
Abstract
Multiple myeloma (MM) arises from abnormal plasma cells (PCs) progressing from precursor states, including monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Understanding this transition and progression to overt MM requires improved non-invasive strategies. We employed a liquid biopsy approach to detect and characterize circulating PCs across disease states in 68 patients (MGUS = 11, SMM = 21, NDMM = 19, RRMM = 17) using multi-channel immunofluorescence staining and machine learning-assisted rare event detection. PCs were identified by CD138 and B-cell maturation antigen (BCMA) expressions, with distinct phenotypic subpopulations stratifying disease states. The D | CD138 | BCMA-Memb phenotype was the most predictive, with incidence increasing from MGUS to SMM and overt MM (p < 0.005). Multivariate modeling distinguished precursors from overt disease with 86% accuracy. Shifts in BCMA and CD45 expression suggested immune cell profile alterations with progression and treatment. These findings underscore PB-based liquid biopsy as a promising tool for MM detection and monitoring, revealing circulating PC heterogeneity.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a hematologic malignancy caused by the clonal proliferation of plasma cells (PCs). In 2024 alone, MM was projected to account for approximately 35,780 new cases and 12,540 deaths in the United States1. The clinical trajectory of MM involves two precursor states: monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Over time, these precursor states can progress to overt symptomatic MM (newly diagnosed; NDMM) or relapsed/refractory MM (RRMM). Diagnosing progression to MM early remains a significant challenge due to its mostly asymptomatic nature in initial stages and the disease’s inherent heterogeneity2,3,4. Moreover, despite recent advancements, such as B-cell maturation antigen (BCMA)-targeted therapies, MM continues to exhibit high relapse rates5,6,7,8. Improved methods are needed to monitor disease progression and treatment efficacy.
Traditionally, MM diagnosis and monitoring rely on bone marrow (BM) evaluation via aspirate and biopsy9. This procedure is invasive with the potential for complications thus being impractical for repetitive disease evaluation. Additionally, it is usually a blind sample from a random site at the iliac crest which does not capture the heterogeneity of the BM due to the patchy myeloma infiltration. In contrast, peripheral blood (PB) liquid biopsy offers a promising, minimally invasive alternative for disease detection and monitoring by analyzing circulating tumor-related analytes released into the blood stream from the BM. For example, cell-free DNA (cfDNA) analysis has shown potential in monitoring minimal residual disease (MRD) and predicting progression-free survival in advanced MM10,11,12. Similarly, the detection of circulating myeloma cells and exosomes in PB have been correlated with disease progression and poor overall survival13,14,15,16,17,18,19 even though circulating myeloma cells are found at a lower incidence in the PB than the BM20,21. PB has the potential to provide dynamic insights into disease burden from the entirety of the BM compartment, progression, and molecular characteristics, enabling real-time surveillance and facilitating personalized treatment strategies.
Traditional methods for detecting circulating myeloma cells, such as flow cytometry and enrichment-based techniques, are limited in sensitivity and may overlook clinically significant subpopulations of circulating myeloma cells, such as CD138-low expressing cells with greater clonogenic potential. To address these limitations, we utilized the High-Definition Single Cell Assay (HDSCA) workflow as a non-enrichment liquid biopsy tool to analyze PB samples from 68 patients across the MM disease spectrum, leveraging an immunofluorescence assay targeting CD138, BCMA, and CD45 to identify a diverse population of tumor-associated circulating PCs and differentiate disease states. The findings highlight the potential of PB analyses to enhance MM detection and monitoring, particularly in precursor stages, such as MGUS and SMM, thus providing a comprehensive, minimally invasive method for disease surveillance.
Results
Patient demographics and clinical baseline
A total of 68 blood draws were collected for this study with 1 sample per patient. All patients enrolled underwent diagnostic evaluation of their disease state: 11 MGUS, 21 SMM, 19 NDMM, and 17 RRMM patients. Patient demographics and clinical data are provided in Table 1 (additional details from clinical workup are provided in Supplementary Table 1).
Identification and enumeration of cells of interest
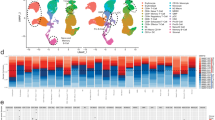
Circulating rare cells of interest were identified and classified based on their biomarker signal expression and localization following our four-channel immunofluorescence staining corresponding to D, CD138, BCMA, and CD45 (Fig. 1). Circulating PCs were defined by the expression of CD138 and/or BCMA.

The composite image and each individual biomarker channels are provided. The cell groups were classified into 12 classes based on the combination of DAPI (D) and 3 markers: CD138, BCMA (membrane [Memb] and perinuclear [Peri] localization), and CD45. A D | CD138 | BCMA | CD45, B D | CD138 | BCMA, C D | BCMA | CD45, D D | CD138 | CD45, E D | CD138, F D | BCMA, G D | CD45, H DAPI-only. BCMA membrane and perinuclear localization examples provided in the left and right columns, respectively. Images taken at 100x magnification. Blue: DAPI, Red: CD138, White: BCMA, Green: CD45.
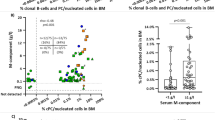
PB samples taken after diagnosis were compared across the disease spectrum to determine the presence of circulating PCs (Fig. 2). Table 2 shows the cell types with significant differences between disease states. MGUS was distinguished from NDMM by total cells (p-value = 0.0150), total CD138+ (p-value = 0.0409), total BCMA+ (p-value = 0.0190), total BCMA-Memb (p-value = 0.0169), D | CD138 (p-value = 0.0081), D | CD138 | BCMA-Memb (p-value = 0.0021), and D | BCMA-Memb|CD45 (p-value = 0.0169) cells. SMM was distinguished from NDMM by total BCMA-Peri (p-value = 0.0294), D | BCMA-Peri|CD45 (p-value = 0.0190) and D | BCMA-Memb|CD45 (p-value = 0.0229) cells.

A Enumeration and B frequency of circulating rare cells based on channel classification. C Boxplot of cellular distribution per disease state for comparisons that are significantly different (p-value < 0.05). All cellular comparisons are provided in Supplementary Fig. 1. Black line is median, boxed area is the interquartile range (IQR), error-bars show the complete range of the data. D: DAPI, 138: CD138, 45: CD45, Peri: perinuclear localization of BCMA, Memb: membrane bound BCMA.
There was one specific cellular phenotype that was the most significant at stratifying the disease states in which incidence increased with disease status: D | CD138 | BCMA-Memb. These cells were able to stratify MGUS from the overt disease states (NDMM [p-value = 0.0021] and RRMM [p-value = 0.0038]), while also presenting at a significantly higher count in SMM than MGUS (p-value = 0.0044). This indicates the potential to monitor transition between precursor states and overt disease using PB. Interestingly, NDMM and RRMM were stratified by total BCMA-Peri (p-value = 0.0043), D | BCMA-Memb|CD45 (p-value = 0.0324), and D | BCMA-Peri|CD45 cells (p-value = 0.0036), all of which NDMM samples had a significantly higher incidence of cells compared to RRMM. This suggests a potential shift in the cellular profile after treatment. Overall, this data indicates there are unique PCs present in the PB across the disease states.
Cellular morphometrics
The heterogeneity of the rare cell populations detected in PB across different disease states was analyzed using cellular morphometrics. Two-dimensional UMAP plots are presented in Fig. 3A, where each rare cell is represented as a single point. These plots illustrate the progression of CD138+ PCs, which increase in abundance as the disease advances. Additional distributions of cellular and nuclear area, eccentricity, and median channel intensities for each disease state are shown in Fig. 3B, further highlighting the morphological diversity and variability of the rare cell populations across disease progression. For example, the observed total BCMA signal intensity, which includes both membrane and perinuclear contributions, reveals three distinct peaks. While the density plot does not distinguish signal localization, imaging suggests that these peaks may correspond to functionally distinct BCMA+ PC subtypes, with a notable transition to higher signal intensity in RRMM. Conversely, observed CD45 signal intensity shifts toward lower expression levels as the disease status becomes more aggressive. Interestingly, the nuclear and cellular areas and eccentricities remain relatively stable, with only minor observable variations from precursor to overt disease states. These findings underscore the morphological and functional complexity of the circulating PC populations as MM progresses.

A Morphology UMAP plot of rare cells by channel-type classification and separated by disease state: MGUS, SMM, NDMM, RRMM. B Probability density distribution plots for select morphometric parameters across channel-type classifications of median CD138 signal intensity, median BCMA signal intensity, median CD45 signal intensity, nuclear area, nuclear eccentricity, cellular area, and cellular eccentricity.
Correlations between the circulating cell subsets and clinical metrics
Correlation analyses were performed to investigate potential clinical associations between circulating rare cells identified in PB samples and the various clinical data metrics, which included the diagnostic parameters (based on BM) and survival information. The significant correlations observed are summarized in Table 3. No significant associations were found for variables, such as gender, ethnicity, or race.
The D | CD138 | BCMA-Memb phenotypic cells were the most commonly correlated PB analyte with most of the BM diagnostic measurements, which lead to the patient diagnosis. Additionally, the D | CD138, D | CD138 | BCMA-memb|CD45, D | CD138 | CD45 phenotypes correlated with disease survival. Interestingly, the total cells and total BCMA+ cells correlated with age, which suggests BCMA phenotypic changes in the immune cell profile independent of disease state as the body ages.
Patient level prediction
To determine if the PB could be used to detect precursor states from overt disease in an individual, we conducted both univariate and multivariate analyses (Supplementary Table 2) which indicated that the D | CD138 PCs were the highest single predictor (accuracy = 79%) and most important cellular phenotype for both decision tree and random forest (accuracy = 86%) in determining progression. When considering stratification of precursor states with only NDMM, the D | CD138 PCs was the highest predictor (accuracy = 80%). For the multivariate model, the decision tree and random forest predicted with 70% accuracy, both indicating the D | BCMA-Memb|CD45 cells as the most important feature. Similarly, we wanted to determine if the distinct precursor states could be predicted by PB analytes. The D | CD138 | BCMA-Memb PCs had a 71% accuracy in predicting the transition from MGUS to SMM for the univariate model and was the most important predictor in the decision tree model (accuracy = 71%). Together these findings highlight the potential utility of PB PCs as predictors of disease with tailored modeling approaches for optimized accuracy based on the clinical question, underscoring the value of heterogenous PCs in early detection and monitoring of disease status.
Discussion
In this proof-of-concept study, we have demonstrated the feasibility of detecting circulating rare cells, including a variety of PCs, in the blood of patients with MM and its precursor conditions. Slide-based immunofluorescence methods similar to what we presented here have detected circulating myeloma PCs in 19, 25, and 80% of MGUS, SMM, and NDMM, respectively22,23,24,25. Using a non-enrichment approach, we have characterized the spectrum of PCs (malignant and non-malignant) and rare cells to show the transition of cells in the PB across disease states with a prediction accuracy of 86% by multivariate analysis. Further, the correlations between PC subtypes detected in the PB and BM flow cytometry measurements support a biologically relevant link between compartments. These exploratory results underscore the transformative potential of the PB liquid biopsy as a non-invasive diagnostic and monitoring tool. We identified cell types that not only stratify disease stages but also correlate with clinical parameters by leveraging multi-channel immunofluorescence and rigorous phenotypic characterization. Unlike traditional diagnostic methods that rely on BM, which are invasive, painful, and associated with potential complications, the PB offers a quick, minimally invasive alternative. By minimizing discomfort and the risks associated with invasive sampling, PB can enhance patient compliance and facilitate more frequent monitoring, which is critical for managing a disease with a high risk of progression and relapse.
Our findings highlight the potential utility of specific circulating cell subsets, particularly D | CD138 | BCMA-Memb phenotypic PCs, in differentiating between precursor states (MGUS and SMM) and overt disease (NDMM and RRMM). This cellular phenotype demonstrated the most significant ability to stratify disease states, with incidence progressively increasing from MGUS to SMM and overt disease. Notably, D | CD138 | BCMA-Memb PCs also distinguished MGUS from SMM, emphasizing their potential utility in identifying subtle transitions within precursor states (p-value = 0.0044). Importantly, the ability to identify myeloma PCs in precursor conditions like MGUS and SMM presents an unprecedented opportunity for early detection and intervention. A prior study found that patients with MGUS could be divided into two groups, one likely to progress to advanced disease and another that remains stable over time26. Univariate and multivariate prediction analyses achieved moderate accuracy (71%), reflecting the phenotypic overlap and biological uncertainty in this early-stage transition, which mirrors the clinical overlap and the challenge of defining progression within precursor states, which underscores a critical area for future research. The transition from these precursor states to symptomatic MM is typically recognized only after significant organ damage or clinical symptoms emerge27,28, but a PB-based approach could shift this paradigm by enabling real-time tracking of disease evolution. Early identification of patients at high risk for progression could allow clinicians to implement preventative or therapeutic measures earlier, potentially delaying or preventing symptomatic MM, ultimately improving patient outcomes by reducing disease burden and preserving organ function. This is especially important in SMM where registrational trials are underway that could lead to an FDA approved treatment that will revolutionize our standard of care approach to precursor states.
It is important to note that other phenotypic PC classifications provided insights into the biological transition. BCMA signal intensity revealed three distinct subtypes, with higher expression in RRMM, while CD45 intensity decreased as disease status became more aggressive, highlighting the functional complexity of circulating PCs in MM. These findings suggest that treatment impacts the cellular profile, with RRMM samples showing a distinct shift compared to treatment-naïve NDMM samples. Such shifts in cellular profiles post-treatment could provide insights into mechanisms of treatment resistance and guide therapeutic decision-making. Correlation analyses further revealed that total cells and BCMA+ PCs were associated with age, indicating BCMA-related phenotypic changes in immune cell profiles that may occur independently of disease state. These findings suggest that age-related immune alterations could contribute to MM progression and warrant further investigation.
Moreover, PB facilitates longitudinal monitoring, providing a dynamic view of disease progression, therapeutic response, and the emergence of resistance. By allowing frequent sampling and analysis of PCs, clinicians can gain insights into the molecular and cellular changes driving the disease, enabling adaptive treatment strategies tailored to individual patient needs. This capability is particularly valuable in MM as a highly heterogeneous disease composed of various molecular subgroups, each characterized by an assortment of genomic alterations with evolving treatment landscapes29,30, including therapies targeting BCMA. Detailed characterization of the genetic, epigenetic, and phenotypic profiles of these myeloma PCs could shed light on the pathways driving disease initiation, progression, and resistance to therapy. Such insights could guide the identification of novel therapeutic targets and inform the development of more effective treatment strategies tailored to specific disease subtypes or progression patterns. Previous studies assessing circulating MM cells typically use flow cytometry and have shown limitations in the utility of the PB compared to BM, especially in detecting MRD31,32,33. The method used here provides an unbiased cellular detection to profile rare cells with the potential for downstream molecular analyses34,35,36, showing potential clinical promise for PB as a tool to assess treatment efficacy and detect early signs of relapse, thus ensuring timely intervention and optimizing therapeutic outcomes.
While a direct comparison to established platforms, such as CellSearch or flow cytometry would provide valuable context, it was not feasible within the scope of this study due to limitations in sample volume, IRB protocol constraints, and distinct pre-analytical requirements for each platform. Nonetheless, our approach offers several advantages that support its complementary and, in some contexts, superior analytical value. Most notably, our method demonstrates increased sensitivity in detecting rare PC subsets, including phenotypically diverse or low-abundance populations that may be missed by conventional marker-restricted approaches. Unlike flow cytometry, which typically requires immediate processing and may compromise cell integrity for downstream analyses, our platform preserves rare cells in a state suitable for single-cell genomic and proteomic profiling, allowing for a broader analytical scope. The platform used here is also antigen-agnostic and adaptable to diverse surface markers, enabling the detection of heterogeneous or low-abundance cell populations that may escape detection by marker-restricted platforms, such as CellSearch. Additionally, the workflow accommodates up to 48 h from blood draw to processing, offering logistical flexibility for multi-center or clinically integrated studies. We are currently planning future studies to assess the degree of overlap and complementarity between technologies, which will further define the unique contributions and potential clinical utility of our assay in the context of PC and rare cell analysis.
Despite its promise, this study has notable limitations that warrant further investigation. The relatively small sample size per disease state limits the generalizability of the findings, emphasizing the need for validation in larger and more diverse patient cohorts. Additionally, while the detection and characterization of myeloma PCs in blood samples represent significant advancements, further refinement of the techniques is essential to enhance assay sensitivity and specificity. Improving these methodologies will be critical to capturing the full spectrum of disease heterogeneity and ensuring robust clinical applicability. By enabling non-invasive, longitudinal tracking of disease dynamics, liquid biopsies may offer a patient-centered approach that enhances care delivery. Future analyses at the cohort level will explore this through serial sampling across patients. Furthermore, the molecular insights gained from these analyses can deepen our understanding of MM biology, paving the way for innovative therapeutic interventions and improved outcomes for patients. These findings underscore the importance of continued research to optimize liquid biopsy techniques and validate their utility across diverse clinical settings.
The main limitation of this study is the moderate follow-up time, which restricts our ability to correlate findings with standard survival metrics, which is a key objective of ongoing and future research. The follow-up time for patients was highly variable, reflecting the real-world nature of the cohort. Specifically, follow-up durations ranged from 0 days (in newly diagnosed patients) to 6 years, depending on the timing of enrollment and disease stage at the time of sample collection. Survival analyses, such as Kaplan-Meier curves or other modeling approaches, will be essential to better assess the prognostic significance of circulating PC phenotypes. Given that survival remains the gold standard for biomarker validation, separating survival analyses from other clinical metrics will be crucial to fully understand the impact of circulating PCs on patient outcomes, paving the way for their use in clinical decision-making. Furthermore, while our study focused on circulating PCs, integrating other liquid biopsy analytes, such as cell-free DNA or extracellular vesicles, could provide a more comprehensive picture of disease evolution. Improved models incorporating additional biomarkers or longitudinal data may enhance diagnostic accuracy, stratify patients based on risk, and identify therapeutic targets, further advancing the personalized care of MM. While our findings are promising, further work is needed to establish sensitivity, specificity, and clinical thresholds in larger, independent cohorts.
Methods
Study design
The study recruited and enrolled patients following an approved protocol (PA18-1073) from the Institutional Review Board (IRB) at The University of Texas MD Anderson Cancer Center (MDACC). All study participants have provided written informed consent in accordance with the principles of the Declaration of Helsinki. Patients underwent standard diagnostic tests, including a BM aspirate and core needle biopsy, bloodwork, and whole-body imaging. The study collected matched PB and BM specimens from 68 patients (11 MGUS, 21 SMM, 19 NDMM, 17 RRMM). Paired BM samples were taken for clinical workup only. RRMM patients have undergone one or more lines of therapy.
Sample acquisition and processing
All blood samples (8 mL) were collected using anti-coagulated preservative tubes (Cell-Free DNA blood collection tube, Streck, La Vista, NE, USA) at MDACC and shipped to the USC Michelson Convergent Science Institute in Cancer (CSI-Cancer) via FedEx overnight at controlled room temperature using validated, temperature-stabilizing containers, and processed within 48 h post-draw, as previously described35,37,38,39. Upon sample arrival, a complete blood cell count was taken and red blood cells lysed. All remaining nucleated cells were plated as a monolayer at approximately 3 million cells per custom cell adhesive glass slide (Marienfeld, Lauda, Germany). Slides were subsequently incubated in 7% BSA, dried, and stored at −80 °C.
Immunofluorescent staining
Two slides per sample were thawed prior to immunofluorescent (IF) staining using an IntelliPATH FLX™ autostainer (Biocare Medical LLC, Irvine, CA, USA) as previously described35. All steps were conducted at room temperature. Slides were fixed with 2% paraformaldehyde for 20 min, blocked with 10% goat serum for 30 min, and incubated with a primary antibody mix consisting of CD138 (2 µg/mL, Exbio, clone A-38, cat# 10-520-C100, Vestec, Czech Republic), CD45 Alexa647 (1.6 µg/mL, AbD Serotec, cat# MCA87A647, Raleigh, NC, USA), and BCMA (2.5 µg/mL, Abcam, cat# ab253242, Cambridge UK) for 1 h. Next, slides were blocked again with 10% goat serum for 1 h prior to incubation with secondary antibodies AlexaFluor555 goat anti-mouse (1:500, Invitrogen, cat# A21127, Waltham, MA, USA), AlexaFluorPLUS488 goat anti-rabbit (1:500, Invitrogen, cat# A32731, Waltham, MA, USA), and DAPI (4’,6-diamidino-2-phenylindole; D) for 40 min. Slides were mounted with a glycerol-based media, coverslipped, and sealed for downstream imaging and technical analysis.
Image acquisition, processing, and cell enumeration
Automated high-throughput fluorescence scanning microscopy at 100X magnification was employed for the imaging of 2304 frames across each slide as previously described35,37. The gain and exposure times for all channels were normalized by the scanner software.
Rare event detection was conducted through a customized algorithm Outlier Clustering Unsupervised Learning Automated Report (OCULAR), which was previously used in analysis of liquid biopsy samples for detection of rare events in carcinomas40,41,42. This method was modified for the purposes of this study in MM disease progression. In short, the approach utilizes principal component analysis (PCA) and hierarchical clustering to identify small clusters of cells or individual cells that significantly deviate from the median cell based on computational distance. To further refine this process and eliminate technical artifacts, we developed a machine learning classification model to determine the biological relevance of cells. A histogram gradient boosting algorithm was implemented with k-fold cross-validation across 1000 iterations to identify the top-performing models. Additionally, hyperparameter grid searches were conducted on morphometric features to pinpoint subsets that optimized model accuracy. Prediction confidence for each cell was used as a threshold to balance sensitivity (accurately identifying biologically relevant events) and specificity (accurately excluding irrelevant events). The model was developed from manually curated data of 19 samples (24,300 individual cells).
Slides with manual cell-level annotations were divided into training and testing sets, while non-annotated slides were utilized in downstream model development to test performance. The cell groups were classified into 12 classes based on the combination of three markers: CD138, BCMA (membrane [Memb] and perinuclear [Peri] localization), and CD45. The channel classification models were developed from manual cellular classifications from 11 samples (2906 individual cells). Cells are characterized by their IF expression in each of the channels and referred to by only their positive markers. The supervised machine learning algorithms were trained to distinguish between groups using the full feature space, allowing it to capture complex, non-linear patterns that may not be evident through traditional threshold-based or univariate statistical analyses.
The OCULAR rare cells detected were automatically curated with the previously mentioned machine learning model set at 90% prediction confidence to reduce noise and subsequently characterized by their IF channel expression. Using the cell count taken at processing and the cell count per slide analyzed, the final cell enumerations were able to be reported as cells per mL.
Statistical analysis
Non-parametric statistical two-sided tests were used to determine correlations between detected rare events in the liquid biopsy samples and clinical parameters. We utilized the Wilcoxon rank sum test (categorical) and Spearman’s rank correlation (continuous and ordinal) to analyze correlations between rare event enumerations and clinical data elements. P-values below 0.05 were considered statistically significant. Each comparison was analyzed independently. Cellular morphometrics were used to investigate the heterogeneity of the rare-event population. We visualized cell heterogeneity via two-dimensional UMAPs (Uniform Manifold Approximation and Projection) and morphometric probability distribution plots.
Data availability
All data discussed in this manuscript are included in the main manuscript text or supplementary materials. The imaging data are available through the BloodPAC Data Commons, Accession ID “BPDC000152” (https://data.bloodpac.org/discovery/BPDC000152).
Code availability
All code developed is standard and easily reproducible.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
Bories, C. & Jagannath, S. Asymptomatic monoclonal gammopathies. Clin. Lymphoma Myeloma Leuk. 14, S78–S86 (2014).
Dutta, A. K. et al. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia 33, 457–468 (2019).
Feng, F. et al. Single-cell multi-omics analysis reveals heterogeneity of malignant plasma cell during progression of multiple myeloma. Blood 144, 6845–6845 (2024).
Ahmed, A. & Killeen, R. B. Relapsed and Refractory Multiple Myeloma, in StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Robert Killeen declares no relevant financial relationships with ineligible companies. (StatPearls, 2025).
Palumbo, A. & Anderson, K. Multiple myeloma. N. Engl. J. Med 364, 1046–1060 (2011).
Razzo, B., Garfall, A. L. & Cohen, A. D. Options at the time of relapse after anti-BCMA therapy. Hematology 2023, 450–458 (2023).
Van Oekelen, O. et al. Interventions and outcomes of patients with multiple myeloma receiving salvage therapy after BCMA-directed CAR T therapy. Blood 141, 756–765 (2023).
Rajkumar, S. V. et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 15, e538–e548 (2014).
Dhakal, B. et al. Assessment of molecular residual disease using circulating tumor DNA to identify multiple myeloma patients at high risk of relapse. Front Oncol. 12, 786451 (2022).
Li, S., Zhang, E. & Cai, Z. Liquid biopsy by analysis of circulating myeloma cells and cell-free nucleic acids: a novel noninvasive approach of disease evaluation in multiple myeloma. Biomark. Res 11, 27 (2023).
Mack, E. K. M. et al. Monitoring multiple myeloma in the peripheral blood based on cell-free DNA and circulating plasma cells. Ann. Hematol. 101, 811–824 (2022).
Krishnan, S. R. et al. Isolation of human CD138+microparticles from the plasma of patients with multiple myeloma1. Neoplasia 18, 25–32 (2016).
Liu, Z. Y. et al. The potential diagnostic power of CD138+microparticles from the plasma analysis for multiple myeloma clinical monitoring. Hematol. Oncol. 37, 401–408 (2019).
Manier, S. et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 129, 2429–2436 (2017).
Wang, X. et al. Recent progress of exosomes in multiple myeloma: pathogenesis, diagnosis, prognosis and therapeutic strategies. Cancers 13, 1635 (2021).
Witzig, T. E. et al. Quantitation of circulating peripheral blood plasma cells and their relationship to disease activity in patients with multiple myeloma. Cancer 72, 108–113 (1993).
Bertamini, L. et al. High levels of circulating tumor plasma cells as a key hallmark of aggressive disease in transplant-eligible patients with newly diagnosed multiple myeloma. J. Clin. Oncol. 40, 3120–3131 (2022).
Vigliotta, I. et al. Circulating multiple myeloma cells (CMMCs) as prognostic and predictive markers in multiple myeloma and smouldering MM patients. Cancers 16, 2929 (2024).
Billadeau, D. D. et al. Clonal circulating cells are common in plasma cell proliferative disorders: a comparison of monoclonal gammopathy of undetermined significance, smoldering multiple myeloma, and active myeloma. Blood 88, 289–296 (1996).
Korthals, M. et al. Molecular monitoring of minimal residual disease in the peripheral blood of patients with multiple myeloma. Biol. Blood Marrow Transpl. 19, 1109–1115 (2013).
Bianchi, G. et al. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia 27, 680–685 (2013).
Kumar, S. et al. Prognostic value of circulating plasma cells in monoclonal gammopathy of undetermined significance. J. Clin. Oncol. 23, 5668–5674 (2005).
Witzig, T. E. et al. Peripheral blood monoclonal plasma cells as a predictor of survival in patients with multiple myeloma. Blood 88, 1780–1787 (1996).
Witzig, T. E. et al. Detection of peripheral blood plasma cells as a predictor of disease course in patients with smouldering multiple myeloma. Br. J. Haematol. 87, 266–272 (1994).
Oben, B. et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat. Commun. 12, 1861 (2021).
Bowcock, S. et al. Presenting symptoms in newly diagnosed myeloma, relation to organ damage, and implications for symptom-directed screening: a secondary analysis from the tackling early morbidity and mortality in myeloma (TEAMM) Trial. Cancers 15, 3337 (2023).
van de Donk, N. W. et al. Diagnosis, risk stratification and management of monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Int J. Lab Hematol. 38, 110–122 (2016).
Manier, S. et al. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 14, 100–113 (2017).
Morgan, G. J., Walker, B. A. & Davies, F. E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 12, 335–348 (2012).
Han, W. et al. Prognostic value of circulating clonal plasma cells in newly diagnosed multiple myeloma. Hematology 26, 510–517 (2021).
Sanoja-Flores, L. et al. Blood monitoring of circulating tumor plasma cells by next generation flow in multiple myeloma after therapy. Blood 134, 2218–2222 (2019).
Wang, N. et al. Enrichment of circulating myeloma cells by immunomagnetic beads combined with flow cytometry for monitoring minimal residual disease and relapse in patients with multiple myeloma. Ann. Hematol. 98, 2769–2780 (2019).
Ndacayisaba, L. J. et al. Enrichment-free single-cell detection and morphogenomic profiling of myeloma patient samples to delineate circulating rare plasma cell clones. Curr. Oncol. 29, 2954–2972 (2022).
Ndacayisaba, L. J. et al. Characterization of BCMA expression in circulating rare single cells of patients with plasma cell neoplasms. Int. J. Mol. Sci. 23, 13427 (2022).
Setayesh, S. M. et al. Targeted single-cell proteomic analysis identifies new liquid biopsy biomarkers associated with multiple myeloma. NPJ Precis Oncol. 7, 95 (2023).
Marrinucci, D. et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys. Biol. 9, 016003 (2012).
Rodriguez-Lee, M. et al. Effect of Blood Collection Tube Type and Time to Processing on the Enumeration and High-Content Characterization of Circulating Tumor Cells Using the High-Definition Single-Cell Assay. Arch. Pathol. Lab Med 142, 198–207 (2018).
Shishido, S. N. et al. Preanalytical Variables for the Genomic Assessment of the Cellular and Acellular Fractions of the Liquid Biopsy in a Cohort of Breast Cancer Patients. J. Mol. Diagn. 22, 319–337 (2020).
Chai, S. et al. Platelet-coated circulating tumor cells are a predictive biomarker in patients with metastatic castrate resistant prostate cancer. Mol. Cancer Res. 19, 2036–2045 (2021).
Narayan, S. et al. Defining a liquid biopsy profile of circulating tumor cells and oncosomes in metastatic colorectal cancer for clinical utility. Cancers 14, 4891 (2022).
Shishido, S. N. et al. Characterization of cellular and acellular analytes from pre-cystectomy liquid biopsies in patients newly diagnosed with primary bladder cancer. Cancers 14, 758 (2022).
Acknowledgements
We thank the patients and their caregivers who consented to this study. We also thank the clinical research staff who contributed to the study. We are grateful to past and current technical staff at CSI-Cancer for processing the samples. This work was funded in whole or in part by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (P.K., R.Z.O.), the NCI U01CA285013 (P.K., J.M., S.N.S.), and USC Norris Comprehensive Cancer Center (CORE) Support 5P30CA014089-40 (P.K., J.M.). This work also received institutional support from the Winnie and James Hart Endowed Fellowship (S.M.S.); the USC Michelson Center Convergent Science Institute in Cancer, Vassiliadis Research Fund, and Hart Family Research Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conceptualization, S.N.S, J.M., and P.K.; computational analysis, J.M.; validation, S.N.S. and J.M.; formal analysis, S.N.S. and J.M.; data curation, S.N.S., M.K., and J.M.; writing—original draft preparation, S.N.S., M.K., and J.M.; writing—review and editing, S.N.S., M.K., J.M., E.E.M., R.Z.O., K.P. and P.K.; data visualization and investigation, S.N.S. and J.M.; project administration, S.N.S. and P.K.; funding acquisition, P.K., E.E.M. and R.Z.O.; subject recruitment, A.U.V., L.M.R., D.B., E.E.M., R.Z.O., and K.P. All authors have read and agreed to the published version of the manuscript. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Corresponding author
Ethics declarations
Competing interests
The HDSCA technology described here is licensed to Epic Sciences. P.K. has ownership in Epic Sciences. E.E.M. is an employee at GSK. All other authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shishido, S.N., Mason, J., Kamal, M. et al. Characterizing circulating rare cells in peripheral blood for detecting and monitoring multiple myeloma and precursor states. npj Precis. Onc. 9, 388 (2025). https://doi.org/10.1038/s41698-025-01175-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01175-2
