isolated reports and anecdotal evidence from our clinical practice (PTEN Multidisciplinary Clinic and Center of Excellence, Cleveland Clinic, Cleveland, Ohio, USA) show that individuals with PHTS can also have other cancers beyond those known to be associated with the syndrome (i.e., non-component cancers or NCC). Importantly, the spectrum of non-component cancers has not been systematically studied in PHTS. In addition, it is not known whether these cancers are incidental (i.e., sporadic) or whether they are bona fide components of PHTS that were missed in earlier studies due to limited patient series. In this study, we report the spectrum of non-component cancers from a prospective international series of pediatric and adult patients with PHTS to inform prospective cancer risk assessment and surveillance recommendations.
Results
Patient characteristics and germline PTEN variant spectrum
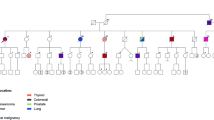
We prospectively accrued 701 research participants with germline PTEN variants from September 1, 2005, through January 6, 2022. Of the 701 research participants, 340 (49%) received cancer diagnoses (any cancer type), with 144 (42%) of those diagnosed with second primary malignant neoplasms (SMN). Of the 340 individuals with PHTS and cancer, 101 (30%) had at least one diagnosis with a non-component cancer (NCC) (Table 1). These cancers include any cancer type other than those known to be associated with PHTS, the latter including breast, thyroid, endometrial, kidney, and colon cancers, as well as melanoma6,11. Amongst those with cancer (n = 340), PHTS patients with NCC (n = 101) showed an overrepresentation of male biological sex compared to patients without NCC (n = 239; 29% versus 15%, P = 0.003). In addition, patients with NCC were of older age at study enrollment compared to patients without NCC (median [IQR] age at consent, 56 [47–64] versus 47 [40–57] years; P = 0.0005). However, there were no differences between those with and without NCC when we compared the ages at cancer diagnosis, and the Cleveland Clinic (CC) score, a semi-quantitative surrogate of overall phenotypic burden (Table 1)5. Interestingly, 71 (70%) of PHTS patients with NCC also had a diagnosis of at least one other PHTS component malignancy (Table 1). These patients had an older age at enrollment compared to PHTS patients diagnosed with NCC in the absence of any other component cancer, although this observation was statistically non-significant (median [IQR] age at consent, 57 [50–64] versus 49 [38–63] years; P = 0.06). Of the 71 PHTS patients diagnosed with both NCC and component cancers, 42 had a full set of cancer diagnosis dates to identify the chronological sequence of cancer occurrence events; 16 (38%) had at least one NCC diagnosed before a component cancer, with a median (IQR) duration of 10 (5–16) years between both events, and 26 (62%) had at least one component cancer diagnosed before an NCC, with a median (IQR) duration of 10 (5–17) years between both events.
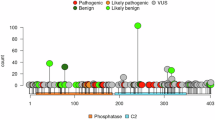
PTEN variants were classified into tier 1 including all pathogenic and likely pathogenic variants (215 variants [63%]), and tier 2 including variants of uncertain significance (VUS) and those with conflicting interpretation of pathogenicity with at least one VUS designation (125 variants [37%]). No differences were observed with respect to the PTEN overall tier 1 versus tier 2 variant classifications, as well as the PTEN variant types between individuals without versus those with NCC (Table 1). Considering only individuals with Tier 1 PTEN variants (n = 215), 63 (29%) had at least one diagnosis with an NCC.
Cancer spectrum and lifetime cancer risks
We observed a wide spectrum of diagnosed cancer types in the NCC category. These malignancies were generally categorized into hematological, gastrointestinal, genitourinary, germ cell, soft tissue, skin, and other cancer types (Table 2). While most cancers occurred in single individuals or in a limited number of individuals, the most overrepresented were non-melanoma skin cancers (41/101 or 40.6%), ovarian cancer (10/101 or 9.9%), prostate cancer (10/101 or 9.9%), soft tissue sarcomas (7/101 or 6.9%), and lung cancer (6/101 or 5.9%). Ovarian cancer cases included malignant epithelial ovarian tumors, malignant ovarian germ cell tumors, and malignant ovarian stromal tumors. There were five individuals who were diagnosed with both basal cell and squamous cell carcinomas, and one individual who was diagnosed with two soft tissue sarcomas (malignant giant cell tumor of tendon sheath of the right hand at age 46, and chondrosarcoma at age 59). Compared with standard population risks, we observed elevated risks of ovarian cancer (age-adjusted SIR 11.4; 95% CI, 5.8–20.3), prostate cancer (SIR 4.1; 95% CI, 1.9–7.8), and soft tissue sarcomas (SIR 10.7; 95% CI, 3.9–23.7). These observations were consistent when only including individuals with PHTS and tier 1 germline PTEN variants, except for ovarian cancer which became insignificant and comparable to the general population (Table 3). We could not evaluate the age-adjusted SIR for non-melanoma skin cancers due to the lack of age-related population-based estimates. The median (IQR) age at diagnosis for ovarian cancer was 49 (46–57) compared to 63 years in the SEER database, 59 (56–61) for prostate cancer versus 67 years in the SEER database, and 46 (28–47) for soft tissue sarcomas compared to 62 years in the SEER database (Table 4).
Independent cases of non-component cancers
In support of these data, we performed a literature survey of clinical cases reported by independent research groups describing non-component cancers in PHTS (Table 5). Of note, we did not limit the NCC to those we observed in our patient series. All cases included individuals with pathogenic and/or pathogenic germline PTEN variants, except for one case with a VUS12, and another case where the exact PTEN variant was not reported13. Relatedly, of the 33 reviewed studies, 16 performed genomic or immunohistochemical studies on tumor specimen derived from individuals with PHTS and NCC. Of these 16 studies, 13 reported somatic loss of heterozygosity or deletion at the PTEN locus and/or loss of PTEN protein expression through immunohistochemical analyses. These data corroborate the role of PTEN in the etiology of at least a subset of these non-component cancers.
Discussion
In this study, we report the spectrum of non-component cancers from the longitudinal follow-up of an international prospective series of pediatric and adult patients with PHTS. Anecdotal evidence from our clinical practice shows that individuals with PHTS can also have other cancers beyond the six component cancers known to be associated with the syndrome. Indeed, independent case reports and case series corroborate this finding12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44.
In our extended series, we found that approximately one-third of patients with PHTS and cancer have at least one NCC. Interestingly, those with NCC showed an overrepresentation of male biological sex. We posited that this was the case since we are comparing this group to PHTS patients with component cancers, known to be enriched with breast cancer and thus female sex. However, we found this to not be the case since most patients with NCC also had a prior or subsequent component cancer (71/101), with equal enrichment of breast cancer between the two groups. In addition, patients with NCC were of older age at study enrollment compared to those without NCC. Since most patients with NCC also had a PHTS component cancer, this observation does not support the hypothesis that these individuals had delayed enrollment due to the lack of diagnosis with cancers typical for PHTS. Indeed, individuals with PHTS and NCC alone tended to have a younger age at enrollment compared to those who had at least one other component cancer. Therefore, we acknowledge that age at enrollment is a complex phenomenon that may be influenced by multiple factors besides phenotypes of interest. Importantly, the ages at cancer diagnosis were not different between patients with PHTS with and without NCC. This corroborates the importance of clinicians being cognizant of other features that would raise a suspicion for a PHTS diagnosis (e.g., macrocephaly, pathognomonic mucocutaneous lesions), particularly in the presence of NCC. Of note, the median ages at diagnosis for ovarian cancer, prostate cancer, and soft tissue sarcomas were younger in individuals with PHTS compared to estimates from the general population. While we cannot propose guidelines for the surveillance of these cancers in the absence of age-related penetrance estimates, we call for more awareness regarding these malignancies, particularly where risk reduction strategies may be implemented. For example, in scope of the elevated risk of endometrial cancer as a component malignancy, and the consideration of hysterectomy with completion of childbearing7, we recommend considering bilateral salpingo-oophorectomy in women for whom a total abdominal hysterectomy is indicated.
Considering only individuals with Tier 1 PTEN variants (n = 215), 63 (29%) had at least one diagnosis with an NCC, which is higher than the 5–10% estimates from European adult individuals with PHTS45,46. However, the latter studies did not systematically study NCC, and it is unclear whether the reported estimates also included individuals with component cancers in addition to the NCC45,46. It is important to note that we focused our comparisons within individuals with PHTS and cancer since it is impossible to ascertain whether participants without cancer at the time of enrollment (particularly children, adolescents, and young adults), will go on to develop cancer later in life. Compared with standard population risks and including individuals with both tier 1 and tier 2 PTEN variants, we observed elevated risks of ovarian cancer, prostate cancer, and soft tissue sarcomas. These cancers were also associated with a younger median age at diagnosis compared to standard population-based estimates, supporting a non-incidental (i.e., sporadic) finding in the context of PHTS. This is also supported by the finding that additional somatic PTEN alterations, loss of heterozygosity at the PTEN locus, and/or loss of PTEN protein expression have been found in at least a subset of various non-component cancer specimens derived from individuals with PHTS (Table 5)12,18,19,20,21,25,27,30,31,36,40,41,44. Relatedly, PTEN somatic alterations and expression changes, as well as downstream PI3K-AKT signaling have been associated with sporadic carcinogenesis in the ovaries, prostate, and soft tissues47,48,49,50,51,52,53,54.
This study has limitations. There is a possibility that patients lost to follow-up may have developed cancer since the time of accrual or the time of their last clinical visit. This may result in an incomplete spectrum and underestimation of NCC frequencies. Relatedly, the heterogeneous spectrum of non-component cancers resulted in a limited number of individuals per cancer type, likely reflecting the lack of capture of these cancers in earlier studies6. The limited number of individuals per cancer type precluded us from performing age-related penetrance estimates within PHTS, and cancer risk estimates of all reported non-component cancers. We also could not evaluate the age-adjusted SIR for non-melanoma skin cancers due to the lack of age-related population-based estimates. Relatedly, there is a possibility that at least a subset of NCC may have arisen secondary to component cancer treatments. However, this scenario is unlikely, especially when the NCC is the first reported cancer and given the wide spectrum of observed NCC. Moreover, it is possible that since these patients receive more frequent and specialized care, providers and patients may be more judicious on working up possible cancer, including NCC. This may have resulted in overdiagnosis of NCC, although these patients do not receive any additional routine screening for NCC under current PHTS surveillance guidelines and are unlikely to have increased detection. In addition, we could not perform functional studies to evaluate somatic PTEN second hits, LOH, and/or loss of expression in tumor specimen from individuals with NCC in our study. However, with awareness regarding the presence of NCC in PHTS, we anticipate independent systematic studies to validate and characterize these risks in other PHTS populations around the world.
According to the presented data, we recommend increased awareness regarding the existence of NCC in individuals with PHTS. The elevated lifetime cancer risks of prostate cancers and soft tissue sarcomas in carriers of tier 1 PTEN variants will inform prospective validation studies to characterize age-related penetrance and accordingly, the appropriate ages for any warranted enhanced surveillance and risk-reducing strategies, in an adequate representative sample of patients.
Methods
Research participants
Research participants were prospectively accrued from September 1, 2005, through January 6, 2022, as part of a prospective follow-up study approved by the Cleveland Clinic institutional review board (IRB protocol 8458)11. The study was conducted in agreement with the principles of the Declaration of Helsinki. All participants provided informed written consent to participate. Participants were evaluated at the PTEN Multidisciplinary Clinic and Center of Excellence at the Cleveland Clinic (Cleveland, Ohio, USA). This study included both pediatric and adult patients with PHTS accrued from community and academic medical centers throughout North and South America, Europe, Australia, and Asia. Inclusion criteria for the Cleveland Clinic PTEN study (IRB 8458) included meeting, at minimum, the relaxed operational diagnostic criteria of the International Cowden Consortium (ICC), meaning individuals fulfilling pathognomonic criteria, or at least 2 criteria; either major or minor55,56. Reported cancer diagnoses were documented through pathology reports or verified cancer genetics visits. For the purposes of this focused study, we prioritized individuals from the parent PTEN study with confirmed germline PTEN testing results and regardless of specific cancer phenotypes. Specialist genetics staff reviewed all checklists, and if necessary, corresponded with the enrolling center to obtain primary documentation of medical records and pathology reports for phenotypic confirmation with patient consent. Baseline information including any (component and non-component) cancer history was recorded at the time of consent. Between July 2021 and July 2022, we obtained updated phenotypic information in those who had not routinely seen us in genetics clinic within 3 years11. We reviewed cancer-related health records of patients with PHTS internal to the Cleveland Clinic health system.
Germline PTEN variant classification
PTEN variants were classified into tier 1 (n = 514/701 or 73%), including all pathogenic and likely pathogenic variants, and tier 2 (n = 187/701 or 27%), including variants of uncertain significance (VUS) and those with conflicting interpretation of pathogenicity (n = 9; at least one classification should be a VUS). PTEN variant classifications were ascertained by clinical genetic testing reports where available, ClinVar database classifications, and/or the ClinGen gene-specific criteria for PTEN variant curation57. Analyses were performed for the entire series of patients (tier 1 and tier 2 PTEN variants) and for those with tier 1 PTEN variants only, separately.
Published case reports and case series
We conducted a literature search to identify case reports, case series, and other studies that report on non-component cancers in PHTS. Our literature search used Ovid MEDLINE and EMBASE databases of all articles published between 2002 and 2023. Search terms (alone or in combination) included: “PTEN Hamartoma Tumor Syndrome”, “Cowden Syndrome”, “Bannayan-Riley-Ruvalcaba Syndrome”, “Proteus syndrome”, “Proteus-like syndrome”, “Lhermitte Duclos”, “PHTS”, “PTEN or PTEN Phosphohydrolase”, “Neoplasms”, “(overgrowth or hamartom* or fibrom* or macrocep*)”, and “(carcin* or cancer* or neoplas* or malign*)”. Identified articles were imported into Covidence, a web-based collaboration software platform that streamlines the literature review process, where studies were deduplicated and screened. The literature review process took place between September 2022 and March 2023. We excluded any article without English translation, those that go back to more than 20 years, and those that cover PHTS component cancers. We also excluded studies from our research group to avoid the reporting of duplicate cases. We only included studies where the germline PTEN variant status was known and included either tier 1 or tier 2 PTEN variants to be consistent with the study design. Abstract screening and full text review were performed in parallel by two independent reviewers. Any discrepancies were resolved through consensus or discussion with a third reviewer. Following this process, an independent PubMed search was performed in September 2023 to cover any articles missed since the time of the Covidence literature search.
Statistical analysis
Clinical and demographic characteristics among participants with germline PTEN variants and cancer (n = 340) are described and comparisons between NCC versus without NCC were tested using a Chi-square test for categorical variables or t-test for continuous variables. In the whole series of 701 participants, we calculated standardized incidence ratios (SIR) using age-specific incidence data from the Surveillance Epidemiology and End Results database (SEER incidence data, November 2022 submission, 1975–2020). We evaluated four different cancer types separately and did not adjust for multiple comparisons. Analyses were performed using R software version 4.2.1 (R Project for Statistical Computing) and OpenEpi version 3.01 (Open Source Epidemiologic Statistics for Public Health) software. All statistical tests were two-sided, and P values < 0.05 were deemed significant.
Data availability
Aggregate data are provided in the associated tables. To protect individual patient information, the Cleveland Clinic Foundation IRB and Legal Department do not permit clinical information to be deposited in a publicly accessible database (by policy). Requests for such limited deidentified data relevant to this paper should be made to the corresponding author L.Y. (yehial@ccf.org). Thereafter, the Legal Department will ask for material transfer and data sharing agreements to be executed.
References
Yehia, L. & Eng, C. In GeneReviews((R)) (eds. Adam, M. P. et al) (1993).
Yehia, L., Keel, E. & Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 71, 103–116 (2020).
Brownstein, M. H., Wolf, M. & Bikowski, J. B. Cowden’s disease: a cutaneous marker of breast cancer. Cancer 41, 2393–2398 (1978).
Starink, T. M. et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin. Genet. 29, 222–233 (1986).
Tan, M. H. et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 88, 42–56 (2011).
Tan, M. H. et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 18, 400–407 (2012).
Daly, M. B. et al. Genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl Compr. Canc Netw. 19, 77–102 (2021).
Bubien, V. et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J. Med. Genet. 50, 255–263 (2013).
Nieuwenhuis, M. H. et al. Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam. Cancer 13, 57–63 (2014).
Hendricks, L. A. J., Hoogerbrugge, N., Schuurs-Hoeijmakers, J. H. M. & Vos, J. R. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin. Genet. 99, 219–225 (2021).
Yehia, L. et al. Longitudinal analysis of cancer risk in children and adults with germline PTEN variants. JAMA Netw. Open 6, e239705 (2023).
Uemura, S. et al. Pancreatic adenocarcinoma with a germline PTEN p.Arg234Gln mutation. Fam. Cancer 17, 255–259 (2018).
Tsunezuka, H., Abe, K., Shimada, J. & Inoue, M. Pulmonary atypical carcinoid in a patient with Cowden syndrome. Interact. Cardiovasc Thorac. Surg. 22, 860–862 (2016).
Mazereeuw-Hautier, J., Assouere, M. N., Moreau-Cabarrot, A., Longy, M. & Bonafe, J. L. Cowden’s syndrome: possible association with testicular seminoma. Br. J. Dermatol 150, 378–379 (2004).
Devi, M., Leonard, N., Silverman, S., Al-Qahtani, M. & Girgis, R. Testicular mixed germ cell tumor in an adolescent with Cowden disease. Oncology 72, 194–196 (2007).
Farhadi, M. R. et al. Intramedullary ependymoma associated with Lhermitte-Duclos disease and Cowden syndrome. Clin. Neurol. Neurosurg. 109, 692–697 (2007).
Tate, G., Suzuki, T. & Mitsuya, T. Mutation of the PTEN gene in a human hepatic angiosarcoma. Cancer Genet Cytogenet 178, 160–162 (2007).
Cho, M. Y. et al. First report of ovarian dysgerminoma in Cowden syndrome with germline PTEN mutation and PTEN-related 10q loss of tumor heterozygosity. Am. J. Surg. Pathol. 32, 1258–1264 (2008).
Barbosa, M., Henrique, M., Pinto-Basto, J., Claes, K. & Soares, G. Prostate cancer in Cowden syndrome: somatic loss and germline mutation of the PTEN gene. Cancer Genet 204, 224–225 (2011).
Bourdeaut, F. et al. Homozygous PTEN deletion in neuroblastoma arising in a child with Cowden syndrome. Am. J. Med. Genet A 155A, 1763–1766 (2011).
Sugihara, T. et al. Cowden syndrome complicated with hepatocellular carcinoma possibly originating from non-alcoholic steatohepatitis (NASH). Hepatol. Res. 41, 189–193 (2011).
Villeneuve, H. et al. Acinic cell carcinoma of the retromolar trigone region: expanding the tumor phenotype in Cowden syndrome? Fam. Cancer 10, 691–694 (2011).
Boespflug, A. et al. Primary lung adenocarcinoma occurring in a PTEN related syndrome (Cowden’s disease): routine EGFR sequencing also highlights two rare somatic mutations S768I and V769L. Lung Cancer 79, 318–320 (2013).
Chandhanayingyong, M. C. et al. Ewing sarcoma in a patient with Cowden syndrome. J. Natl Compr. Canc Netw. 13, 1310–1314 (2015).
Langer, S. W. et al. Cowden syndrome and concomitant pulmonary neuroendocrine tumor: a presentation of two cases. Case Rep. Med 2015, 265786 (2015).
Matsumoto, K., Nosaka, K., Shiomi, T., Matsuoka, Y. & Umekita, Y. Tumor-to-tumor metastases in Cowden’s disease: an autopsy case report and review of the literature. Diagn. Pathol. 10, 172 (2015).
Taylor, A. et al. Malignant peripheral nerve sheath tumor in Cowden syndrome: a first report. J. Neuropathol. Exp. Neurol. 74, 288–292 (2015).
Hagelstrom, R. T. et al. Breast cancer and non-Hodgkin lymphoma in a young male with Cowden syndrome. Pediatr. Blood Cancer 63, 544–546 (2016).
Molloy, C., Cahill, R., Gallagher, D., Murphy, P. & Quinn, J. Early-onset chronic lymphocytic leukaemia in a young man with Cowden syndrome. Ann. Hematol. 95, 1205–1206 (2016).
Neychev, V. et al. Neuroendocrine tumor of the pancreas as a manifestation of Cowden syndrome: a case report. J. Clin. Endocrinol. Metab. 101, 353–358 (2016).
Loffler, M. W. et al. First case report of malignant peritoneal mesothelioma and oral verrucous carcinoma in a patient with a germline PTEN mutation: a combination of extremely rare diseases with probable further implications. BMC Med. Genet. 19, 144 (2018).
Lopez, C., Abuel-Haija, M., Pena, L. & Coppola, D. Novel germline PTEN mutation associated with Cowden syndrome and osteosarcoma. Cancer Genomics Proteom. 15, 115–120 (2018).
Mizuno, S. et al. Multiple keratotic papules and plaques on the trunk in Cowden’s disease with MALT lymphoma. J. Dermatol 45, 238–240 (2018).
Rushin, L. et al. Is malignant germ-cell tumor associated with Cowden syndrome? Clin. Genitourin. Cancer 17, e429–e432 (2019).
Won, H. S. et al. PTEN mutation identified in patient diagnosed with simultaneous multiple cancers. Cancer Res. Treat. 51, 402–407 (2019).
Yauy, K. et al. Ovarian clear cell carcinoma in Cowden syndrome. J. Natl Compr. Canc Netw. 17, 7–11 (2019).
Galli, E. et al. Burkitt lymphoma as fourth neoplasia in a patient affected by Cowden syndrome with a novel PTEN germline pathogenic variant. Mediterr. J. Hematol. Infect. Dis. 12, e2020034 (2020).
Greidinger, A. et al. Neuroendocrine tumors are enriched in Cowden syndrome. JCO Precis. Oncol. 4, PO.19.00241 (2020).
Miguelote, S., Silva, R., Fougo, J. L., Barbosa, L. E. & Araujo Teixeira, J. P. Cowden syndrome is a risk factor for multiple neoplasm: a case report. World J. Surg. Oncol. 18, 211 (2020).
Fiala, E. M. et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat. Cancer 2, 357–365 (2021).
Leibowitz, M. S. et al. Neuroblastoma and cutaneous angiosarcoma in a child with PTEN hamartoma tumor syndrome. Pediatr. Blood Cancer 69, e29656 (2022).
Kouzuki, K. et al. Immature teratoma of the ovary associated with Cowden syndrome. Pediatr. Blood Cancer 69, e29555 (2022).
Pena-Couso, L. et al. Considerations on diagnosis and surveillance measures of PTEN hamartoma tumor syndrome: clinical and genetic study in a series of Spanish patients. Orphanet J. Rare Dis. 17, 85 (2022).
Prieto, R., Hofecker, V. & Corbacho, C. Coexisting lipomatous meningioma and glioblastoma in Cowden syndrome: a unique tumor association. Neuropathology 43, 110–116 (2023).
Hendricks, L. A. J. et al. Genotype-phenotype associations in a large PTEN Hamartoma Tumor Syndrome (PHTS) patient cohort. Eur. J. Med. Genet. 65, 104632 (2022).
Hendricks, L. A. J. et al. Cancer risks by sex and variant type in PTEN hamartoma tumor syndrome. J. Natl Cancer Inst. 115, 93–103 (2023).
Nero, C., Ciccarone, F., Pietragalla, A. & Scambia, G. PTEN and gynecological cancers. Cancers (Basel) 11, 1458, https://doi.org/10.3390/cancers11101458 (2019).
Martins, F. C. et al. Clinical and pathological associations of PTEN expression in ovarian cancer: a multicentre study from the Ovarian Tumour Tissue Analysis Consortium. Br. J. Cancer 123, 793–802 (2020).
Kwabi-Addo, B. et al. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc. Natl Acad. Sci. USA 98, 11563–11568 (2001).
Jamaspishvili, T. et al. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 15, 222–234 (2018).
Stefano, S. & Giovanni, S. The PTEN tumor suppressor gene in soft tissue sarcoma. Cancers (Basel) 11, 1169, https://doi.org/10.3390/cancers11081169 (2019).
Movva, S. et al. Multi-platform profiling of over 2000 sarcomas: identification of biomarkers and novel therapeutic targets. Oncotarget 6, 12234–12247 (2015).
Yin, L. et al. Mutational analysis of p53 and PTEN in soft tissue sarcoma. Mol. Med. Rep. 5, 457–461 (2012).
Cote, G. M., He, J. & Choy, E. Next-generation sequencing for patients with sarcoma: a single center experience. Oncologist 23, 234–242 (2018).
Eng, C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J. Med. Genet. 37, 828–830 (2000).
Pilarski, R. & Eng, C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J. Med. Genet. 41, 323–326 (2004).
Mester, J. L. et al. Gene-specific criteria for PTEN variant curation: recommendations from the ClinGen PTEN Expert Panel. Hum. Mutat. 39, 1581–1592 (2018).
Acknowledgements
We are grateful to all our patients and families who have participated in research studies for more than two decades. We thank the clinical research team at the PTEN Multidisciplinary Clinic and Center of Excellence at the Cleveland Clinic for administrative support. We are also grateful for our funding sources, including the American Cancer Society (RPG-02-151-01-CCE and Clinical Research Professorship), the National Institute of Neurological Disorders and Stroke (U54NS092090), National Institute of Child Health and Human Development (R01HD105049), Breast Cancer Research Foundation, Doris Duke Distinguished Clinical Scientist Award, the Ambrose Monell Foundation, PTEN Research Foundation and the Zacconi Center of PTEN Research Excellence (all to C.E.). L.Y. is an Ambrose Monell Foundation Cancer Genomic Medicine Project Staff at the Cleveland Clinic Genomic Medicine Institute. G.P. is a Crile Research Fellow at the Cleveland Clinic Genomic Medicine Institute. C.E. is the Sondra J. and Stephen R. Hardis Chair of Cancer Genomic Medicine at the Cleveland Clinic and an American Cancer Society Clinical Research Professor. The funders played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
L.Y. and C.E. conceptualized and designed the project. L.Y., G.P., A.T., D.L., and J.J. collected the data. L.Y. and G.P. performed the data analyses. L.Y., G.P., S.P., Y.N., and C.E. interpreted the data. L.Y. and S.P. performed the statistical analyses. C.E. supervised the project. L.Y. drafted the original version of the manuscript. All authors drafted, critically revised, and gave final approval of the paper. C.E., the senior author of this manuscript, passed away on the 13th day of August 2024. C.E. was involved in the preparation and revision of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yehia, L., Plitt, G., Tushar, A.M. et al. Extended spectrum of cancers in PTEN hamartoma tumor syndrome. npj Precis. Onc. 9, 61 (2025). https://doi.org/10.1038/s41698-025-00847-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00847-3
