
Abstract
This study aimed to identify core DNA methylation sites linked to urothelial bladder cancer (UBC) and to develop prognostic and early diagnostic models using multi-sample datasets from tissue, blood, and urine. By applying six machine learning algorithms, we constructed a diagnostic signature based on four DNA methylation sites and a prognostic signature involving ten sites. The diagnostic model showed high accuracy in detecting bladder cancer in both tissue and urine samples, while the prognostic model effectively predicted survival outcomes. Further analyses revealed associations with survival, enriched pathways, immune infiltration, genetic mutations, and responses to immunotherapy and chemotherapy. Cellular experiments, including Q-PCR, WB, Co-IP, and ChIP, demonstrated that C1QTNFNF6 significantly influences UBC prognosis and that TET2 promotes demethylation of C1QTNFNF6, elevating its expression and accelerating tumor progression. These findings present eleven key methylation sites with strong clinical applicability for diagnosis and prognosis in UBC, and reveal an epigenetic mechanism involving TET2 and C1QTNFNF6 that drives disease development.
Similar content being viewed by others

Introduction
Urothelial bladder cancer (UBC) is a highly prevalent malignancy with significant mortality rates (approximately 614,000 new cases and 220,000 deaths in 2022), posing substantial challenges to global public health1. UBC can be classified into nonmuscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC) based on whether the tumor has invaded the bladder muscle layer. These two types of cancer have completely different prognostic features. For example, the overall 5-year survival rate of NMIBC patients is 70–98%, but more than 50% of NMIBC patients will experience tumor recurrence2.
Cystoscopy is the current standard in diagnosing bladder cancer, but it is an invasive and costly procedure. Urinary markers are non-invasive but most of the available markers are limited by its diagnostic performance. Moreover, the biological heterogeneity of UBC significantly complicates efforts to predict disease progression and response to therapy, making accurate prognosis assessment elusive. Therefore, there is an urgent need for non-invasive, cost-effective alternatives that can enhance early detection, precise prognosis, and tailored treatment strategies3.
Recent advances in molecular biology have highlighted the potential of DNA methylation as an early biomarker for UBC. DNA hypermethylation in the promoter regions of tumor suppressor genes is considered one of the earliest detectable events in UBC development. For instance, methylation of tumor suppressor genes such as P16INK4a and TP53 leads to their functional loss, thereby facilitating the malignant transformation and proliferation of UBC cells4,5. However, the diagnostic performance of existing biomarkers is often insufficient, and they necessitate further validation. Given these limitations in existing biomarkers, innovative approaches are needed. Machine learning techniques, combined with high-throughput sequencing, offer an unprecedented opportunity to harness these data and construct accurate diagnostic and prognostic models. By integrating genetic variants, immune components, and therapeutic responses, these models can provide more accurate predictions and facilitate personalized medicine approaches.
In this study, we aim to identify core DNA methylation sites associated with UBC and develop prognostic and early diagnostic models using advanced machine learning techniques. We also focus on elucidating the underlying mechanisms of UBC progression, particularly the roles of C1QTNF6 and TET2, which have been implicated in cancer biology but remain poorly understood in the context of UBC. Our findings are expected to provide novel insights into the molecular basis of UBC, improve early detection, and guide personalized treatment strategies, ultimately contributing to better patient outcomes. This work may also inform future studies on the role of epigenetic modifications in cancer progression and therapy response.
Results
Identification of key DNA methylation sites in bladder cancer through multi-omics analysis of tissue, blood, and urine samples
DNA methylation alterations frequently occur in the early stages of cancer, with stable expression in tumor tissues and detectability in bodily fluids. To identify key DNA methylation sites associated with UBC, we conducted a multi-omics analysis of DNA methylation levels in three sample types: cancer tissue, peripheral blood, and urine.
Urine sample analysis
Using the GSE120288 dataset, which compares DNA methylation levels between UBC patients and healthy controls, we identified 358 differentially methylated sites (gene-level average, DES) with upregulation and 8,009 with downregulation (P < 0.05; Fig. 1A, Supplementary Data S1-2). These findings suggest that urine DNA methylation profiles may serve as potential biomarkers for UBC detection.

A Differentially expressed genes at the DNA methylation level in the GSE120288 dataset. B Differentially expressed genes at the DNA methylation level in the TCGA-BLCA dataset. C Differentially expressed genes at the mRNA level in the TCGA-BLCA dataset. D, E Common differentially expressed genes at the DNA methylation level across tissue, blood, and urine samples. F Methylation-related genes (MRGs) identified by the intersection of all differentially expressed genes at both the DNA methylation and mRNA levels.
Tissue sample analysis
Analysis of the TCGA-BLCA dataset, comprising UBC tissue samples, revealed 19,806 DES (P < 0.05; Fig. 1B, Supplementary Data S3-4), including 2466 upregulated and 17,340 downregulated sites. These extensive DNA methylation alterations highlight their potential role in tumorigenesis and disease progression.
Blood sample analysis
Previously reported DNA methylation data from blood samples of UBC patients identified 8888 DES (P < 0.05, Supplementary Data S5), with 5803 upregulated and 3,085 downregulated sites. The presence of consistent methylation changes across different sample types suggests their potential use as minimally invasive biomarkers.
Integrated multi-omics analysis
To identify robust DNA methylation markers, we focused on DES that were consistently present across all three sample types (tissue, urine, and blood). Furthermore, considering the regulatory role of DNA methylation in gene expression, we analyzed differential mRNA expression levels in UBC using the TCGA-BLCA dataset. A total of 9495 differentially expressed mRNA targets were identified (P < 0.05; Fig. 1C, Supplementary Data S6), with 5980 upregulated and 3515 downregulated.
A four-quadrant Venn diagram analysis was employed to integrate DNA methylation and mRNA expression data, identifying key targets that met the following criteria: (1) abnormal DNA methylation, (2) abnormal downstream mRNA levels, and (3) concordance with DNA methylation-regulated mRNA expression patterns (Fig. 1D-F). This analysis identified 173 key DES (Supplementary Data S7) that were consistently dysregulated across all three sample types, exhibiting concordant mRNA expression changes.
Development of a DNA methylation-site model for bladder cancer diagnosis
Selection of differentially methylated sites
We identified 173 key differentially expressed genes (Supplementary Data S7) that consistently exhibited abnormal DNA methylation levels across all three sample types, accompanied by corresponding mRNA expression changes. Upon analyzing the 3757 DNA methylation sites corresponding to these genes (Supplementary Data S8), we performed differential methylation analysis on cancer and control samples from the TCGA-BLCA database using the R package “ChAMP,” yielding corresponding P-value and logFC values. The differential threshold was set to: P.adj < 0.05, |logFC | > 0.25, and Δβ ≥ 0.15. Further screening for DNA methylation sites located in promoter regions (TSS200 & TSS1500) and CpG islands led to the identification of 17 significant methylation sites.
Correlation analysis between genes and methylation sites
Based on the aforementioned 17 differentially methylated sites, we matched the corresponding six genes and analyzed their correlation using the Spearman method. The correlation heatmap generated using the R package “ggplot2” (Fig. 2A) revealed that sites with significant negative correlations between methylation levels and gene expression (Spearman ρ < −0.3, p < 0.05) were retained, resulting in 10 sites (cg10395685, cg16536329, cg03234732, cg23037403, cg02165355, cg24843380, cg26673012, cg17985646, cg07994987, cg07593839).

A Correlation heatmap showing the relationship between DNA methylation levels and gene expression for six genes. The color intensity represents the Spearman correlation coefficient (ρ), with darker colors indicating stronger correlations. Significant negative correlations (Spearman ρ < −0.3, p < 0.05) are highlighted. B-1 LASSO regression coefficients for the selected methylation sites. The graph shows the coefficients as a function of the log(lambda), with the optimal lambda (λ.min) indicated by the red vertical line. B-2 Cross-validation error rates for the LASSO regression model. The red dashed line indicates the optimal lambda (λ.min), which minimizes the mean squared error. C-1 Receiver Operating Characteristic (ROC) curves for individual methylation sites. The area under the curve (AUC) values are provided for each site, indicating their diagnostic performance. C-2 Decision curve analysis (DCA) for the logistic regression model incorporating four methylation sites vs ALL model with seven methylation sites. C-3 ROC curve for the logistic regression model using four methylation sites. The AUC value is 0.952, indicating excellent diagnostic performance. C-4 Box plots comparing model scores between normal and tumor samples in the TCGA-BLCA dataset. C-5 Box plots comparing expression levels of specific methylation sites (cg10395685, cg16536329, cg23037403, cg02165355) between normal and tumor samples across different groups (g1-g5). D-1 ROC curves for individual methylation sites in the validation dataset (GSE120288). The AUC values are provided for each site, demonstrating their diagnostic performance. D-2 ROC curve for the logistic regression model using four methylation sites in the validation dataset. The AUC value is 0.972, indicating excellent diagnostic performance. D-3 Decision curve analysis (DCA) for the logistic regression model in the validation dataset. D-4 Box plots comparing model scores between normal and tumor samples in the GSE120288 dataset. D-5 Box plots comparing expression levels of specific methylation sites (cg10395685, cg16536329, cg23037403, cg02165355) between normal and tumor samples across different groups (g1-g5) in the validation dataset.
Construction of the diagnostic model
A diagnostic model for UBC was constructed using Lasso regression analysis combined with a logistic regression model. Utilizing the R software glmnet package, we selected lambda.min (0.00016) as λ for model construction. The graphical representation of gene coefficients (Fig. 2B) and the cross-validation error plot (Fig. 3) indicated that seven feature variables were ultimately selected (cg10395685, cg16536329, cg23037403, cg02165355, cg26673012, cg17985646, cg07994987).

A SVM-RFE analysis. B Lasso regression analysis. C Random forest analysis. D Boruta feature selection analysis. E XGBoost analysis. F PAMR analysis. G A total of 62 MRGs were supported by more than one algorithm. K Univariate Cox regression analysis identified 11 significant MRGs (P < 0.05).
ROC analysis of individual sites
ROC analysis was performed on the aforementioned seven methylation sites using the R package “pROC” to plot ROC curves (Fig. 4). The results showed that sites cg10395685, cg16536329, cg23037403, and cg02165355 had AUC values > 0.8, designating them as key sites for inclusion in the logistic regression model.

A Validation of the prognostic signature across multiple datasets: Kaplan-Meier analysis in (A-1) TCGA-BLCA (P < 0.001), A-2 GSE13507 (P < 0.005), and (A-3) GSE176307 (P = 0.04). B Prediction of chemotherapy response for multiple agents, including AICAR, axitinib, bexarotene, bicalutamide, bortezomib, cyclopamine, dasatinib, and docetaxel. C ESTIMATE algorithm analysis showing that higher-risk bladder cancer patients had lower immune-related scores (P < 0.001), including: (C-1) Immune score; (C-2) Stromal score; (C-3) ESTIMATE score. D Distribution of the top 20 mutated genes in the high- and low-risk groups.
Evaluation of the diagnostic model
A logistic regression model was constructed using the glm function for cg10395685, cg16536329, cg23037403, and cg02165355, yielding regression coefficients for each site. The diagnostic efficacy of the model was demonstrated using the model’s ROC curve and decision curve analysis (DCA). The ROC curve area was 0.957, indicating good diagnostic capability. The DCA curve results showed that as the risk threshold increased, the benefits of the Logistic Model gradually surpassed the All strategy (a diagnostic model incorporating seven methylation sites), particularly at medium and high-risk thresholds, where the Logistic Model’s benefits were significantly higher than other strategies, indicating its effectiveness as a predictive model (Fig. 2C).
Validation of the diagnostic model
The diagnostic model was validated using the GSE120288 methylation dataset, with ROC curves and DCA curves plotted accordingly (Fig. 2D). The results showed that the ROC curve area was greater than 0.9, indicating good diagnostic capability. The DCA curve results indicated that before the high-risk threshold of 0.6, the standardized net benefit of the logistic regression model was higher than the All strategy, suggesting good performance at low thresholds. Additionally, the ROC curve areas for individual sites were all greater than 0.8, indicating their good diagnostic potential.
Differential analysis of model scores
Model scores were calculated for both datasets based on the model’s regression coefficients, and the expression of model scores in tumor and control groups was analyzed using the Wilcoxon test. Box plots generated using the R package “ggplot2” showed that model scores were significantly higher in the tumor group than in the normal group (p < 0.05) in both datasets, further confirming the model’s diagnostic value. The results indicated that in the TCGA-BLCA dataset, cg10395685, cg16536329, cg23037403, and cg02165355 were significantly more highly expressed in the tumor group than in the control group (P < 0.05). In the GSE120288 dataset, cg10395685, cg16536329, cg23037403, and cg02165355 also showed significantly higher expression in the tumor group than in the control group (P < 0.05), with expression trends consistent with those in the TCGA-BLCA dataset.
Machine-learning-based identification of key DNA methylation level and their transcriptional associations in bladder cancer
Identifying the functional roles of 173 critical DNA methylation sites in UBC poses a considerable challenge due to their sheer number, making comprehensive experimental validation infeasible in terms of both time and cost. To address this, we employed a deep learning framework incorporating six machine learning algorithms to reduce dimensionality and identify key targets.
Given the extensive transcriptomic data available for UBC—including clinical prognosis, tumor recurrence, drug resistance, and immune components—we focused on evaluating the transcriptional impact of these DNA methylation sites. As DNA methylation directly modulates gene expression, transcriptomic analysis provides crucial insights into its biological significance. Using the TCGA-BLCA dataset, we systematically assessed transcriptomic alterations associated with the 173 DNA methylation sites and ultimately identified 11 key sites with significant transcriptional associations.
To ensure rigorous feature selection, we implemented six distinct machine learning algorithms, identifying:
37 genes via SVM-RFE,
16 genes via Lasso,
121 genes via Random Forest,
30 genes via Boruta,
22 genes via XGBoost, and
36 genes via PAMR (Fig. 2A-F).
By integrating these results, we identified a core set of 62 methylation-related genes (MRGs, Supplementary Data S9) that were consistently selected by at least two algorithms (Fig. 2G).
To further refine these candidates, we performed univariate Cox regression analysis (Fig. 3K, Supplementary Data S10) on the 62 MRGs. This analysis identified 11 key genes—CNTD1, PYCR1, C1QTNF6, PITX1, GEMIN6, GPS2, RPP21, PLS1, COMMD7, RPS6KA1, and CTHRC1—that were significantly associated with UBC prognosis (P < 0.05). These findings underscore their potential roles in tumor progression and clinical outcomes, suggesting that these DNA methylation sites may serve as critical biomarkers for prognostic assessment.
Development and validation of a prognostic model for bladder cancer based on DNA methylation-related genes
We constructed a prognostic model incorporating 10 DNA MRGs that accurately predict multiple prognostic outcomes in UBC. To develop this model, we utilized two datasets: TCGA-BLCA as the training set and GSE13507 as the validation set. The model was constructed using union COX-Lasso analysis, which integrates the strengths of COX regression in survival analysis with the variable selection capabilities of LASSO (Supplementary Data S11). This approach outperforms either COX or LASSO analysis alone by first selecting optimal variables with LASSO and then constructing a survival model with COX regression.
The final prognostic signature included 10 MRGs, with the risk score formula defined as:
Performance Evaluation of the Prognostic Model
-
1.
Survival Analysis in UBC
Kaplan-Meier (KM) analysis revealed that the prognostic signature significantly predicted overall survival (OS) in both the training and validation cohorts. Patients with higher risk scores exhibited worse OS (P < 0.001 in the training set; P = 0.005 in the validation set; Fig. 4A). The two cohorts were stratified into nonmuscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC) based on T stage. The TCGA-BLCA cohort included 4 NMIBC patients and 369 MIBC patients, while the GSE13507 cohort included 103 NMIBC patients and 62 MIBC patients. The prognostic model maintained its predictive efficacy for UBC prognosis in both subgroups across the two cohorts (P < 0.05, Supplementary Fig. 1).
-
2.
Immunotherapy and Chemotherapy Sensitivity Analysis
In an immunotherapy cohort (GSE176307), higher risk scores were associated with worse OS, consistent with immune-related characteristic analysis (Fig. 3A). Additionally, the risk score predicted sensitivity to several chemotherapy drugs, including aicar, axitinib, bexarotene, bicalutamide, bortezomib, cyclopamine, dasatinib, and docetaxel, using the R package “pRRophetic” (Fig. 3B). Notably, docetaxel is a first-line treatment for UBC, bicalutamide is widely used for prostate cancer, and axitinib is a novel therapy for advanced renal cell carcinoma.
-
3.
Immune-Related Characteristics and Mutation Profiles
Immune-related characteristic analysis revealed that patients with higher risk scores exhibited a stronger anti-immune state, while those with lower risk scores had a better immune state. The ESTIMATE algorithm showed that higher risk scores were associated with worse immune-related scores (P < 0.001), including immune score, stroma score, and ESTIMATE score (Fig. 3C). Mutation analysis identified the top 20 mutated genes in both groups, with TP53, TIN, and KMT2D being the most frequent (Fig. 3D). GSEA analysis identified 14 potentially different hallmark pathways, with the top four being epithelial-mesenchymal transition, inflammatory response, apical junction, and myogenesis.
Demethylation of C1QTNF6 induces upregulation and promotes proliferation and invasion in bladder cancer
We first evaluated the expression and DNA methylation status of 11 specific genes in UBC (5637 cell line) and normal bladder epithelial cells (SV-HUC-1) using quantitative PCR (Q-PCR) and DNA methylation analysis. Transcriptomic analysis revealed upregulation of C1QTNF6, PITX1, GEMIN6, COMMD7, and CTHRC1 in UBC tissues, while PYCR1 expression was downregulated (Fig. 5A). DNA methylation analysis identified differential methylation of PYCR1, RPP21, and C1QTNF6. Notably, C1QTNF6 exhibited demethylation in cancer tissues, while methylation was observed in normal tissues. In contrast, PYCR1 and RPP21 were methylated in cancer and demethylated in normal tissues (Fig. 5B). Combining Q-PCR and DNA methylation data, only C1QTNF6 demonstrated significant demethylation and upregulation at both the transcriptional and protein levels, as confirmed by Western blotting (Fig. 5C).

A qPCR analysis of 11 target gene expression levels. B DNA methylation levels of the 11 target genes in SV-HUC-1 versus 5637 cell lines. C Western blotting analysis confirming C1QTNF6 upregulation in bladder cancer cell lines (P < 0.01). D Downregulation of C1QTNF6 expression in the 5637 cell line (P < 0.01). E, F Functional assays demonstrating that C1QTNF6 downregulation reduces proliferation (E, CCK-8 assay, P < 0.001) and weakens invasion ability (F, Transwell assay, P < 0.01). G DNA methylation levels of C1QTNF6 were increased (P < 0.05). I Upregulation of C1QTNF6 DNA methylation suppressed its expression (Western blotting, P < 0.01). J, K Increased DNA methylation of C1QTNF6 inhibited proliferation (J, CCK-8 assay, P < 0.001) and invasion (K, Transwell assay, P < 0.01) in bladder cancer cell lines.
Next, we investigated the functional consequences of C1QTNF6 modulation. Downregulation of C1QTNF6 expression via siRNA resulted in reduced proliferation and migration of UBC cells (Fig. 5D-F). Conversely, increasing the DNA methylation level of C1QTNF6 using dimethyl sulfate suppressed its expression and inhibited cell proliferation and invasion. Upregulation of DNA methylation led to a significant reduction in both C1QTNF6 mRNA and protein levels (Fig. 6G-I), as well as a decrease in cell proliferation and migration (Fig. 5J, K). CCK-8 assays and Transwell migration assays further confirmed the suppressive effects of C1QTNF6 DNA methylation on UBC cell proliferation and invasion.

A TET2 expression was elevated in the 5637 cell line. B, C Knockdown of TET2 led to reduced C1QTNF6 expression at both the DNA methylation, mRNA levels and protein levels. D, E TET2 downregulation resulted in decreased proliferation (D, CCK-8 assay, P < 0.001) and reduced invasion ability (E, Transwell assay, P < 0.01).
TET2 mediates active DNA demethylation of C1QTNF6
The TET family of proteins are key players in active DNA demethylation. Given the significant upregulation of C1QTNF6 in UBC, we hypothesized that TET enzymes might mediate its demethylation. Q-PCR analysis revealed that TET2 expression was significantly elevated in UBC cell lines (Fig. 7A). Knockdown of TET2 using siRNA increased the methylation of C1QTNF6 (Fig. 6B, C) and suppressed both its mRNA and protein expression, as confirmed by Western blotting and Q-PCR (Fig. 6B, C). Functional assays showed that TET2 inhibition decreased UBC cell proliferation and invasion (Fig. 6D, E). These findings suggest that TET2 facilitates the DNA demethylation of C1QTNF6, leading to its upregulation and promoting cancer cell proliferation and invasion.

A Co-immunoprecipitation (co-IP) and western blotting assays demonstrated direct interaction between C1QTNF6 and TET2 proteins. B ZENBU genome browser screenshot illustrating the colocalization of C1QTNF6 and TET2 ChIP-seq signals with DNA methylation levels. C Overlap between C1QTNF6 (blue) and TET2 (red) ChIP-seq peaks. D Genome-wide distribution of TET2 and C1QTNF6 ChIP-seq peaks. E Total read count per million mapped reads for TET2 and C1QTNF6 ChIP-seq peaks. F, G Distribution of TET2 and C1QTNF6 ChIP-seq signals around transcription start sites (TSS, 63 kb) (F) and CpG islands (CGI, 63 kb). I Comparison of C1QTNF6 and TET2 loop CUT&Tag signals showing a strong positive correlation (r = 0.71, P < 0.05) at TSS regions, with 96% of C1QTNF6 peaks overlapping with TET2 peaks.
Physical interaction and genome-wide colocalization of C1QTNF6 and TET2
To explore the molecular interaction between C1QTNF6 and TET2, we performed co-immunoprecipitation (co-IP) assays followed by Western blotting. The results demonstrated a direct physical interaction between C1QTNF6 and TET2 proteins in the 5637 UBC cell line (Fig. 7A), suggesting that C1QTNF6 may be part of a DNA demethylation complex involving TET enzymes.
To further investigate the potential colocalization of C1QTNF6 and TET2 in chromatin, we performed ChIP-seq for both proteins in the 5637 cell line. ChIP-seq yielded 40,509,430 and 37,792,610 reads for C1QTNF6 and TET2, respectively, mapped to the hg38 human genome (Fig. 7B, C). When C1QTNF6 and TET2 were overexpressed, additional reads were generated, with 58,835,410 and 65,457,824 reads, respectively (Fig. 7D). Analysis identified 1660 peaks for C1QTNF6 and 2149 peaks for TET2 (Fig. 7E), with a significant overlap of 932 peaks (56.1%) from C1QTNF6 and 43.4% of TET2 peaks overlapping, suggesting substantial colocalization at specific genomic regions.
Further analysis revealed that TET2 reads were enriched around C1QTNF6 ChIP peaks, with this enrichment significantly reduced following C1QTNF6 knockdown (P < 0.001, T-test). Heatmap analysis indicated that both C1QTNF6 and TET2 were highly enriched at transcription start sites (TSS) and CpG islands (Fig. 7F, G), suggesting that both proteins colocalize at key regulatory regions of the genome.
Finally, correlation analysis of C1QTNF6 and TET2 loop CUT&Tag signals showed a positive correlation (r = 0.71, P < 0.05) at TSS regions, with 96% overlap between C1QTNF6 and TET2 peaks (Fig. 7I). These data further support the conclusion that C1QTNF6 and TET2 colocalize and cooperate at gene regulatory regions, potentially influencing DNA demethylation and gene expression in UBC.
Discussion
Cell-free DNA (cfDNA) methylation analysis has emerged as a promising non-invasive approach for identifying and validating epigenetic biomarkers with diagnostic and prognostic potential in various cancers6. Recent studies have demonstrated the feasibility of cfDNA methylation-based liquid biopsies for early cancer detection, such as in metastatic prostate cancer7, pancreatic cancer8, and gastric cancer9. In UBC, the FDA-approved Bladder EpiCheck assay targeting 13 cfDNA methylation sites has shown high specificity (88%) and negative predictive value (94.4%) for non-muscle-invasive UBC10. However, these studies primarily focused on single-sample types or limited methylation sites, highlighting the need for comprehensive multi-sample analyses11. Our study addresses this gap by integrating tissue, blood, and urine samples to identify core DNA methylation sites associated with UBC.
The translational impact of liquid biopsy markers in bladder cancer management is significant. For instance, a recent study by Pilala et al. 12 demonstrated that decreased methylation of the MIR145 core promoter in pre-treatment cfDNA was associated with short-term disease progression and poor overall survival in patients with muscle-invasive bladder cancer (MIBC) after radical cystectomy. This study highlighted the potential of cfDNA methylation as a minimally invasive and independent predictor of MIBC treatment outcome. Another study by Bettegowda et al. 13 showed that circulating tumor DNA (ctDNA) was detectable in a high percentage of patients with various advanced cancers, including bladder cancer, and could provide insights into the mechanisms underlying resistance to targeted therapies. These studies underscore the potential of liquid biopsy markers to improve the diagnosis, prognosis, and treatment of bladder cancer.
This study offers several key advantages over existing research. Our multi-sample approach (tissue, blood, and urine) enhances the representativeness and applicability of the identified methylation sites14. Integrating multiple machine learning algorithms increases the reliability and generalizability of our diagnostic and prognostic models15. Additionally, our comprehensive analysis provides a holistic view of UBC management, offering non-invasive, accurate diagnostic tools and robust prognostic models to guide treatment decisions and improve patient outcomes16.
The diagnostic model based on five DNA methylation sites (COMMD7, CPS2, PITX1, PLS1, and RPS6KA1) exhibited high accuracy in distinguishing UBC patients from healthy individuals in both tissue (ROC: 0.99) and urine samples (ROC: 0.85). The strong correlation between the risk score derived from these methylation sites and the TNM staging of UBC highlights its potential for non-invasive diagnosis and staging. Additionally, the prognostic model incorporating 10 DNA methylation-related transcriptome molecules demonstrated robust predictive ability in both the training and test sets (KM analysis, P < 0.001 and P = 0.005, respectively). High risk scores were associated with poor immune status, immune tolerance, and chemotherapy resistance, as well as the activation of adverse prognostic pathways such as TP53 mutations17. Collectively, these models offer valuable tools for early detection and personalized treatment planning in UBC, potentially improving patient outcomes18.
A key finding of this study is the elucidation of the role of C1QTNF6 in UBC progression. We demonstrated that TET2 facilitates the demethylation of C1QTNF6, promoting cancer progression. This is the first study to reveal the direct interaction between TET2 and C1QTNF6, confirmed by Co-IP and ChIP-seq experiments. This finding underscores the importance of TET2-mediated demethylation in UBC and highlights C1QTNF6 as a potential therapeutic target, potentially informing the development of novel epigenetic therapies.
Despite its strengths, this study has limitations. The analysis of C1QTNF6 was limited to a single UBC cell line (5637), and the functional roles of the other 10 methylation sites require further validation in larger and more diverse cohorts. Future work will focus on expanding our sample size and exploring additional sample types to validate the clinical utility of our models. Additionally, we plan to develop a cfDNA methylation-based liquid biopsy protocol for early.
Based on UBC tissue, blood, and urine samples, along with an integrated algorithm of six machine learning methods, 11 key DNA methylation sites were identified, and diagnostic and prognostic models for UBC were developed, showing promising applications in a variety of clinical outcomes. Furthermore, TET2 is involved in the demethylation of C1QTNF6, upregulating the expression levels of C1QTNF6 and inducing the malignant progression of UBC.
Methods
Data source
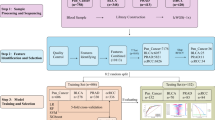
DNA methylation data for UBC were sourced from the TCGA19 and GEO datasets20. TCGA - BLCA offered DNA methylation data from 416 cancer tissues and 21 normal tissues, along with mRNA expression and prognosis data of 406 cancer cases. Blood DNA methylation data from UBC patients were retrieved from a related study21. The GSE120288 dataset provided urine DNA methylation data from 12 patients (6 cancer vs. 6 normal). For validation, the GSE13507 dataset contributed mRNA expression and prognosis data (188 cancer vs. 68 normal), while the GSE111933 dataset provided DNA methylation data for verifying the diagnostic model (46 cancer vs. 46 normal).
Differential expression analysis
Differential expression analysis was carried out using the R package “limma”, with significance set at P < 0.05. This method was applied to both DNA methylation and mRNA expression data22.
Machine learning
To develop a robust prognostic signature, six machine - learning algorithms were utilized: Support Vector Machines (SVM)23, Lasso Regression (via the “glmnet” package)24, Random Forest (with the “randomForest” package)25, Boruta (using the “Boruta” package)26, XGBoost (employing the “XGBoost” package)27, and pamr (from the “pamr” package)28. Genes identified by at least two algorithms were selected for subsequent model construction.
Identification of potential DNA methylation and transcriptional molecules
The process entailed four steps: 1. Differential methylation and mRNA expression data were extracted from urine, blood, and tissue samples. 2. Genes showing both differential methylation and mRNA expression were chosen for further analysis. 3. The six - algorithm machine - learning approach was used to pinpoint potential genes. 4. A prognostic signature was constructed using the TCGA - BLCA dataset with a combined COX - Lasso analysis29. This approach, which considered both survival status and survival time, provided more accurate results compared to traditional Lasso analysis.
Construction and validation of diagnostic and prognostic models
The diagnostic model for UBC, based on DNA methylation, was constructed via Lasso regression analysis and logistical regression analysis. The TCGA - BLCA dataset served as the training set, and the GSE11933 dataset was the testing set. Model effectiveness was validated using clinical characteristics and diagnostic outcomes of BLCA patients.
For the prognostic model, the TCGA-BLCA dataset was the training set, and GSE13507 was the testing set. Kaplan - Meier curves were employed to assess model performance. Additionally, the prognostic signature was evaluated in pan-cancer datasets from TCGA.
Enrichment analysis and immune-related characteristics
Patients in the training set were classified into high- and low-risk groups according to the prognostic signature. Gene Set Enrichment Analysis (GSEA) was performed to identify differentially enriched hallmark pathways between the two group30. Immune-related characteristics were evaluated using CIBERSORT to predict 22 immune cell components and ESTIMATE to calculate stromal and immune scores31. Tumor Mutation Burden (TMB) was analyzed with the “maftools” package, and the top 20 mutated genes were determined32.
Immunotherapy and chemotherapy sensitivity analysis
Data from the GSE176307 dataset, which included UBC patients who had received immunotherapy, were utilized to analyze the prognostic model’s predictive value for immunotherapy sensitivity. A Ridge regression model, constructed using the “pRRophetic” package, predicted drug sensitivity (IC50) based on tumor gene expression33 and drug sensitivity data from the Genomics of Drug Sensitivity in Cancer (GDSC) database34.
Cell culture and DNA methylation assay
UBC (5637) and bladder epithelial (SV - HUC - 1) cell lines were obtained from the Authenticated Cell Culture Compilation of China. DNA methylation levels were analyzed by bisulfite sequencing. Genomic DNA was treated with bisulfite, converting unmethylated cytosine to uracil while leaving methylated cytosine intact35. Specific primers targeting 11 loci were designed for PCR amplification, followed by sequencing and data analysis.
Quantitative PCR
RNA extraction
Total RNA was isolated using TRIzol reagent (Invitrogen#15596026, USA) following the manufacturer’s protocol. Briefly, cells were lysed in TRIzol, and RNA was extracted with chloroform, precipitated with isopropanol, and washed with 75% ethanol. RNA pellets were dissolved in RNase-free water, and concentration/purity was measured using a NanoDrop spectrophotometer (A260/A280 ratio ≥ 1.8).
Reverse transcription
cDNA was synthesized from 1 µg of total RNA using the PrimeScript RT Reagent Kit (Takara#RR037A, Japan) with oligo(dT) primers and random hexamers in a 20 µL reaction volume, following the manufacturer’s protocol. Reactions included 5× PrimeScript Buffer, RNase Inhibitor, and Reverse Transcriptase, incubated at 37 °C for 15 min and 85 °C for 5 s.
qPCR
Reactions were performed in triplicate using the SYBR Green-based Q-PCR Kit (Bioer#AQ131-01, Hangzhou, China) on an Eppendorf Mastercycler X226488N platform. Each 20 µL reaction contained 10 µL 2× SYBR Green Master Mix, 0.5 µM forward/reverse primers, 2 µL cDNA (diluted 1:10), and nuclease-free water. Cycling conditions: 95 °C for 3 min; 40 cycles of 95 °C for 10 s, 60 °C for 30 s; followed by a melt curve analysis. Relative gene expression was normalized to GAPDH and calculated using the 2 − ΔΔCt method36.
Western blotting analysis
Proteins were extracted using RIPA buffer (Beyotime#P0013B, Shanghai, China) supplemented with protease inhibitors (Roche#04693132001, Switzerland). Lysates were centrifuged (12,000 × g, 15 min, 4 °C), and supernatants were quantified via BCA assay (Thermo Fisher#23225, USA). Samples (20 µg protein/lane) were separated by 10% SDS-PAGE, transferred to PVDF membranes37 (Millipore#IPVH00010, USA) and blocked with 5% non-fat milk. Membranes were probed overnight at 4 °C with primary antibodies: anti-C1QTNF6 (1:1000, Abcam#ab36898, UK), anti-TET2 (1:1000, Abcam#ab309481, UK), and anti-GAPDH (1:5000, Proteintech#60004-1-Ig, Wuhan, China). HRP-conjugated secondary antibodies (Proteintech, Wuhan, China) were used, and signals were detected with ECL reagent (Thermo Fisher#34577, USA). Band intensities were quantified using ImageJ (NIH).
CCK-8 and transwell assays
Cells (3 × 10³/well) were seeded in 96-well plates. 10 µL CCK-8 reagent (Beyotime#No. C0037, Shanghai, China) was added to each well, incubated for 2 h at 37 °C, and absorbance was measured at 450 nm.
For invasion assays38, 5 × 10⁴ cells in serum-free medium were seeded into Matrigel-coated inserts (Corning#354480, USA). Complete medium (10% FBS) was added to the lower chamber. After 24 h, non-invading cells were removed, and migrated cells were fixed (4% PFA), stained (0.1% crystal violet), and counted under a microscope.
Co-immunoprecipitation (Co-IP)
Cells were lysed in IP buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, protease inhibitors). Lysates (500 µg) were incubated overnight at 4 °C with 2 µg of target antibody or IgG control (Proteintech#B900610, Wuhan, China), followed by 2 h with Protein A/G beads (Beyotime#P2179M, Wuhan, China). Beads were washed 3× with IP buffer, boiled in 2× Laemmli buffer, and analyzed by Western blotting39.
ChIP-sequencing
Chromatin was crosslinked with 1% formaldehyde, quenched with glycine, and sonicated (Bioruptor) to 200–500 bp fragments. Lysates were immunoprecipitated with anti-TET2 antibody (1:1000, Abcam#ab309481, UK) or control IgG (Proteintech#B900610, Wuhan, China). DNA was purified, and libraries were prepared using the NEBNext Ultra II DNA Kit (NEB#E7645S). Sequencing was performed on Illumina NovaSeq (150 bp paired-end). Reads were aligned to the human hg38 genome using Bowtie2, peaks were called with Model-based Analysis of ChIP-Seq 2 (MACS2) with a threshold of false discovery rate (FDR) < 0.05, and visualized with Next-generation Sequencing Plot (Ngsplot)40.
Statistical analysis
All statistical analyses were performed with R software (version 4.3.2) and GraphPad Prism (version 10.0). Two-sided P-values < 0.05 were considered statistically significant unless otherwise specified. Continuous variables are presented as mean ± standard deviation (SD); categorical variables are summarized as frequencies and percentages.
Ethical review and approval were waived in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and institutional requirements.
Data availability
The original data presented in this study were included in the article. All data were retrieved from public databases.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Su, S. et al. Preoperative serum gamma-glutamyltransferase as a prognostic biomarker in patients undergoing radical cystectomy for bladder cancer. Front. Oncol. 11, 648904 (2021).
Tan, W. S. et al. Intermediate-risk non-muscle-invasive bladder cancer: updated consensus definition and management recommendations from the International Bladder Cancer Group. Eur. Urol. Oncol. 5, 505–516 (2022).
Ye, F. et al. DNA methylation modification map to predict tumor molecular subtypes and efficacy of immunotherapy in bladder cancer. Front Cell Dev. Biol. 9, 760369 (2021).
Wan, M., Meng, H. & Li, H. Potential role of TWIST1 and its methylation in bladder urothelial carcinoma. Transl. Cancer Res 13, 6070–6086 (2024).
De La Cruz-Sigüenza, D. A. et al. The non-vesicle cell-free DNA (cfDNA) induces cell transformation associated with horizontal DNA transfer. Mol. Biol. Rep. 51, 174 (2024).
Dillinger, T. et al. Identification of tumor tissue-derived DNA methylation biomarkers for the detection and therapy response evaluation of metastatic castration resistant prostate cancer in liquid biopsies. Mol. Cancer 21, 7 (2022).
Li, S. et al. Genome-wide analysis of cell-free DNA methylation profiling for the early diagnosis of pancreatic cancer. Front. Genet. 11, 596078 (2020).
Qi, J. et al. Plasma cell-free DNA methylome-based liquid biopsy for accurate gastric cancer detection. Cancer Sci. 115, 3426–3438 (2024).
D’Andrea, D. et al. Diagnostic accuracy, clinical utility and influence on decision-making of a methylation urine biomarker test in the surveillance of non-muscle-invasive bladder cancer. BJU Int 123, 959–967 (2019).
Papadimitriou, M. A. et al. Preoperative Cell-Free DNA (cfDNA) in Muscle-Invasive Bladder Cancer Treatment Outcome. Clin. Chem. 69, 399–410 (2023).
Pilala, K. M. et al. MIR145 core promoter methylation in pretreatment cell-free DNA: a liquid biopsy tool for muscle-invasive bladder cancer treatment outcome. JCO Precis. Oncol. 8, e2300414 (2024).
Bettegowda, C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra24 (2014).
Dadhania, V. et al. Meta-analysis of the luminal and basal subtypes of bladder cancer and the identification of signature immunohistochemical markers for clinical use. EBioMedicine 12, 105–117 (2016).
Broderick, T., Gelman, A., Meager, R., Smith, A. L. & Zheng, T. Toward a taxonomy of trust for probabilistic machine learning. Sci. Adv. 9, eabn3999 (2023).
Erbe, R., Gore, J., Gemmill, K., Gaykalova, D. A. & Fertig, E. J. The use of machine learning to discover regulatory networks controlling biological systems. Mol. Cell 82, 260–273 (2022).
Hu, W., Chen, S., Thorne, R. F. & Wu, M. TP53, TP53 target genes (DRAM, TIGAR), and autophagy. Adv. Exp. Med. Biol. 1206, 127–149 (2019).
Sherwani, S. et al. Estrogenized HSA induced high-affinity autoantibodies in breast cancer-Novel biomarker for early detection. Front. Oncol. 14, 1493320 (2024).
Kim, T., Issa, D. & Onyshchenko, M. Analyzing TCGA data to identify gene mutations linked to hepatocellular carcinoma in Asians. Gastrointest. Tumors 9, 43–58 (2022).
Clough, E. et al. NCBI GEO: archive for gene expression and epigenomics data sets: 23-year update. Nucleic Acids Res. 52, D138–D144 (2024).
Jordahl, K. M. et al. Mediation by differential DNA methylation of known associations between single nucleotide polymorphisms and bladder cancer risk. BMC Med. Genet. 21, 228 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Yu, R., Kong, X. & Li, Y. Optimizing the diagnostic algorithm for pulmonary embolism in acute COPD exacerbation using fuzzy rough sets and support vector machine. COPD 20, 1–8 (2023).
Wang, R., Shen, J., Chen, Y., Gao, J. & Yao, J. Fatty acid metabolism-related signature predicts survival in patients with clear cell renal carcinoma. Aging 14, 9969–9979 (2022).
Pan, P. et al. Significance of platelets in the early warning of new-onset AKI in the ICU by using supervise learning: a retrospective analysis. Ren. Fail 45, 2194433 (2023).
Tigga, N. P. & Garg, S. Efficacy of novel attention-based gated recurrent units transformer for depression detection using electroencephalogram signals. Health Inf. Sci. Syst. 11, 1 (2023).
Wang, L., Duan, S. B., Yan, P., Luo, X. Q. & Zhang, N. Y. Utilization of interpretable machine learning model to forecast the risk of major adverse kidney events in elderly patients in critical care. Ren. Fail 45, 2215329 (2023).
Shilpashree P. S., et al. Grading the severity of damage to the perijunctional actomyosin ring and zonula occludens-1 of the corneal endothelium by ensemble learning methods. J. Ocul. Pharmacol. Ther. (2023).
Xia, L., Nan, B. & Li, Y. Debiased lasso for stratified Cox models with application to the national kidney transplant data. Ann. Appl Stat. 17, 3550–3569 (2023).
Sithara, S., Crowley, T., Walder, K. & Aston-Mourney, K. Identification of reversible and druggable pathways to improve beta-cell function and survival in Type 2 diabetes. Islets 15, 2165368 (2023).
Hou, Z. et al. Co-expression prognostic-related genes signature base on propofol and sevoflurane anesthesia predict prognosis and immunotherapy response in glioblastoma. Ann. Med. 55, 778–792 (2023).
Liu, L. et al. Overexpression of POLA2 in hepatocellular carcinoma is involved in immune infiltration and predicts a poor prognosis. Cancer Cell Int. 23, 138 (2023).
Zhang, Y. et al. Landscape of PCOS co-expression gene and its role in predicting prognosis and assisting immunotherapy in endometrial cancer. J. Ovarian Res. 16, 129 (2023).
Yang, L. et al. ELF3 promotes gemcitabine resistance through PKMYT1/CDK1 signaling pathway in gallbladder cancer. Cell Oncol. 46, 1085–1095 (2023).
Pataer, P., Zhang, P. & Li, Z. Single methylation sensitive restriction endonuclease-based cascade exponential amplification assay for visual detection of DNA methylation at single-molecule level. Anal. Chem. 96, 13335–13343 (2024).
Adhikari, B., Tellez-Isaias, G., Jiang, T., Wooming, B. & Kwon, Y. M. Development of real-time PCR assay for quantitative detection of Clostridium septicum. Poult. Sci. 103, 103681 (2024).
Kroon, C. et al. Blind spots on western blots: assessment of common problems in western blot figures and methods reporting with recommendations to improve them. PLoS Biol. 20, e3001783 (2022).
James, N., Kini, S., Pai, S., Shenoy, N. & Kabekkodu, S. P. Comparative evaluation of corneal storage medias used as tooth avulsion medias in maintaining the viability of periodontal ligament cells using the cell counting kit-8 assay. Clin. Cosmet. Investig. Dent. 14, 87–94 (2022).
Lin, J. S. & Lai, E. M. Protein-protein interactions: co-immunoprecipitation. Methods Mol. Biol. 1615, 211–219 (2017).
Sanz, L. A. & Chédin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 14, 1734–1755 (2019).
Acknowledgements
This study was financially supported by the public welfare projects of Huzhou Science and Technology Bureau (2024GZ84), Zhejiang Province's Elite and 'Leading Swan' Research and Development Project (2025C02058), Zhejiang Provincial Natural Science Foundation of China (LTGY24H160039).
Author information
Authors and Affiliations
Contributions
S.J.W. wrote the draft manuscript; W.R.J. and Z.Y.J. edited the manuscript, L.Z.J. analyzed the data and D.G.Q. draw the figures. All authors revised the manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, J., Li, Z., Wang, R. et al. Bladder cancer diagnostic and prognostic models from DNA methylation by multi algorithm machine learning. npj Precis. Onc. 9, 409 (2025). https://doi.org/10.1038/s41698-025-01195-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01195-y
