
Abstract
Cervical cancer represents a significant global health challenge, with complex cellular and molecular mechanisms driving its progression from HPV infection to invasive malignancy. This study employed an integrated approach combining single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (stRNA-seq) to comprehensively characterize the tumor microenvironment (TME) across different stages of cervical cancer development. Through analysis of samples from normal cervix, HPV-infected normal cervix, high-grade squamous intraepithelial lesions (HSIL), and invasive cervical cancer, we identified distinct cellular populations and their dynamic changes during disease progression. Our findings revealed significant heterogeneity in immune cell populations, particularly highlighting the role of SPP1+ macrophages that were substantially enriched in cervical cancer compared to precancerous and normal tissues. Cell-cell communication networks and spatial mapping demonstrated that SPP1+ macrophages interact extensively with immune cells through the SPP1-CD44 signaling axis. This interaction contributes to an immunosuppressive microenvironment through modulation of T cell function and promotion of tumor cell survival. Furthermore, high expression of SPP1 correlated with advanced tumor stages and poor overall survival in cervical cancer patients, highlighting its potential as a prognostic biomarker. Our comprehensive characterization of the cellular landscape and intercellular communication networks in cervical cancer progression provides valuable insights for the development of targeted therapeutic strategies aimed at modulating the TME, particularly through disruption of the SPP1-CD44 axis. These findings establish a foundation for more effective personalized approaches to improve clinical outcomes in cervical cancer patients.
Similar content being viewed by others


Introduction
Cervical cancer remains a significant global health challenge, ranking as the 8th most common cancer among women worldwide, with an estimated 660,000 new cases and 350,000 deaths in 2022 alone1,2. HPV infection is recognized as the primary etiological factor in the development of cervical cancer, with high-risk HPV types contributing to the majority of cases3. Despite advances in screening and vaccination, the progression from HPV infection to cervical cancer involves complex biological processes that are not fully understood. Understanding the cellular and molecular mechanisms underlying this progression is crucial for developing targeted therapeutic strategies and improving patient outcomes4.
The tumor microenvironment (TME) plays a critical role in cancer progression, immune evasion, and treatment resistance5. In cervical cancer, the TME consists of malignant cells, cancer-associated fibroblasts, and various immune cells, including dendritic cells, B cells, T cells, monocytes, neutrophils, natural killer cells, and macrophages, collectively modulating tumor progression6. Recent studies have highlighted the importance of immune cell infiltration in cervical cancer, with distinct differences in the immune landscape between early-onset and late-onset disease7. However, a comprehensive understanding of the cellular heterogeneity and intercellular communication networks within the cervical cancer TME remains limited8. Advancements in scRNA-seq and stRNA-seq have revolutionized our ability to dissect the complex cellular heterogeneity and spatial organization within the TME9. These technologies enable high-resolution characterization of cell populations, their functional states, and interactions within tumor tissues.
Macrophages represent a key component of the cervical cancer TME, exhibiting remarkable heterogeneity and plasticity10. Traditional classification into M1 (pro-inflammatory) and M2 (anti-inflammatory) phenotypes has been challenged by recent single-cell studies revealing much more complex macrophage populations. Among these, SPP1+ macrophages have emerged as particularly important in shaping the tumor immune landscape7. These macrophages, characterized by high expression of secreted phosphoprotein 1 (SPP1, also known as osteopontin), have been identified across multiple cancer types, such as including hepatocellular carcinoma, glioblastoma, often correlating with poor prognosis and immune suppression11,12,13.
The SPP1-CD44 signaling axis has recently been identified as a pivotal pathway in tumor-immune interactions14. CD44, a cell-surface glycoprotein involved in cell-cell interactions, adhesion, and migration, serves as a receptor for SPP1 among other ligands. In various cancer types, this interaction has been shown to contribute significantly to tumor progression, metastasis, and immune evasion8,15. SPP1-CD44 signaling promotes tumor stemness, chemoresistance, and creates an immunosuppressive microenvironment by modulating T cell function and macrophage polarization15,16. In recent studies, single-cell transcriptomics revealed that the SPP1-CD44 axis mediates critical interactions between tumor cells and immune cells, potentially driving therapy resistance in multiple cancers17,18.
In cervical cancer specifically, the role of the SPP1-CD44 axis remains incompletely characterized. Our study combines scRNA-seq and stRNA-seq approaches to comprehensively profile cellular heterogeneity and intercellular communication networks across different stages of cervical cancer progression, from normal cervix to invasive carcinoma. By integrating multiple computational analytical approaches, we aim to elucidate the dynamic changes in cellular composition, functional states, and interaction patterns driving disease progression.
Our analysis has revealed significant enrichment of SPP1+ macrophages in cervical cancer samples compared to normal and precancerous tissues. These macrophages exhibit distinct transcriptional signatures and form specific cellular interaction networks with tumor cells and other immune cells via the SPP1-CD44 axis. Notably, SPP1+ macrophages showed enhanced interactions with epithelial tumor cells and T lymphocytes through SPP1-CD44 signaling, potentially contributing to immune suppression and tumor promotion. Spatial transcriptomics further confirmed the co-localization of SPP1+ macrophages with exhausted T cells in the TME, highlighting their role in creating immunosuppressive niches. Furthermore, our findings demonstrate that high expression of SPP1 correlates with advanced tumor stage and poorer overall survival in cervical cancer patients, suggesting its potential value as a prognostic biomarker. The SPP1-CD44 axis emerged as a critical pathway in tumor-promoting cellular interactions, representing a promising target for therapeutic intervention. These insights into the complex cellular and molecular landscape of cervical cancer progression provide a foundation for developing novel targeted therapies aimed at modulating the TME and improving clinical outcomes in cervical cancer patients.
Results
ScRNA-seq Profiling of Gradual Progress from Normal to Cervical Cancer
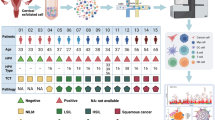
The study design flowchart is illustrated in Fig. 1A. The quality control of scRNA-seq was shown in Supplementary Fig. 1A–C. To elucidate the cellular composition and heterogeneity at various stages of cervical cancer progression, we conducted an extensive analysis integrating scRNA-seq data. We identified and annotated various cell types using this data. Figure 1B illustrates the expression of representative markers in different cell types, including mast cells (TPSAB1), NK/T cells (CD3E), epithelial cells (EPCAM), B cells (CD19), endothelial cells (PECAM1), neutrophils (NCF1), smooth muscle cells (ACTA2), myeloid cells (CD68), fibroblasts (COL1A1), and plasma cells (MZB1).

A Study design of this study. B Expression of marker genes for different cell types: mast cells (TPSAB1), NK/T cells (CD3E), epithelial cells (EPCAM), B cells (CD19), endothelial cells (PECAM1), neutrophils (NCF1), smooth muscle cells (ACTA2), myeloid cells (CD68), fibroblasts (COL1A1), and plasma cells (MZB1). C Functional enrichment analysis showing significant biological processes and pathways for each cell type. D UMAP plot visualizing the clustering of different cell types. E Proportions of cell types across conditions.
Functional enrichment analysis revealed significant biological processes and pathways associated with the identified cell types (Fig. 1C), which align with our cell type annotations. Notably, processes such as alpha-beta T cell activation, T cell receptor signaling pathway, neutrophil chemotaxis, and myeloid leukocyte activation were enriched, highlighting the important role of the immune landscape in the microenvironment.
The Uniform Manifold Approximation and Projection (UMAP) plot in Fig. 1D visualizes the clustering of different cell types. To understand the impact of HPV infection and disease progression on cellular composition, we compared the proportions of different cell types across various conditions: cervical cancer with HPV infection (CA_HPV), high-grade squamous intraepithelial lesion with HPV infection (HSIL_HPV), normal samples with HPV infection (N_HPV), and uninfected normal samples (NO_HPV) (Fig. 1E). Specifically, there was a notable increase in myeloid cells and NK/T cells in cancer samples and HPV-infected samples compared to normal samples without HPV infection. This suggests an immune response and reshaping of the immune microenvironment associated with HPV infection and tumorigenesis. Conversely, the proportion of epithelial cells decreased in cancerous conditions, reflecting the potential disruption of normal epithelial architecture during the progression to malignancy.
Dynamic changes in immune cell populations during cervical cancer development
We performed comprehensive profiling of immune cell populations across different stages of cervical cancer development. UMAP clustering of T cells revealed distinct subpopulations, including CD4 + T cells (exhausted PDCD1 + , exhausted CTLA4 + , and naive), CD8 + T cells (GZMB+ and GZMK+ CTLs), and Th17 cells (Fig. 2A, B). Notably, we observed a significant decrease in Th17 cells accompanied by an increase in exhausted CD4 + T cells in CA_HPV compared to other conditions (Fig. 2C).

A UMAP visualization of T cell subclusters, colored by cell type, showing distinct populations of CD4 + T cells (exhausted PDCD1 + , exhausted CTLA4 + , and naive), CD8 + T cells (GZMB+ and GZMK+ CTLs), and Th17 cells. B Dot plot showing expression of signature genes across T cell subclusters. C Stacked bar chart depicting the proportion of T cell subclusters across different disease stages. D UMAP visualization of B cell subclusters, colored by cell type. E Dot plot showing expression of signature genes across B cell subclusters. F Stacked bar chart depicting the proportion of B cell subclusters across different disease stages. G Heatmap showing the tissue distribution preference of B cell subclusters across different disease stages. H Feature plots showing expression of representative neutrophil markers (CSF3R, FCGR3B, NAMPT). I UMAP visualization of neutrophil subclusters, colored by cell type. J Stacked bar chart depicting the proportion of neutrophil subclusters across different disease stages. K Heatmap showing the tissue distribution preference of neutrophil subclusters across different disease stages.
B cell analysis identified three main subpopulations: plasma cells, memory B cells, and naive B cells (Fig. 2D, E). The distribution analysis revealed that memory and naive B cells were predominantly present after HPV infection, while plasma cells showed reduced proportion (Fig. 2F, G).
Neutrophil characterization revealed seven distinct subpopulations based on marker gene expression (Fig. 2H, I). These subpopulations showed differential distribution across disease stages (Fig. 2J, K). Notably, CXCL8+ and LCN2+ neutrophils were enriched in HPV-negative samples, while LGALS3+ and SPP1+ neutrophils were more prevalent in CA_HPV samples. Functional enrichment analysis revealed significant pathways associated with each neutrophil subpopulation (Supplementary Fig. 1D). SPP1_neutrophils were enriched in pathways related to negative regulation of immune system processes, while LGALS3_neutrophils were involved in stress response to metal ion pathways. CytoTRACE analysis has further illuminated the maturity status of neutrophil subpopulations, specifically indicating that the SPP1_neutrophil subset exhibits a less mature phenotype (Supplementary Fig. 1E). These results indicate the disability to anti-tumor of SPP1_neutrophils in TME.
Characterization and Functional Dynamics of Macrophage Subtypes in Cervical Cancer
In this study, we performed a comprehensive to explore the heterogeneity and functional dynamics of macrophage subtypes. Using UMAP clustering, we identified 6 macrophage subtypes: SPP1+ macrophages (SPP1_mac), CXCL9+ macrophages (CXCL9_mac), VCAN+ macrophages (VCAN_mac), IGKC+ macrophages (IGKC_mac), CD1E+ macrophages (CD1E_mac), and DNASE1L3+ macrophages (DNASE1L3_mac) (Fig. 3A). Each subtype exhibited distinct marker gene expression patterns, as illustrated in the dot plot (Fig. 3B). The distribution of these macrophage subtypes varied significantly across different conditions (Fig. 3C, D). Notably, SPP1+ macrophages showed significantly higher in CA_HPV compared to other conditions, indicating their potential role in the progression of cervical cancer. CXCL9+ macrophages also exhibited a significant increase in HSIL_HPV, while other subtypes, including IGKC + , VCAN + , CD1E + , and DNASE1L3+ macrophages, were mainly enriched in N_HPV and NO_HPV conditions.

A UMAP plot showing the clustering of macrophage subtypes identified in cervical cancer samples. B Dot plot illustrating the expression of marker genes for each macrophage subtype. C Bar plot displaying the distribution of macrophage subtypes across different conditions. D Heatmap showing the distribution preference across macrophage subtypes. E GO enrichment analysis for each macrophage subtype. F Violin plots displaying the expression levels of key genes involved in the ERK1/ERK2 pathway across macrophage subtypes. G UMAP plot focusing on SPP1+ macrophages. H, I Pseudotime trajectory analysis of SPP1+ macrophages across different conditions. J The inflammatory response pathway scored in SPP1+ macrophages along the pseudotime trajectory.
GO enrichment analysis further elucidated the biological processes associated with each macrophage subtype (Fig. 3E). For instance, SPP1+ macrophages were significantly associated with the ERK1/ERK2 cascade, indicating their potential crucial role in modulating the immune response within the TME, potentially contributing to tumor growth and progression19. Other macrophage subtypes displayed an immune-activated landscape, participating in B and T cell activation and immune signaling transduction.
Violin plots demonstrated the expression levels of key genes involved in the ERK1/ERK2 pathway across macrophage subtypes (Fig. 3F). SPP1+ macrophages exhibited universally elevated expression of genes such as GPNMB and APOE. This suggests that targeting these pathways or genes in SPP1+ macrophages could be a potential therapeutic strategy to alter the immune landscape in cervical cancer.
We furtherly focus on the SPP1+ macrophages (Fig. 3G). Pseudotime trajectory analysis highlighted the heterogeneity and functional dynamics of SPP1+ macrophages during cervical cancer progression (Fig. 3H, I). This dynamic shift underscores the complexity of macrophage roles in the TME and their potential impact on disease outcomes.
Further analysis of the inflammation response pathway revealed a significant downregulation in SPP1+ macrophages (Fig. 3J). This finding underscores the suppression of the pro-inflammatory response in SPP1+ macrophages within the TME, highlighting their potential as targets for therapeutic intervention aimed at modulating inflammation and immune responses in HPV-related cervical cancer.
Dissection the heterogeneity of epithelial and tumor cells
To explore the heterogeneity of epithelial cells in the TME of cervical cancer samples, we employed InferCNV analysis to detect copy number variations (CNVs) across cell populations, using the normal epithelial cells in NO_HPV as a reference. The analysis revealed distinct CNV profiles in tumor epithelial cells, particularly the gain CNV events on Chromosome 1. Furthermore, we identified tumor cells in CA_HPV based on K-means clustering (Fig. 4A, Supplementary Fig. 1F, G, Supplementary Table 1).

A Heatmap showing copy number variation (CNV) profiles of epithelial cells, with normal epithelial cells (NO_HPV) as reference. B UMAP visualization distinguishing normal epithelial cells from tumor epithelial cells. C CytoTRACE analysis of epithelial cell populations. D GSEA showing enriched pathways in tumor cells compared to normal epithelial cells. E Distribution of cells along the pseudotime axis. F, G Cell density plot along the pseudotime trajectory of epithelial cells across different cell and disease stages. H Branched heatmap showing gene expression dynamics along the pseudotime trajectory at branch point 1.
UMAP dimensionality reduction clearly distinguished normal epithelial cells from tumor epithelial cells in CA_HPV samples (Fig. 4B). CytoTRACE analysis demonstrated enhanced stemness characteristics in tumor epithelial cells compared to their normal counterparts (Fig. 4C). Differential expression analysis identified distinct transcriptional profiles between normal and tumor epithelial populations (Supplementary Fig. 2A). GSEA revealed significant enrichment of pathways related to epithelial cell differentiation, proliferation, and migration in tumor cells. Conversely, immune-related pathways including humoral immune response, adaptive immune response, and phagocytosis recognition were downregulated in tumor cells compared to normal epithelial cells (Fig. 4D).
Comprehensive metabolic pathway analysis revealed significant metabolic reprogramming between normal and tumor epithelial cells in cervical cancer. ScMetabolism analysis demonstrated distinct metabolic signatures between these populations (Supplementary Fig. 1H). Tumor epithelial cells exhibited significant upregulation of glycolysis/gluconeogenesis and amino sugar/nucleotide sugar metabolism pathways. Conversely, normal epithelial cells showed enhanced activity in multiple metabolic pathways, including fatty acid metabolism, TCA cycle, and oxidative phosphorylation. Metabolic flux analysis using scFEA revealed differential metabolic activities between normal and tumor epithelial cells (Supplementary Fig. 1I). Notably, tumor cells displayed elevated metabolic flux in several pathways, including glucose uptake (Glucose-in -> Glucose) and various nucleotide metabolism pathways (dCDP -> dCMP). Detailed analysis of glucose metabolism demonstrated significantly higher glucose uptake (Supplementary Fig. 1J) and glucose-to-G6P conversion (Supplementary Fig. 1K) in tumor cells compared to normal epithelial cells. The hallmark glycolysis pathway analysis further confirmed the metabolic shift, showing markedly higher glycolytic activity in tumor epithelial cells (Supplementary Fig. 1L). This enhanced glycolytic phenotype aligns with the Warburg effect commonly observed in cancer cells, supporting their increased energy demands and biosynthetic requirements for rapid proliferation.
Pseudotime trajectory analysis was employed to elucidate the transition relationships between normal and tumor epithelial cells (Fig. 4E, Supplementary Fig. 2B). When stratified by HPV infection status, the analysis revealed distinct progression patterns across different conditions (Supplementary Fig. 2C). The pseudotime distribution analysis demonstrated that tumor cells predominantly occupied later pseudotime points (15–20), while normal epithelial cells were enriched in earlier stages (Fig. 4F). Notably, cells from HSIL_HPV samples exhibited similar pseudotime distributions to CA_HPV cells, suggesting comparable molecular characteristics (Fig. 4G).
A branched heatmap analysis was performed to visualize the expression dynamics of key genes along the pseudotime trajectory for branch point 1. This analysis revealed distinct gene expression profiles and associated biological processes (Fig. 4H). The pre-branch stage was enriched in immune response and activation, including pathways related to humoral immune response, complement activation, and phagocytosis, suggesting active immune surveillance in early-stage cells. As cells transition into the branch, we observed a shift from initial expression of differentiation-associated genes to subsequent upregulation of EMT markers. This transition from differentiated to dedifferentiated states aligns with typical cancer progression patterns, where cells gradually lose their specialized epithelial characteristics and acquire more mesenchymal features. These trajectory patterns, combined with the HPV status analysis, suggest a model where HPV infection may compromise immune surveillance mechanisms while promoting cellular dedifferentiation. The similar molecular characteristics between HSIL_HPV and CA_HPV cells indicate that HPV-induced changes in cellular programs may establish a pre-malignant state that facilitates tumor progression.
Distinct molecular subtypes reveal intratumoral heterogeneity in cervical cancer
Single-cell transcriptomic analysis of CA_HPV tumor cells revealed three distinct tumor cell subpopulations (Epi_C1, Epi_C2, and Epi_C3) through UMAP dimensionality reduction (Fig. 5A). Dot plot visualization demonstrated distinctive marker gene expression patterns across these subpopulations, highlighting their molecular heterogeneity (Fig. 5B). Further investigation of cell cycle states revealed characteristic proliferation patterns, wherein Epi_C1 exhibited predominant G1 phase occupancy, while Epi_C2 and Epi_C3 demonstrated enhanced proliferative activity with increased proportions of cells in S and G2/M phases (Fig. 5C).

A UMAP visualization of tumor cell subclusters (Epi_C1, Epi_C2, and Epi_C3). B Dot plot showing expression of signature genes across tumor cell subclusters. C Bar chart showing the cell cycle phase distribution across tumor cell subclusters. D CytoTRACE analysis of tumor cell subclusters. E Heatmap of differentially expressed genes across tumor cell subclusters. F GO enrichment analysis of signature genes for each tumor cell subcluster. G Heatmap showing pathway activity scores across tumor cell subclusters. H Heatmap showing transcription factor activity scores across tumor cell subclusters.
CytoTRACE analysis elucidated a clear differentiation hierarchy among these tumor subpopulations, with Epi_C3 displaying the highest differentiation potential (Fig. 5D). This hierarchical organization was further substantiated by differential expression analysis of cluster-specific marker genes, as visualized in the heatmap (Fig. 5E).
Gene Ontology analysis revealed distinct functional characteristics associated with each tumor subpopulation (Fig. 5F). Specifically, Epi_C1 demonstrated enrichment in pathways related to keratinocyte differentiation and cytokine signaling regulation. Epi_C2 exhibited significant enrichment in processes governing granulocyte chemotaxis and migration, while Epi_C3 showed distinctive enrichment in pathways responding to mechanical stimuli and metal ion stress.
Comprehensive pathway activity analysis unveiled subtype-specific signaling patterns (Fig. 5G). Notably, Epi_C1 exhibited elevated activity in Androgen, Hypoxia, TNFα, JAK-STAT, and VEGF signaling pathways. Epi_C2 showed high activation of Estrogen and WNT pathways, whereas Epi_C3 demonstrated enhanced activation of MAPK, NFκB, and TGFβ pathways. Additionally, transcription factor activity analysis revealed differential regulatory networks governing these tumor subpopulations (Fig. 5H). Specifically, Epi_C1 was characterized by high activity of MXD1, ELK3, IRF7, SP1, and several other transcription factors including GRHL1, TEAD1, and FOXO3. Epi_C2 showed distinct activation of ZNF415 STAT1, while Epi_C3 exhibited elevated activity of transcription factors including EGR1, BCLAF1, and cell cycle regulators such as MYC and ZNF580. These findings provide a detailed molecular characterization of intratumoral heterogeneity in cervical cancer, offering potential therapeutic targets for subtype-specific interventions.
Differences in intercellular communication across conditions reveal the pivotal role of SPP1+ macrophages in the TME
To investigate the differential cell–cell communication in the TME across different stages of HPV infection, we employed CellChat analysis. The dot plots generated revealed distinct patterns of incoming and outgoing interaction strengths among various cell types in different stages of HPV (Fig. 6A). Notably, in the CA_HPV stage, myeloid cells exhibited significantly stronger outgoing interaction strengths compared to other conditions. A comparative analysis of differential interactions revealed significant changes in both the number and strength of interactions among various cell types (Fig. 6B, Supplementary Fig. 3A). Myeloid cells showed notable differences in their interaction profiles, suggesting a shift in the cellular communication landscape as the disease progresses from HSIL_HPV to CA_HPV. The heatmaps illustrate the relative changes in interaction counts and strengths, with myeloid cells displaying overall increased interactions with other cell types, except for plasma cells, in the CA_HPV stage.

A Dot plots showing incoming and outgoing interaction strengths among cell types across different disease stages. B Heatmaps comparing differential interaction counts and strengths between HSIL_HPV and CA_HPV stages. C Dot plots showing communication probabilities between SPP1+ macrophages and epithelial. D Dot plots showing communication probabilities between SPP1+ macrophages and CD8 + T cells. E Dot plots showing communication probabilities between SPP1+ macrophages and exhausted CD4 + T cells. F Violin plots showing expression of key ligands (SPP1, FN1) and receptors (SDC1, CD44) across cell types and disease stages.
Given the importance of macrophages in the TME, we specifically examined the interactions involving SPP1+ macrophages (SPP1_mac), which has high abundance in CA_HPV. The dot plots highlight the communication probabilities between SPP1_mac and various epithelial cell types, both tumor and normal (Fig. 6C). In the CA_HPV stage, SPP1_mac showed enhanced interactions with epithelial tumor cells via several ligand-receptor pairs, especially FN1-SDC1 and FN1-SDC4, indicating a potential role in promoting tumor progression. Further analysis of SPP1_mac interactions with immune cells, such as CD8+ cytotoxic T lymphocytes (CTLs) and exhausted CD4 + T cells, revealed significant communication via ligand-receptor pairs such as FN1-CD44 and SPP1-CD44, which manifest specific upregulated signaling in CA_HPV (Fig. 6D, E). These interactions suggesting that SPP1_mac may contribute to immune modulation and tumor immune evasion in TME. Violin plots depicting the expression levels of key ligands (SPP1, FN1) and receptors (SDC1, CD44) across different cell types and HPV stages provided further insights into the cellular sources and targets of these interactions (Fig. 6F). SPP1 expression was predominantly observed in myeloid cells, while FN1 was expressed across multiple cell types, including fibroblasts and endothelial cells. Receptors SDC1 and CD44 were highly expressed in epithelial and myeloid cells, respectively, underscoring their involvement in cell-cell communication within the TME. The enhanced interactions between SPP1_mac and epithelial tumor cells, as well as immune cells, highlight the complex interplay within the TME and suggest potential targets for therapeutic intervention.
Spatial analysis of tumor microenvironment in cervix cancer
Using the cell2location framework, we deconvoluted the spatial transcriptomic data to infer the cell abundance of various cell types within the CA_HPV TME (Fig. 7A, Supplementary Fig. 3B). MISTy analysis was further employed to identify spatial niches and analyze cell-cell interactions based on spatial distances. The spatial distribution maps showed the co-localization in the juxta_2 view. Consistent with our cell communications analysis by CellChat, which displayed the SPP1+ macrophages forming spatial niches with CD4-exhausted T cells expressing CTLA4 and PDCD1, and CD8+ cytotoxic T lymphocytes (CTLs) expressing GZMB and GZMK in the TME (Fig. 7B, C, Supplementary Fig. 3C, D).

A Spatial distribution map showing cell type abundance in cervical cancer tissue sections using the cell2location framework. B, C Spatial niches and cell-cell interactions identified by MISTy analysis, showing SPP1+ macrophages forming spatial niches with CD4-exhausted T cells and CD8+ cytotoxic T lymphocytes. D Spatial expression maps of SPP1, FN1, and CD44 in cervical cancer tissue sections. E–H Signaling flow and abundance of specific ligand-receptor interactions explored using Commot. I, J Kaplan-Meier survival curve for cervical cancer patients based on SPP1 and FN1 expression levels. K Box plot showing SPP1 expression across different pathological T stages of cervical cancer.
We furtherly focus on the key ligands and receptors expression. Spatial distribution maps of SPP1, FN1, and CD44 expression levels further confirmed their prominent expression in the tumor communication hotspot regions, correlating with the identified interaction networks (Fig. 7D, Supplementary Fig. 3E).
Using Commot, we explored the signaling flow and abundance of specific ligand-receptor interactions within the spatial transcriptomic data. The network diagram highlighted the complex communication between various cell types, with SPP1_mac emerging as a central player in the TME (Fig. 7E–H, Supplementary Fig. 3E–I). Key interactions involving SPP1, FN1, and CD44 were identified, indicating their roles in cell-cell communication. These findings underscore the importance of these molecules in the spatial organization and communication within the TME.
Survival analysis indicated that high expression levels of SPP1 and FN1 were associated with poorer overall survival in patients with CA_HPV, as shown by the Kaplan-Meier plots (Fig. 7I, J). Additionally, box plots of SPP1 expression across different pathological T stages revealed a significant increase in SPP1 expression with advancing tumor stages (Fig. 7K). These results suggest that SPP1 and FN1 could serve as potential prognostic biomarkers and therapeutic targets in CA_HPV.
Discussion
Our comprehensive analysis has provided profound insights into the cellular and molecular landscape of cervical cancer progression. By elucidating the dynamic changes in cellular composition, functional states, and intercellular communication networks, our findings have significant implications for understanding the pathogenesis of cervical cancer and developing targeted therapeutic strategies.
Cervical cancer is characterized as an immune-infiltrated but immunosuppressive cancer type, primarily due to the modulation of the TME by HPV20. HPV infection plays a crucial role in immune evasion strategies employed by cervical cancer cells. These include downregulation of major histocompatibility complex (MHC) genes, inhibition of the cGAS pathway, and recruitment of immunosuppressive cells to the TME20. Our data indicate that HPV-positive cervical cancer samples exhibited higher proportions of myeloid cells, including SPP1+ macrophages, compared to HPV-negative samples, suggesting that HPV infection may drive the recruitment and polarization of these immunosuppressive myeloid cells.
SPP1 (secreted phosphoprotein 1, also known as osteopontin) is a secreted non-collagenous glycoprotein highly expressed in tumor and stromal cells, with elevated levels in serum, plasma, and tumor tissues associated with poor prognosis in multiple cancer types21. Our analysis revealed that SPP1 was overexpressed in cervical cancer, consistent with previous findings showing SPP1 overexpression in numerous cancers, validating our findings of SPP1’s prognostic value22.
Macrophages in the TME can exhibit a spectrum of functional states, traditionally categorized as M1 (pro-inflammatory and anti-tumor) and M2 (anti-inflammatory and pro-tumor) phenotypes23. However, recent single-cell studies have revealed that this dichotomy is oversimplified, and macrophage populations are much more complex24. Recent research has revealed that the CXCL9:SPP1 (CS) macrophage polarization paradigm significantly outperforms the conventional M1/M2 classification in prognostic assessment of head and neck squamous cell carcinoma (HNSCC)25. Emerging scRNA-seq data reveal that SPP1+ macrophages dominate the immunosuppressive TME across various malignancies, including Hepatocellular carcinoma (HCC), glioblastoma (GBM), and colorectal cancer12,13,26. In the context of cervical cancer, we observed an activation of ERK1/ERK2 pathway in SPP1+ macrophages, which been implicated in several immunosuppressive processes, including the regulation of cytokine production, cell survival, and cellular metabolism27. Our data indicate that SPP1+ macrophages leverage this pathway to exhibit a unique transcriptional signature characterized by upregulated APOE (involved in lipid metabolism) and GPNMB (linked to lysosomal dysfunction), which related to synergistically impair antigen presentation by dendritic cells (DCs) and cytokine secretion to enhance tumor cell survival28,29,30,31. The increased abundance of SPP1+ macrophages in cervical cancer compared to precancerous and normal tissues further supports their role in cervix cancer progression. This novel finding not only advances our understanding of macrophage heterogeneity in tumor biology but also unveils promising therapeutic opportunities by targeting this specific cellular subset.
The interaction between SPP1 and CD44 significantly also impairs anti-tumor immunity in cervical cancer. The interactions via SPP1-CD44 between SPP1+ macrophages and CTLs/exhausted CD4 + T cells may contribute to the immunosuppressive environment by inhibiting CTL function and promoting T cell exhaustion32. Analysis of intratumoral T cells has revealed that SPP1-CD44 signaling hinders sustained T cell proliferation, with a negative correlation observed between SPP1 expression in malignant cells and CD44 expression in T cells with T cell proliferation scores16. This ligand-receptor interaction also contributes to the formation of an immunosuppressive microenvironment that facilitates tumor evasion from immune surveillance. Beyond its effects on tumor and immune cells in our study, the SPP1-CD44 axis influences the broader TME by modulating stromal components. SPP1 interacts with various components of the extracellular matrix and contributes to the formation of a pro-tumorigenic stroma that supports cancer progression33,34. The downstream signaling events induce cytoskeletal reorganization and upregulation of matrix metalloproteinases, facilitating tumor cell invasion through the extracellular matrix33. Additionally, SPP1-CD44 signaling promotes epithelial-mesenchymal transition through activation of the JAK2/STAT3 pathway, further enhancing metastatic potential35. In cervical cancer, this may results in increased tumor fibrosis and altered matrix stiffness, which further promotes tumor cell invasion and restricts immune cell infiltration.
Therefore, the SPP1-CD44 axis emerged as a critical pathway in tumor-promoting cellular interactions, representing a promising target for therapeutic intervention18. Multiple approaches could be employed to target this axis, including anti-SPP1 RNA aptamers like OPN-R336, or CD44-targeting agents such as RG735637,38. Similarly, for the FN1-CD44 axis which also observed in our study, targeting FN1 has shown promise in disrupting the tumor-supportive extracellular matrix39,40. Although there are fewer reports, based on our results, we speculate that the small molecule inhibitor RGDS peptide (Fibronectin inhibitor) for FN1 may have potential efficacy in inhibiting FN1-mediated signaling pathways and reducing tumor growth and metastasis. Therefore, the SPP1-CD44 axis represents a critical communication pathway in the cervical cancer TME, orchestrating complex interactions between tumor cells, macrophages, and other immune components that collectively promote tumor progression and immune evasion. Therapeutic targeting of this axis, through direct inhibition of ligand-receptor interactions or modulation of downstream signaling pathways, holds considerable promise for improving treatment outcomes in cervical cancer patients. As our understanding of the SPP1-CD44 axis continues to evolve, further refinement of targeting strategies may lead to more effective and personalized therapeutic approaches for cervical cancer.
Although our study provides valuable insights into the SPP1-CD44 signaling axis in cervical cancer progression, we acknowledge several important limitations, primarily concerning our sample size. The current analysis was performed using a limited cohort of samples, with only 9 specimens across different cervical pathology states. The SPP1-CD44 signaling patterns we observed may represent only a subset of potential interaction networks present across a broader patient population. Previous research has demonstrated considerable variability in immune cell infiltration and molecular profiles among cervical cancer patients, suggesting our findings may not comprehensively represent this heterogeneity. The limited sample diversity affects our capacity to draw conclusions about the relationship between SPP1-CD44 signaling and response to different therapeutic modalities. The involvement of this axis in immune checkpoint inhibitor resistance and chemotherapy response, as suggested by our data and other studies15,41, necessitates investigation in larger patient populations with diverse treatment histories. To address these limitations, future studies should incorporate larger, more diverse patient cohorts with comprehensive clinical annotation. Multi-center collaborative efforts would enable the collection of sufficient samples across all disease stages and treatment responses.
Our integrative analysis of scRNA-seq and stRNA-seq data provides a comprehensive understanding of the cellular and molecular landscape of cervical cancer, highlight the critical role of SPP1+ macrophages and the ERK1/ERK2 cascade in shaping the immune landscape of cervical cancer. By elucidating the dynamic changes in cellular composition, functional states, and intercellular communication networks, our study offers valuable insights into the mechanisms driving cervical cancer progression and identifies potential targets for therapeutic intervention. Future studies should focus on developing specific inhibitors of this pathway and evaluating their efficacy in preclinical models of cervical cancer, with the ultimate goal of translating these findings into novel therapeutic approaches for patients.
Methods
Data collection
The scRNA-seq (single-cell RNA sequencing) dataset retrieved from GSE20865342 comprises two uninfected normal samples (GSM6360680, GSM6360681), two normal samples with HPV infection (GSM6360682, GSM6360683), two HSIL samples with HPV infection (GSM6360684, GSM6360685), and three cervical cancer samples with HPV infection (GSM6360686, GSM6360687, GSM6360688).
The stRNA-seq (spatial transcriptome RNA sequencing) data of the two cervical cancer samples from GSE20865442 (GSM6360691, GSM6360692) were analyzed using the 10x Space Ranger platform.
The bulk RNA-seq information sourced from TCGA-CESC underwent a transformation process where raw gene expression counts were converted to Transcripts Per Million (TPM) values and subsequently normalized through log2(TPM + 1) conversion.
Standardized analysis of scRNA-seq and stRNA-seq data
Prior to integration, quality control was conducted on the scRNA-seq data. Cells with low UMI counts ( <200), gene expression outside the range of 200 to 5000, and high levels of mitochondrial-derived UMI counts ( >15%) were removed to eliminate potential noise and artifacts. The scRNA-seq data was then integrated using Harmony43(v1.2.1). The stRNA-seq data was preprocessed using the sctransform method, which normalizes the data, removes technical variation, and identifies highly variable genes within each cell. All standardized analyses were performed according to the official guidelines of Seurat44 (v4.4.0), except where otherwise specified.
Pseudotime trajectory and cell stemness analysis
The trajectory analysis was performed using Monocle 245 (v2.16.0) package to reveal cell-state transitions. Additionally, the CytoTRACE46 (v0.3.3) package was used to infer cell stemness and differentiation potential.
Enrichment analysis and gene set scoring
The functional enrichment analysis and the gene set enrichment analysis47 (GSEA) were performed using the clusterProfiler48 package (v4.6.2). Gene set scoring was conducted using the “AddModuleScore” function in Seurat. The background gene set for GO (Gene Ontology, GO) (c5.go.bp.v7.4.symbols) was downloaded from the Molecular Signatures Database (MSigDB) (Supplementary Data 1). Additionally, the HALLMARK_INFLAMMATORY_RESPONSE and HALLMARK_GLYCOLYSIS gene sets from MSigDB were used (Supplementary Table 2, 3).
Copy number alteration analysis
The infercnv49 (v0.1.6.0) R package was used to infer the copy number alterations of epithelial cells in tumor samples. By leveraging gene expression data, inferCNV can estimate the underlying genomic alterations that lead to the observed expression profiles in epithelial cells of tumor samples. Subsequently, K-means clustering was performed for tumor cell identification.
Transcription factors analysis by pySCENIC
The regulation of gene expression is a fundamental process that dictates cellular identity, function, and response to environmental cues. Transcription factors (TFs) are key players in this regulatory landscape, orchestrating the precise expression of target genes in a context-dependent manner. The pySCENIC50,51 (v0.12.1) software was used to infer the TF regulatory network from scRNA-seq data.
Cells metabolism inference of scRNA-seq data
To evaluate the cells’ metabolic features from scRNA-seq data, we used the scMetabolism52 (v0.2.1) R package to assess overall metabolic activity based on KEGG metabolic pathways. Additionally, we employed the scFEA53 (v1.1.2) algorithm to infer the direction of metabolic signaling flux.
Cells communication analysis of scRNA-seq data
The advent of scRNA-seq has revolutionized our understanding of cellular heterogeneity and the intricate communication networks among cells. To analyze cell-to-cell interactions, we used the CellChat54 (v1.6.1) R package to infer and visualize cell communication networks from scRNA-seq data.
Cell type deconvolution of stRNA-seq data by cell2location
The cell2location is an innovative computational framework designed to leverage spatial transcriptomic data alongside scRNA-seq datasets. We used cell2location55 (v0.1.3) with cervix cancer scRNA-seq data as a reference to infer the cell abundance of each spot in the stRNA-seq data. The model was trained for 30,000 epochs, with the parameters set to “N_cells_per_location=20” and “detection_alpha=20”.
Spatial niche and cells distance analysis
MISTy (Multi-view Interpretable Spatio-Temporal) is a modeling approach that analyzes cell-cell interactions through different spatial contexts (views), enabling a comprehensive understanding of the spatial niche and cellular interactions within a sample. We used the MISTyR56 (v1.6.1) package to identify spatial niches in stRNA-seq data based on the spatial distance between cells.
Spatial cells interactions and signaling flow
Deciphering cell-to-cell communication signal flow is important for investigating the tumor microenvironment (TME). Using Commot57 (v0.0.3), we explored the abundance of specific ligand and receptor interactions in stRNA-seq data and inferred the signaling direction.
Statistical Analysis
The statistical framework for this study was implemented using Python and R (v4.2.2) programming environments. Hypothesis testing was performed with method-specific approaches: categorical variable comparisons utilized Fisher’s exact test, multi-group contrasts employed one-way ANOVA, non-parametric evaluations leveraged the Wilcoxon rank-sum procedure, and survival outcomes were analyzed via log-rank testing. Statistical significance thresholds were established at P < 0.05.
Data availability
The datasets utilized for the bioinformatics analysis in our study are available from the TCGA database (https://www.cancer.gov/ccg/research/genome-sequencing/tcga) and GEO (https://www.ncbi.nlm.nih.gov/geo/).
Code availability
All code generated for analysis is available from the github repository: https://github.com/BioinfoXP/spp1_cc_code/tree/master.
References
Stelzle, D. et al. Estimates of the global burden of cervical cancer associated with HIV. Lancet Glob. Health 9, e161–e169 (2021).
Guida, F. et al. Global and regional estimates of orphans attributed to maternal cancer mortality in 2020. Nat. Med. 28, 2563–2572 (2022).
de Sanjose, S. et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol. 11, 1048–1056 (2010).
Cohen, P. A., Jhingran, A., Oaknin, A. & Denny, L. Cervical cancer. Lancet 393, 169–182 (2019).
Hao, B. et al. Single-cell RNA sequencing analysis revealed cellular and molecular immune profiles in lung squamous cell carcinoma. Transl. Oncol. 27, 101568 (2023).
Sheng, B. et al. Single-cell RNA sequencing of cervical exfoliated cells reveals potential biomarkers and cellular pathogenesis in cervical carcinogenesis. Cell Death Dis. 15, 130 (2024).
Chen, Q., Deng, D., Zhu, H. & Li, S. Single-cell transcriptomics unveils multifaceted immune heterogeneity in early-onset versus late-onset cervical cancer. World J. Surg. Oncol. 23, 12 (2025).
Li, C., Guo, L., Li, S. & Hua, K. Single-cell transcriptomics reveals the landscape of intra-tumoral heterogeneity and transcriptional activities of ECs in CC. Mol. Ther. - Nucleic Acids 24, 682–694 (2021).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Xu, L. et al. Tumor-associated macrophage subtypes on cancer immunity along with prognostic analysis and SPP1-mediated interactions between tumor cells and macrophages. PLOS Genet. 20, e1011235 (2024).
Wu, J., Shen, Y., Zeng, G., Liang, Y. & Liao, G. SPP1+ TAM subpopulations in tumor microenvironment promote intravasation and metastasis of head and neck squamous cell carcinoma. Cancer Gene Ther. 31, 311–321 (2024).
Liu, Y. et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J. Hepatol. 78, 770–782 (2023).
Wei, J. et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Invest. 129, 137–149 (2018).
Liu, F., Zhang, J., Gu, X., Guo, Q. & Guo, W. Single-cell transcriptome sequencing reveals SPP1-CD44-mediated macrophage–tumor cell interactions drive chemoresistance in TNBC. J. Cell. Mol. Med. 28, e18525 (2024).
Tong, W. et al. Spatial transcriptomics reveals tumor-derived SPP1 induces fibroblast chemotaxis and activation in the hepatocellular carcinoma microenvironment. J. Transl. Med. 22, 840 (2024).
Cheng, M. et al. Immunosuppressive role of SPP1-CD44 in the tumor microenvironment of intrahepatic cholangiocarcinoma assessed by single-cell RNA sequencing. J. Cancer Res. Clin. Oncol. 149, 5497–5512 (2023).
Li, G. et al. PPARG/SPP1/CD44 signaling pathway in alveolar macrophages: mechanisms of lipid dysregulation and therapeutic targets in idiopathic pulmonary fibrosis. Heliyon 11, e41628 (2025).
Zhang, J. et al. Single-cell and spatial transcriptomics reveal SPP1-CD44 signaling drives primary resistance to immune checkpoint inhibitors in RCC. J. Transl. Med. 22, 1157 (2024).
Lavoie, H., Gagnon, J. & Therrien, M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 21, 607–632 (2020).
Cao, G. et al. Single-cell dissection of cervical cancer reveals key subsets of the tumor immune microenvironment. EMBO J. 42, e110757 (2023).
Lamort, A.-S., Giopanou, I., Psallidas, I. & Stathopoulos, G. T. Osteopontin as a link between inflammation and cancer: the thorax in the spotlight. Cells 8, 815 (2019).
Zhao, K., Ma, Z. & Zhang, W. Comprehensive analysis to identify SPP1 as a prognostic biomarker in cervical cancer. Front. Genet. 12, 732822 (2022).
Mantovani, A., Sozzani, S., Locati, M., Allavena, P. & Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 (2002).
Revel, M., Sautès-Fridman, C., Fridman, W.-H. & Roumenina, L. T. C1q+ macrophages: passengers or drivers of cancer progression. Trends Cancer 8, 517–526 (2022).
Bill, R. et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 381, 515–524 (2023).
Qi, J. et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 13, 1742 (2022).
Lucas, R. M., Luo, L. & Stow, J. L. ERK1/2 in immune signalling. Biochem. Soc. Trans. 50, 1341–1352 (2022).
Chen, K. et al. Identification of a novel subtype of SPP1 + macrophages expressing SIRPα: implications for tumor immune evasion and treatment response prediction. Exp. Hematol. Oncol. 13, 119 (2024).
Bonacina, F. et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat. Commun. 9, 3083 (2018).
Tsou, P. & Sawalha, A. H. Glycoprotein nonmetastatic melanoma protein B: a key mediator and an emerging therapeutic target in autoimmune diseases. FASEB J. 34, 8810–8823 (2020).
Wallings, R. L. et al. ASO-mediated knock-down of GPNMB in mutant-GRN and in Grn-deficient peripheral myeloid cells disrupts lysosomal function and immune responses. Mol. Neurodegener. 20, 41 (2025).
Klement, J. D. et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest. 128, 5549–5560 (2018).
Glaviano, A. et al. Harnessing the tumor microenvironment: targeted cancer therapies through modulation of epithelial-mesenchymal transition. J. Hematol. Oncol. J. Hematol. Oncol. 18, 6 (2025).
Yang, Y. et al. Tumor-associated-fibrosis and active collagen-CD44 axis characterize a poor-prognosis subtype of gastric cancer and contribute to tumor immunosuppression. J. Transl. Med. 23, 123 (2025).
Huang, Z. et al. SPP1-mediated M2 macrophage polarization shapes the tumor microenvironment and enhances prognosis and immunotherapy guidance in nasopharyngeal carcinoma. Int. Immunopharmacol. 147, 113944 (2025).
Mi, Z. et al. RNA aptamer blockade of osteopontin inhibits growth and metastasis of MDA-MB231 breast cancer cells. Mol. Ther. 17, 153–161 (2009).
Menke-van der Houven van Oordt, C. W. et al. First-in-human phase I clinical trial of RG7356, an anti-CD44 humanized antibody, in patients with advanced, CD44-expressing solid tumors. Oncotarget 7, 80046–80058 (2016).
Zhang, S. et al. Targeting chronic lymphocytic leukemia cells with a humanized monoclonal antibody specific for CD44. Proc. Natl. Acad. Sci. USA 110, 6127–6132 (2013).
Zhou, Y., Shu, C. & Huang, Y. Fibronectin promotes cervical cancer tumorigenesis through activating FAK signaling pathway. J. Cell. Biochem. 120, 10988–10997 (2019).
Meng, M. et al. Cancer/testis-45A1 promotes cervical cancer cell tumorigenesis and drug resistance by activating oncogenic SRC and downstream signaling pathways. Cell. Oncol. 47, 657–676 (2024).
Matsubara, E. et al. The significance of SPP1 in lung cancers and its impact as a marker for protumor tumor-associated macrophages. Cancers 15, 2250 (2023).
Guo, C. et al. Spatiotemporally deciphering the mysterious mechanism of persistent HPV-induced malignant transition and immune remodelling from HPV-infected normal cervix, precancer to cervical cancer: integrating single-cell RNA-sequencing and spatial transcriptome. Clin. Transl. Med. 13, e1219 (2023).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 16, 284–287 (2012).
Tirosh, I. et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Bravo González-Blas, C. et al. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods 20, 1355–1367 (2023).
Wu, Y. et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov. 12, 134–153 (2022).
Alghamdi, N. et al. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome Res. 31, 1867–1884 (2021).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2020).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
Tanevski, J., Flores, R. O. R., Gabor, A., Schapiro, D. & Saez-Rodriguez, J. Explainable multiview framework for dissecting spatial relationships from highly multiplexed data. Genome Biol. 23, 97 (2022).
Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant numbers 82071695, 82203630, and 82471640], the Young Scientific and Technological Talent Innovation Project of Lanzhou [grant number 2023-QN-79], and the Gansu Province Traditional Chinese Medicine Research Topics [grant number GZKZ-2022-8].
Author information
Authors and Affiliations
Contributions
Conceptualization, P.X., J.Z.; methodology and data analysis, P.X.; writing—original draft preparation, P.X., J.Z.; writing—review and editing, P.X., J.Z., R.S., and D.W.; visualization, P.X.; supervision, R.S. and D.W.; funding acquisition, R.S. and D.W. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study does not involve research with human participants, as it is based on publicly available data. Therefore, ethical approval and informed consent are not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xia, P., Zhou, J., Shen, R. et al. Deciphering the cellular and molecular landscape of cervical cancer progression through single-cell and spatial transcriptomics. npj Precis. Onc. 9, 158 (2025). https://doi.org/10.1038/s41698-025-00948-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00948-z
This article is cited by
-
Advancements in single-cell sequencing for cervical cancer research
Molecular and Cellular Biochemistry (2025)
