
Abstract
The aggressive subtype of hepatocellular carcinoma (HCC) is associated with a poor prognosis, and histopathological biopsy is the current method used for its diagnosis and tumour microenvironment analysis. Hence, we constructed a radiomics-based artificial intelligence model with robust predictive performance and explored the underlying biological characteristics by analysing mRNA data. The predictive performance was validated in two external centres, yielding areas under the curve ranging from 0.79 to 0.84, and their ability to predict progression-free survival (PFS) was evaluated. Radiogenomics analysis revealed that the high-risk group exhibited increased cell proliferation and tumour immune suppression. KIT inhibitors may serve as potential therapeutic drugs, whereas ADAM9 and PTK2B are key genes influencing patient prognosis. The artificial intelligence model developed from MRI has emerged as a dependable method for predicting aggressive HCC, with further biological exploration offering the potential to augment its clinical utility.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC) is the most common primary malignant tumour of the liver and is one of the leading causes of cancer death, with high rates of recurrence and mortality1. According to the 5th edition of the WHO classification of tumours of the digestive system, hepatocellular carcinoma is classified into a conventional (or nonspecific) type and a range of histological variants2. The macrotrabecular-massive (MTM) subtype constitutes approximately 10–15% of all hepatocellular carcinomas3. Compared with typical hepatocellular carcinoma, MTM-HCC presents higher histological grading and staging, leading to a worse prognosis4. Another recently discovered novel pathological phenomenon involves endothelial cells surrounding clusters of tumour cells, dividing the tumour into independent spheroid-like units called vessels encapsulating tumour clusters (VETC)5. This type of metastasis involves a tumour metastasis mechanism independent of the epithelial‒mesenchymal transition theory, referred to as the VETC metastasis mode6,7, which often leads to more frequent recurrence and shorter overall survival in patients8. Studies have revealed that HCC with the VETC pattern is largely classified as MTM-HCC5,9,10,11, implying a biological correlation between MTM-HCC and VETC formation; hence, investigating MTM-HCC with the VETC pattern holds significant value.
Currently, several studies have utilized medical imaging to construct models for the preoperative prediction of MTM-HCC or VETC-HCC, all of which yield satisfactory results9,12,13,14. Feng et al.15 further predicted the immune infiltration pattern of the MTM subtype via a CT radiomics model. Notably, in a study based on CT features by Feng et al.16, patients with high predictive scores in both the MTM model and the VETC model had the worst prognosis. Similar results were reported by Kurebayashi et al.17, who reported that VETC/MTM-positive HCC could further stratify HCCs into those associated with a worse prognosis. Accurately predicting the prognosis of MTM-VETC HCC is highly valuable. Increasing evidence from research has demonstrated that radiomics combined with transcriptomics can reveal the intrinsic biological characteristics of the research target18,19,20. To our knowledge, no study has used medical imaging information to predict HCC that simultaneously possesses MTM and the VETC-pattern and to investigate the biological inclinations of associated imaging information at the transcriptomic level.
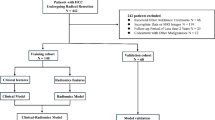
The purpose of this study was to construct a machine learning model based on contrast-enhanced MRI for the preoperative prediction of HCC with both the MTM subtype and VETC pattern and to explore the underlying biological characteristics by analysing bulk RNA sequencing data. The entire analysis process is illustrated in Fig. 1.

a Data preparation for constructing the predictive models. Patient clinical and MRI data were collected, regions of interest in the images were segmented, and radiomics features were extracted and filtered. b Construction and testing of the models, validation of the ability of the models to predict MTM-VETC HCC in two external validation sets, and exploration of the ability of the models to predict prognosis in the outcome set. c Results of radiogenomics analysis. Exploring the biological characteristics between model groups through enrichment analysis and immune cell infiltration analysis, identifying hub genes for the high-risk group predicted by the model, exploring their potential therapeutic targets, and studying their impact on patient prognosis. MTM-VETC macrotrabecular-massive subtype and vessels encapsulating the tumour cluster pattern, HCC hepatocellular carcinoma.
Results
Patient characteristics
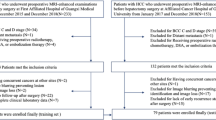
A total of 266 patients with a mean age of 57 years ± 12 (standard deviation) (218 males, 48 females) met the inclusion criteria (Fig. 2). Table 1 summarizes the clinical and pathological characteristics of the patients in the training and external validation sets. Most patients from Institutions I and II had cirrhosis (144 of 236 [61%]). This information was not available for the TCGA cohort. For the Liver Imaging Reporting and Data System (LI-RADS) category, patients classified as LR-5 or LR-M accounted for more than half of the cohort (60.8–76.7%). The inter-reader agreements (Cohen’s kappa) for LI-RADS categorization were 0.957, 0.941, and 0.902 for the training set, vali-set1 and vali-set2, respectively (Table S1). Approximately 14–28% of patients were classified as having HCC with the MTM subtype and VETC pattern (Supplementary Fig. 1), and these patients were more likely to have elevated serum α-fetoprotein (AFP) levels than non-MTM-VETC hepatocellular carcinoma patients were (p < 0.05).

Flowchart of patient selection.
Performance of the predictive models
A total of 9 radiomics features were selected for the eXtreme Gradient Boosting (XGBoost) algorithm to construct the radiomics model (Supplementary Tables 2, 3). The radiomics score of patients with the MTM-VETC subtype was significantly greater than that of patients with the non-MTM-VETC subtype (mean, 60.1 ± 23.1 vs. 17.8 ± 20.3; p < 0.001). Multivariate analysis revealed that an elevated serum AFP level and radiomics score are independent predictive factors for the MTM-VETC subtype (Fig. 3a, Table 2), leading us to integrate these two variables to construct a clinical–radiomics model (Fig. 3b), described by the formula Y = (−4.589) + 1.452 × high serum AFP level + 0.057 × radiomics model score. Since the serum AFP level was the only statistically significant risk factor among the clinical/laboratory data in the univariate regression analysis, we constructed a clinical–LIRADS model by integrating the LI-RADS category and AFP level.

a Forest plot of predictors for the MTM-VETC subtype in the training set. b The clinical–radiomics model presented with a nomogram scaled by the proportional regression coefficient of each predictor. Performance of the predictive models for predicting the MTM-VETC subtype with receiver operating characteristic curve analysis in the training set (c), external validation set 1 (d), and external validation set 2 (e). AFP α-fetoprotein, PLT platelet, ALT alanine aminotransferase, AST aspartate aminotransferase, TBIL total bilirubin, ALB albumin, mALBI modified albumin-bilirubin, PT prothrombin time, OR odds ratio.
Receiver operating characteristic (ROC) curve analysis revealed that the AUCs ranged from 0.76 to 0.91 for the radiomics model and from 0.79 to 0.92 for the clinical–radiomics model in predicting the MTM-VETC subtype. The predictive power was clearly superior to that of the baseline models (AUCs of the LIRADS model: 0.51–0.61; AUCs of the clinical–LIRADS model: 0.67–0.76) (Fig. 3c–e, Table 3). The prediction performance results revealed significant differences in the AUCs between the baseline models (LIRADS model and clinical–LIRADS model) and the radiomics-based models (radiomics model and clinical–radiomics model) in the training set (p < 0.05). Through the radar plot, we found that the clinical–radiomics model demonstrated the best overall performance (Fig. 4a–c). Decision curve analysis revealed that the clinical–radiomics model provided greater net benefit than did the radiomics model and the other two baseline models across threshold probabilities of 15–60% in all three datasets (Fig. 4d–f). The calibration curves further revealed that the clinical–radiomics model exhibited superior calibration accuracy compared with the radiomics and the baseline models, with its predicted probabilities more closely matching the observed outcomes (Fig. 4g–j). The precision‒recall curves revealed that the clinical–radiomics model outperformed the other three models (Supplementary Fig. 2a–c). In the outcome set, the median follow-up period was 16 months. Patients in the low-score group predicted by both the radiomics model (p = 0.003, log-rank test) and the clinical–radiomics model (p = 0.04, log-rank test) had longer PFS than did those in the high-score group (Fig. 4k, l).

The radar plot visualizes the overall performance of the predictive models on the training set (a), external vali-set1 (b), and external vali-set2 (c). Decision curve analysis for the predictive models in the training set (d) and the two validation sets (d–f). g–j Calibration curves of the predictive models in the three datasets. Kaplan‒Meier curves of progression-free survival in the outcome set based on the median of the radiomics model score (k) and clinical–radiomics score (l).
Biologic functions and immune cell infiltration associated with the radiomics-based model
By comparing gene expression in the low- and high-risk groups via the clinical–radiomics model, we identified 288 differentially expressed genes (DEGs), as shown in Supplementary Table 4. By analysing the DEGs, we identified distinct immune microenvironmental features between the low- and high-risk groups. Gene Ontology (GO) term analysis results indicated that the DEGs were enriched mostly in immune-related pathways (Fig. 5a). In addition, gene set enrichment analysis (GSEA) results revealed the activation of immune-related pathways, such as the interferon α/γ response and inflammatory response, in the high-risk group. Therefore, we applied CIBERSORTx and single-sample gene set enrichment analysis (ssGSEA) to analyse differences in immune cell infiltration between the low- and high-risk groups. We found that HCCs in the high-risk group presented relatively greater infiltration of M2 macrophages, resting CD4+ memory T cells, resting NK cells, and activated NK cells (Fig. 5f), along with reduced NK-T-cell infiltration (Fig. 5g).

a GO analysis revealed that differentially expressed genes (DEGs) between the high- and low-risk groups were enriched mostly in immune-related pathways. The results of gene set enrichment analysis (GSEA) comparing the high-risk group to the low-risk group (b) demonstrate the activation of pathways, including cell proliferation-related gene sets (c) and the suppression of metabolic pathways (d). e Spearman correlation between the clinical–radiomics score and the Ki-67 labelling index of patients from Institution I and Institution II. f, g The proportions of immune cells in the high- and low-risk groups. Significant differences between the two subgroups were assessed via the Wilcoxon test (ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
GSEA also revealed activation of cell proliferation-related gene sets21, such as genes related to the G2M checkpoint, E2F targets, IL6–JAK–STAT3 signalling, and PI3K–AKT–MTOR signalling, in the high-risk group (Fig. 5b, c). Moreover, the GSEA results revealed that metabolic pathways, including bile acid and fatty acid metabolism, were suppressed in the high-risk group (Fig. 5d). Consequently, we analysed the relationship between the clinical–radiomics score and Ki-67, an immunohistochemical marker indicative of tumour proliferation, and found a significant positive correlation between them (ρ = 0.31, p < 0.001) (Fig. 5e).
Exploring the hub genes of the radiomics-based model and their clinical application value
Weighted gene coexpression network analysis (WGCNA) revealed that the ‘light yellow’ module had the strongest correlation with the clinical–radiomics model score (Fig. 6a). We performed PPI analysis on the genes within the module and identified 26 hub genes, including MAK16, LSM1, ADAM9, and PTK2B, among others (Supplementary Fig. 3a–d, Supplementary Tables 5, 6). To identify potential therapeutic target pathways in the high-risk group, we conducted Connectivity Map (CMap) analysis on the hub genes and present the top 1% ranked compounds in Fig. 6b. The counts in the results represent the frequency of the same mechanisms of action among the selected compounds. We observed a higher frequency and relatively higher normalized connectivity score for KIT inhibitors in terms of the mechanism of action. Consequently, they are regarded as potential therapeutic target pathways for patients in high-risk groups.

a Gene modules related to the high- and low-risk groups obtained via WGCNA. b Top 1% ranked compounds, along with their corresponding mechanisms of action identified through CMap analysis. Kaplan‒Meier curves of progression-free survival in the TCGA set (c) and GEO set (d) based on the median A–P (ADAM9 and PTK2B) score.
To investigate the impact of hub genes on the prognosis of HCC patients. We gathered the expression levels of the hub genes and clinical information from 341 patients with HCC in the TCGA. Univariate and multivariate Cox regression analyses were conducted among the hub genes (Supplementary Table 7). As depicted in Supplementary Fig. 4, two genes (ADAM9 and PTK2B) significantly influenced the PFS of HCC patients. We subsequently computed a prognostic index for all samples via the following formula: A–P score = expression level of ADAM9 × (0.14988) + expression level of PTK2B × (-0.30710). When the median A–P score was used as the cut-off value, low-score patients exhibited better PFS than high-score patients did (p < 0.001, log-rank test; Fig. 6c). The role of the A–P score was then validated via the GSE14520 (n = 242) HCC dataset. As presented in Fig. 6d, patients in the low-score subgroup had a significantly better prognosis than those in the high-score subgroup did, which was consistent with the results from the TCGA dataset (p = 0.006, log-rank test).
Discussion
In this study, we developed radiomics and clinical–radiomics models to predict the MTM subtype with the VETC pattern in HCC patients preoperatively based on clinical information and contrast-enhanced MRI. Additionally, we constructed baseline models, including the LI-RADS model and the clinical–LI-RADS model based on LI-RADS categorization, which offers standardized imaging terminology for HCC and enables precise stratification of HCC probability and overall malignancy22,23,24. In comprehensive model assessments, the clinical–radiomics model showed marginally superior performance over the radiomics model and clearly outperformed the baseline models. Furthermore, both the radiomics and the clinical–radiomics models could be applied to stratify patient prognosis. In further exploratory research, we discovered the relevant connections between metabolic pathways and immune infiltration in the tumour microenvironment of high-risk patients, as predicted by the clinical–radiomics model. After that, we identified hub genes in the high-risk group to explore potential therapeutic target pathways and their impact on prognosis.
A total of 9 radiomics features were selected; interestingly, radiomics features related to HBP were not selected. Yu et al.12 developed a radiomics model based on contrast-enhanced MRI HBP images to predict the VETC pattern and reported that the peritumoural radiomics model outperformed the intratumoural model, a finding that was consistent with observations in a study by Chong et al.25. Peritumoral hypointensity during the HBP has consistently been shown to be a strong predictor of microvascular invasion (MVI)26, and we speculate that valuable imaging information resides in the peritumoural area rather than the intratumoural area during the hepatobiliary phase. These findings seem to explain our research results. To mitigate the bias in model learning caused by sample imbalance, we adjusted the weights of positive and negative samples via the weighting parameter provided by the XGBoost model, which enabled the model to achieve good predictive performance and robustness. However, the generally lower MTM-VETC positivity rate and variations in positivity rates among the three datasets inevitably lead to a lower F1 score for the models and suboptimal performance on the precision‒recall curves. Our results indicate that elevated serum AFP levels are an independent predictor of the MTM-VETC subtype, which is consistent with previous reports5,15,27.
The enrichment analysis revealed differences in immune-related pathways between the high- and low-risk groups, particularly the suppression of bile acid metabolism, along with the upregulation of inflammatory responses and IL6–JAK–STAT3 signalling, in the high-risk group. Existing evidence suggests that bile acid in HCC leads to tumour immune suppression, resulting in a poor prognosis28,29. Studies have shown that bile acids promote M2 polarization of macrophages, inhibiting the aggregation of NK-T cells30,31. Therefore, we further investigated the immune infiltration patterns of the high- and low-risk groups. We found a significant increase in the relative abundance of M2 macrophages and a significant decrease in NK-T-cell abundance in HCC samples from the high-risk group compared with those from the low-risk group, which is consistent with previous research results. Additionally, the results suggested the activation of the IL6–JAK–STAT3 signalling pathway in high-risk patients. Evidence indicates that bile acids activate NF-κB, release proinflammatory cytokines such as TNF-α and IL-6, activate the IL-6–JAK–STAT3 pathway, inhibit cancer apoptosis, and promote the development of liver cancer24. Furthermore, cell proliferation-related gene sets were upregulated in the high-risk group, implying that HCC in the high-risk group presented increased proliferative capacity, and Ki-67, a well-established marker reflecting tumour proliferative capacity32,33, was positively correlated with the clinical–radiomics model score, which was used to validate these findings.
The tumour microenvironment is collectively influenced by many genes; thus, coexpressed gene analysis was used to select hub genes in the high-risk group. Hub genes were employed in CMap analysis, revealing that inhibitors targeting signalling pathways such as KIT inhibitors represent potential targeted treatment pathways for patients in high-risk groups. The results also indicate that, among several mechanisms of action with a relatively high frequency, sorafenib is the most frequently selected compound for treatment and has been shown to significantly improve the survival of patients with HCC with the VETC pattern34. Finally, we explored the impact of the hub genes on the prognosis of HCC patients. Among the hub genes, ADAM9 and PTK2B were identified as independent risk factors influencing the prognosis of HCC patients. PTK2B, as an interacting protein of TBK1, can enhance the immune response pathway STING-TBK1 signalling35. ADAM9, discovered in recent years as a cancer gene, is overexpressed in many cancers and is associated with tumour invasiveness and poor prognosis36,37,38,39. Sorafenib can inhibit the expression of ADAM9 RNA and protein, leading to the upregulation of membrane-bound major histocompatibility complex class I-related chain A, thereby enhancing natural killer cell activity against HCC40.
MTM-VETC HCC is characterized by high malignancy, strong invasiveness, and poor prognosis. To date, research on this subtype remains limited, and its diagnosis still depends on postoperative pathology. Moreover, this subtype is associated with a significantly higher recurrence rate after surgical resection than other HCC subtypes41,42. To address these challenges, we developed a model that enables the preoperative identification of patients at high risk for MTM-VETC HCC. Using our clinical–radiomics model, we stratified HCC patients according to their predicted risk of harbouring the MTM-VETC subtype. Patients with a clinical–radiomics score greater than 0.15 were identified as being at high risk for MTM-VETC HCC. This subgroup exhibited features of an immunosuppressive tumour microenvironment and enhanced proliferative capacity and was associated with a relatively poor prognosis. For these patients, a comprehensive therapeutic strategy combining surgical resection with immunotargeted therapies is recommended. Specifically, immunotherapies targeting M2 macrophage polarization and NK-T-cell depletion, along with targeted agents such as KIT inhibitors or drugs against ADAM9 and PTK2B, may provide substantial clinical benefit. Conversely, patients with a clinical–radiomics score less than 0.15 were considered to have a low risk of the MTM-VETC subtype. These tumours typically demonstrate stronger antitumour immune responses and are associated with more favourable outcomes. For such patients, surgical resection followed by routine surveillance via CT or MRI is considered an appropriate management approach.
Our study has several limitations. First, the model was constructed using retrospectively collected data from patients undergoing surgical treatment at multiple institutions, which may introduce selection bias. Second, the complexity and lack of algorithm standardization limit the use of radiomics markers. Future work will involve the development of prediction models based on deep learning. Third, the sample imbalance resulting from the lower positivity rate of MTM-VETC HCC restricts the predictive ability of the model, and the limited sample size of the dataset necessitates larger sample sizes for radiogenomics analysis in future studies. Finally, many experiments are needed to validate our transcriptomic analysis results.
In summary, the clinical–radiomics model, which integrates serum AFP levels and MRI-derived radiomics features, has a robust ability to preoperatively predict HCC patients with the MTM subtype and VETC pattern. In addition, this model enables effective prognostic risk stratification among HCC patients. Furthermore, our radiogenomics analysis revealed two key findings: (1) an immunosuppressive tumour microenvironment and overactivation of cell proliferation pathways likely contribute to the poor prognosis observed in high-risk patients, and (2) KIT inhibitors, such as sorafenib, may represent promising therapeutic agents, whereas ADAM9 and PTK2B have emerged as critical genes associated with patient outcomes. Overall, radiogenomics analysis provides a powerful approach to elucidate tumour biological behaviour, predict therapeutic responsiveness, and inform future prospective validation studies.
Methods
Study subjects
This study was approved by the Institutional Review Board of The First People’s Hospital of Shunde (approval number: KYLS20230409), which served as the lead ethics committee. The approval was acknowledged by The First People’s Hospital of Foshan through an expedited review process. The study protocol adhered strictly to the ethical principles outlined in the revised Declaration of Helsinki (2013). Given the retrospective nature of the study, the requirement for informed consent was waived. HCC patients who underwent curative hepatectomy from August 2017 to December 2022 were retrospectively collected to build the predictive models, consisting of a training set from The First People’s Hospital of Shunde (institution I) and two external validation sets from The First People’s Hospital of Foshan (institution II) and The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/projects/TCGA-LIHC). The inclusion criteria were as follows: (a) preoperative contrast-enhanced MRI performed within 2 weeks and (b) available pathological tissue slides with pathological confirmation of HCC. Exclusion criteria were: (a) receipt of other treatments before surgery and (b) poor image quality with obvious artifacts. Following the criteria, 266 patients were enroled for the radiomic analysis.
The outcomes set included 110 patients from the above two medical institutions, who not only met the aforementioned criteria but also had regular postoperative imaging examinations and follow‒up data available. Progression‒free survival (PFS) was defined as the duration from treatment initiation to the onset of progression or death. The genomics set included 29 patients from TCGA who were selected for radiogenomic analysis, and the validation set included 242 HCC samples (GSE14520) with bulk‒RNA seq and clinical prognosis data from the Gene Expression Omnibus (GEO) were used to validate radiogenomic analysis results.
Clinical and laboratory data
Clinical data, including age, sex, preoperative prothrombin time, preoperative serum α-fetoprotein, platelet count, alanine aminotransferase, aspartate aminotransferase, total bilirubin and albumin were recorded. The mALBI grade is assigned based on the result of the formula: mALBI score = (log10 bilirubin [μmol/L] × 0.66) + (albumin [g/L] × −0.0852). A score of −2.60 or lower is grade 1, −2.27 to −2.60 is grade 2a, equal to or higher than −2.27 to −1.39 is grade 2b, and −1.39 or higher is grade 343.
Histopathologic examination
Pathological slides of tumour tissues were reviewed by a pathologist (X.Z., 20 years of experience) to determine the MTM subtype and VETC pattern. The MTM subtype is defined as HCC growth with a macrotrabecular pattern (≥ 6–10 cells thick) covering more than 50% of the tumour area3. The VETC pattern refers to sinusoid-like vessels (CD34+) encapsulating individual tumour clusters and forming cobweb-like networks5. The sections were counterstained with haematoxylin‒eosin, and the largest one was considered the index tumour for subsequent analysis if a patient presented with multiple lesions. Immunohistochemistry was conducted using a commercially available monoclonal mouse anti-human Ki-67 antibody. The Ki-67 labelling index (LI) was determined by calculating the percentage of immunoreactive cells among 1000 malignant cells (×400), and scoring was carried out in the region within the tumour with the highest number of positive nuclei (hot spot)44. The HCC patients were classified into two groups: the MTM-VETC group (both MTM- and VETC-positive) and the non-MTM-VETC group (both MTM- and VETC-negative or only one is positive).
Image analysis
All MRI scans were independently reviewed by two radiologists (Y.C. and Z.C., with 6 and 7 years of experience, respectively) to perform LI-RADS categorization (American College of Radiology, version 2018). Any discrepancies were resolved by consultation with a senior radiologist (Z.L., with 10 years of experience in abdominal MRI). The three readers were aware of the diagnosis of HCC but were blinded to all other clinical and pathological information, including the MTM subtype and VETC pattern status. Please refer to the Supplementary Table 8 for details on the MRI equipment, settings, and scanning methods used in this study.
Image preprocessing and radiomic feature extraction
Two radiologists (Y.C., Z.C.; with 6 and 7 years of experience, respectively), blinded to clinicopathologic data, conducted tumour segmentation using 3DSlicer software (version 5.2.2; https://www.slicer.org/). Volumes of interest were derived by Regions of Interest manually outlining for the tumour by layer on the T1WI pre-enhancement, arterial phase, portal venous phase, delayed phase, hepatobiliary phase images, and T2WI. All VOIs were verified by a senior radiologist (Z.L.), and any discrepancies were resolved through consensus. Two radiologists (Y.C., Z.C.) randomly selected 30 patients for the repeated delineation of target areas to assess intra- and inter-observer reproducibility after 2 weeks.
Image preprocessing was conducted using the SimpleITK v2.3.1 package (https://github.com/SimpleITK/SimpleITK). Initially, N4 bias field correction was applied to the images to correct low-frequency intensity non-uniformity present in MRI images. Subsequently, the voxel dimensions were resampled to 1 × 1 × 1 mm (x-, y-, and z-axes). Finally, the image signal intensity was normalized and resampled into 64 bins. Radiomic features were extracted from each VOI using PyRadiomics v3.1.0 (https://github.com/AIM-Harvard/pyradiomics). 106 radiomic features were extracted from each segmentation, as summarized in Supplementary Table 9, resulting in a total of 636 features for each patient. The z-score method was employed for the standardization of radiomic feature values.
Model building and testing
Initially, we used the LI-RADS classification to develop the baseline models. The LI-RADS model was first derived via logistic regression using the LI-RADS category and was further expanded into the clinical–LIRADS model with the addition of clinical and laboratory features. Radiomics features with intra- and interobserver reproducibility scores greater than 0.75 were selected for comparing the MTM-VETC and non-MTM-VETC groups. Univariable logistic regression was used to identify statistically significant variables among radiomics features with a p-value less than 0.05. Then, we utilized Pearson correlation analysis (with a Pearson correlation coefficient >0.75) and least absolute shrinkage and selection operator regression with 10-fold cross-validation to limit the multicollinearity of the variables. The selected features were integrated into an XGBoost model for the construction of the radiomics model. To address the issue of imbalanced samples in the training set where the MTM-VETC positivity ratio is small (22 of 162 [14%]), the “scale pos weight” parameter inherent to the XGBoost model was used to adjust the weights of the MTM-VETC positivity and non-MTM-VETC positivity samples. Uni- and multivariable logistic regression analyses were used to construct the clinical–radiomics model.
The predictive ability of each model was evaluated through receiver operating characteristic curve analysis, the area under the curve of each model was computed, and the results were compared via the DeLong test. The overall performance of the models was demonstrated through a radar plot. Calibration curves and decision curve analysis were employed to further assess the goodness of fit and clinical utility of the models. Additionally, precision‒recall curves were generated to compare the performances of the different models. Two external sets were used for validation.
Radiogenomic analysis
To explore the specific biological characteristics of the high- and low-risk groups via the clinical–radiomics model. The genomics was set using the cut-off value corresponding to the maximum Youden index. DEGs were identified via the edgeR package with the criteria of |log2 (fold change)|> 1 and a false discovery rate (FDR) < 0.05. The clusterProfiler package was employed for GO enrichment analysis and GSEA with hallmark gene sets, where p values, adjusted via the Benjamin‒Hochberg method, less than 0.05 were deemed statistically significant. The relationships between Ki-67 expression levels and clinical–radiomics model scores were studied via Spearman correlation analysis.
Two methods were employed to evaluate immunocyte infiltration in the tumour microenvironment. Twenty-two infiltrating immune cell types were quantified via CIBERSORTx (https://cibersortx.stanford.edu/). This method uses a deconvolution algorithm to compute the proportions of these 22 immune cell types in each sample, and the total estimated values for all immune cell types in each sample sum to 100%. In the ssGSEA algorithm, the relative abundances of 28 immune cell types were calculated. This algorithm determines the enrichment scores of these 28 immune cell types in each sample based on their gene sets.
To identify hub genes associated with the low- and high-risk groups, we utilized the WGCNA method to construct coexpressed gene modules. Initially, we calculated the median absolute deviation of all genes normalized by fragments per kilobase of transcript sequence per million base pairs sequenced and selected the top 25% ranked genes for further analysis. Then, the Pearson correlation coefficient between adjacent genes in the gene expression matrix was computed to construct a similarity matrix. The similarity matrix was subsequently transformed into an unsigned adjacency matrix by exponentiating the correlation values, with the exponent, also known as the soft threshold, set to 10 (Supplementary Fig. 5). Next, the adjacency matrix was converted into a topological overlap matrix (TOM), where 1–TOM is used as the distance for gene clustering to generate a dynamic pruning tree for module identification. Modules with a p-value less than 0.05 and the highest correlation coefficient values were identified as hub modules. Genes in the hub modules were selected for protein‒protein interaction (PPI) network construction via the Search Tool for the Retrieval of Interacting Genes (STRING) online database (http://string-db.org/). Twelve topological analysis methods embedded in the CytoHubba plugin of Cytoscape were utilized to identify hub genes. Genes repeatedly ranked in the top 10 across the 12 algorithms were identified as hub genes45.
We used CMap to predict potential therapeutic target pathways for the hub genes in the high-risk group. CMap is a gene expression database based on postdrug intervention that is utilized to identify potential therapeutic compounds through the similarity of expression profiles46. Each compound in the CMap analysis results corresponds to a normalized connectivity score, with a smaller value indicating that the compound is more effective in reversing the altered expression of hub genes in high-risk groups. We filtered compounds from the CMap analysis results with false discovery rate (FDR) values less than 0.05 and then selected the top 1% of compounds with the smallest normalized connectivity scores among those with scores less than 0.
Finally, to investigate the impact of the hub genes on prognosis, we performed univariate and multivariate Cox regression analyses to identify genes with independent prognostic factors among the hub genes and construct a prognostic model accordingly. Kaplan‒Meier (KM) survival analysis was employed to validate the predictive ability of the prognostic genes. We sought an external validation set (GSE14520) for further confirmation of the prognostic efficacy of the prognostic genes.
Statistical analysis
Age was compared between groups via the Kruskal‒Wallis H test. Categorical variables were compared via the χ2 test. A kappa statistic of 0.8–1.0, 0.6–0.79, 0.40–0.59, 0.2–0.39, and 0–0.19 was considered excellent, good, moderate, fair, and poor agreement, respectively. Within the outcome set, patients were categorized into low- or high-score groups according to the median value of the radiomics model score and clinical–radiomics model score. Survival curves were generated via the Kaplan‒Meier method, and a subsequent log-rank test was performed. Statistical analyses were performed via R software (version 4.3.2; https://www.r-project.org/) and Python (version 3.9.13; https://www.pyhon.org/).
Data availability
Clinical and imaging data for the TCGA cohort are available through The Cancer Imaging Archive (https://www.cancerimagingarchive.net/), whereas digitized histopathology images and RNA-seq data are accessible via The Cancer Genome Atlas (https://portal.gdc.cancer.gov/projects/TCGA-LIHC). For the GEO cohort, clinical data and RNA-seq data are available from the Gene Expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi). However, clinical and imaging data from patients at Shunde Hospital of Southern Medical University and The First People’s Hospital of Foshan are not publicly available owing to privacy/ethical restrictions but can be provided by the corresponding author upon reasonable request.
Code availability
The codes and pipelines used to analyse sequencing data are as follows: https://github.com/IvanWannaRunAway/VETC-MTM-HCC-Radiogenomics-workflow.
References
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2, 16018 (2016).
Nagtegaal, I. D. et al. The 2019 WHO classification of tumours of the digestive system. Histopathology 76, 182–188 (2020).
Ziol, M. et al. Macrotrabecular-massive hepatocellular carcinoma: A distinctive histological subtype with clinical relevance. Hepatology 68, 103–112 (2018).
Jeon, Y., Benedict, M., Taddei, T., Jain, D. & Zhang, X. Macrotrabecular Hepatocellular Carcinoma: An Aggressive Subtype of Hepatocellular Carcinoma. Am. J. Surg. Pathol. 43, 943–948 (2019).
Renne, S. L. et al. Vessels Encapsulating Tumor Clusters (VETC) Is a Powerful Predictor of Aggressive Hepatocellular Carcinoma. Hepatology 71, 183–195 (2020).
Zhou, H.-C. et al. Dual and opposing roles of the androgen receptor in VETC-dependent and invasion-dependent metastasis of hepatocellular carcinoma. J. Hepatol. 75, 900–911 (2021).
Hanley, K. L. & Feng, G.-S. A new VETC in hepatocellular carcinoma metastasis. Hepatol. Baltim. Md 62, 343–345 (2015).
Zhang, P. et al. The presence of vessels encapsulating tumor clusters is associated with an immunosuppressive tumor microenvironment in hepatocellular carcinoma. Int. J. Cancer 151, 2278–2290 (2022).
Rhee, H. et al. Gadoxetic acid-enhanced MRI of macrotrabecular-massive hepatocellular carcinoma and its prognostic implications. J. Hepatol. 74, 109–121 (2021).
Fang, J.-H. et al. A novel vascular pattern promotes metastasis of hepatocellular carcinoma in an epithelial-mesenchymal transition-independent manner. Hepatol. Baltim. Md 62, 452–465 (2015).
Rhee, H. et al. Hepatocellular carcinoma with irregular rim-like arterial phase hyperenhancement: more aggressive pathologic features. Liver Cancer 8, 24–40 (2018).
Yu, Y. et al. Gd-EOB-DTPA-enhanced MRI radiomics to predict vessels encapsulating tumor clusters (VETC) and patient prognosis in hepatocellular carcinoma. Eur. Radiol. 32, 959–970 (2022).
Cha, H. et al. Comparison of imaging findings of macrotrabecular-massive hepatocellular carcinoma using CT and gadoxetic acid-enhanced MRI. Eur. Radiol. 33, 1364–1377 (2023).
Mulé, S. et al. Multiphase Liver MRI for Identifying the Macrotrabecular-Massive Subtype of Hepatocellular Carcinoma. Radiology 295, 562–571 (2020).
Feng, Z. et al. CT Radiomics to Predict Macrotrabecular-Massive Subtype and Immune Status in Hepatocellular Carcinoma. Radiology 221291. https://doi.org/10.1148/radiol.221291 (2022).
Feng, Z. et al. Preoperative CT for Characterization of Aggressive Macrotrabecular-Massive Subtype and Vessels That Encapsulate Tumor Clusters Pattern in Hepatocellular Carcinoma. 300, 11 (2021).
Kurebayashi, Y. et al. Immunovascular classification of HCC reflects reciprocal interaction between immune and angiogenic tumor microenvironments. Hepatology 75, 1139–1153 (2022).
Li, G. et al. An MRI radiomics approach to predict survival and tumour-infiltrating macrophages in gliomas. Brain J. Neurol. 145, 1151–1161 (2022).
Beig, N. et al. Radiogenomic-Based Survival Risk Stratification of Tumor Habitat on Gd-T1w MRI Is Associated with Biological Processes in Glioblastoma. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 26, 1866–1876 (2020).
Wang, X. et al. Radiomics predicts the prognosis of patients with locally advanced breast cancer by reflecting the heterogeneity of tumor cells and the tumor microenvironment. Breast Cancer Res. 24, 20 (2022).
Oshi, M. et al. G2M checkpoint pathway alone is associated with drug response and survival among cell proliferation-related pathways in pancreatic cancer. Am. J. Cancer Res. 11, 3070–3084 (2021).
EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 69, (2018).
A. et al. Evidence Supporting LI-RADS Major Features for CT- and MR Imaging-based diagnosis of hepatocellular carcinoma: a systematic review. Radiology 286, (2018).
Ja, M. et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 68, (2018).
Chong, H. et al. Peritumoral Dilation Radiomics of Gadoxetate Disodium-Enhanced MRI Excellently Predicts Early Recurrence of Hepatocellular Carcinoma without Macrovascular Invasion After Hepatectomy. J. Hepatocell. Carcinoma 8, 545–563 (2021).
Kim, K. A. et al. Prediction of microvascular invasion of hepatocellular carcinoma: usefulness of peritumoral hypointensity seen on gadoxetate disodium-enhanced hepatobiliary phase images. J. Magn. Reson. Imaging 35, 629–634 (2012).
Calderaro, J. et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 67, 727–738 (2017).
Cai, J., Sun, L. & Gonzalez, F. J. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 30, 289–300 (2022).
Conde de la Rosa, L. et al. STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway. J. Hepatol. 74, 1429–1441 (2021).
Ma, C. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 (2018).
Sun, R. et al. Loss of SIRT5 promotes bile acid-induced immunosuppressive microenvironment and hepatocarcinogenesis. J. Hepatol. 77, 453–466 (2022).
Nielsen, T. O. et al. Assessment of Ki67 in Breast Cancer: Updated Recommendations From the International Ki67 in Breast Cancer Working Group. J. Natl. Cancer Inst. 113, 808–819 (2021).
Uxa, S. et al. Ki-67 gene expression. Cell Death Differ. 28, 3357–3370 (2021).
Fang, J.-H. et al. Vessels That Encapsulate Tumor Clusters (VETC) Pattern Is a Predictor of Sorafenib Benefit in Patients with Hepatocellular Carcinoma. Hepatology 70, 824–839 (2019).
Lin, Y. et al. PTK2B promotes TBK1 and STING oligomerization and enhances the STING-TBK1 signaling. Nat. Commun. 14, 7567 (2023).
Oria, V. O., Lopatta, P. & Schilling, O. The pleiotropic roles of ADAM9 in the biology of solid tumors. Cell. Mol. Life Sci. 75, 2291–2301 (2018).
Lin, Y.-S. et al. ADAM9 functions as a transcriptional regulator to drive angiogenesis in esophageal squamous cell carcinoma. Int. J. Biol. Sci. 17, 3898–3910 (2021).
Huang, Y.-K. et al. Inhibition of ADAM9 promotes the selective degradation of KRAS and sensitizes pancreatic cancers to chemotherapy. Nat. Cancer https://doi.org/10.1038/s43018-023-00720-x (2024).
Tong, Y. et al. Proteogenomic insights into the biology and treatment of pancreatic ductal adenocarcinoma. J. Hematol. Oncol. J. Hematol. Oncol. 15, 168 (2022).
Kohga, K. et al. Sorafenib inhibits the shedding of major histocompatibility complex class I-related chain A on hepatocellular carcinoma cells by down-regulating a disintegrin and metalloproteinase 9. Hepatology 51, 1264–1273 (2010).
Liu, K. et al. Vessels that encapsulate tumour clusters vascular pattern in hepatocellular carcinoma. JHEP Rep. 5, 100792 (2023).
Calderaro, J., Ziol, M., Paradis, V. & Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 71, 616–630 (2019).
Kudo, M. Newly Developed Modified ALBI Grade Shows Better Prognostic and Predictive Value for Hepatocellular Carcinoma. Liver Cancer 11, 1–8 (2022).
Hu, X.-X. et al. Whole-tumor MRI histogram analyses of hepatocellular carcinoma: Correlations with Ki-67 labeling index. J. Magn. Reson. Imaging 46, 383–392 (2017).
Chaudhary, R. K. et al. Identification of signature genes and drug candidates for primary plasma cell leukemia: An integrated system biology approach. Comput. Biol. Med. 162, 107090 (2023).
Lamb, J. et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006).
Acknowledgements
This research was supported by the Guangdong Medical Science and Technology Research Fund (A2024022); Project of Administration of Traditional Chinese Medicine of Guangdong Province of China (20241312); Foshan City's self-funded scientific and technological innovation projects (2320001006750); Scientific Research Start Plan of Shunde Hospital, Southern Medical University (SRSP2021021; SRSP2023008). We gratefully acknowledge the contributions of the TCGA (The Cancer Genome Atlas) and GEO (Gene Expression Omnibus) databases for providing open-access data, which were essential for the completion of this study.
Author information
Authors and Affiliations
Contributions
Y.C., Z.L., and Q.H. were responsible for the overall study design. Y.C., Z.C., C.L., R.Z., X.C., X.L., W.L., C.Z., X.G. and X.Z. supervised the data collection. Y.C., Z.C., C.L. and X.C. performed the data analysis. Y.C., Z.C., C.L., and R.Z. drafted the manuscript. B.G., H.C., F.O., Z.L., and Q.H. were responsible for manuscript editing. All the authors read, discussed, and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Y., Cai, Z., Luo, C. et al. Integrated analysis of radiomics and RNA reveals the biological basis and therapeutic implications of aggressive hepatocellular carcinoma subtypes. npj Precis. Onc. 10, 28 (2026). https://doi.org/10.1038/s41698-025-01233-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01233-9
