
Abstract
Bone metastasis is a major cause of morbidity and mortality in breast cancer, yet effective prognostic models and targeted therapies remain limited. Here, a machine learning (ML)-driven multi-omics framework integrating epithelial–mesenchymal transition (EMT) and nucleotide metabolism (NM) signatures is presented to uncover prognostic biomarkers and guide rational drug discovery. Using gene expression omnibus (GEO) and the cancer genome atlas-breast invasive carcinoma (TCGA-BRCA) bone metastasis datasets, applied the least absolute shrinkage and selection operator (LASSO) ML to identify NM-associated hub genes, revealing peroxiredoxin 4 (PRDX4) as a key risk-associated gene. Multi-level analyses demonstrated that PRDX4 expression correlates with immune cell infiltration, microsatellite instability (MSI), tumor mutational burden (TMB), EMT activation, and poor overall survival. Consensus clustering stratified patients into distinct EMT–NM molecular subgroups with divergent clinical outcomes, immune checkpoint expression, and tumor stemness scores, providing a foundation for precision patient stratification. To accelerate translational impact, we performed drug repurposing and molecular docking, identifying Docetaxel as a high-affinity PRDX4-targeting compound with favorable binding energetics. Together, this work demonstrates how ML-driven multi-omics analysis can bridge biomarker discovery and drug design, guiding multitarget and multi-drug strategies to improve outcomes in bone metastatic breast cancer.
Similar content being viewed by others

Introduction
Rising incidence and high mortality rates make breast cancer (BC) a major global health challenge. Epidemiological studies indicate that BC is the most common malignancy among women worldwide, and the bone is the third most frequent site of metastasis after lung and liver involvement1,2. Bone metastasis (BM) substantially affects patients’ quality of life and survival, occurring in nearly 70–85% of patients with advanced-stage disease. Despite its clinical importance, there is currently no universally accepted prognostic model specifically designed to predict BM risk or to guide therapeutic decision-making in breast cancer3,4.
The underlying molecular mechanisms that drive BC progression include epithelial–mesenchymal transition (EMT) and nucleotide metabolism (NM), both of which are strongly implicated in tumor growth, invasion, and metastasis. Invasive capacity that facilitates local invasion and dissemination to distant sites5,6. Aberrations in NM pathways, as reported in many studies, are associated with tumor progression and chemoresistance, suggesting that these pathways are promising therapeutic targets7,8. In addition, EMT and NM lead to the dissemination of BC cells into and colonization of bones, where the tumor cells manage to grow and multiply under a bone supportive niche9.
Gene Expression Omnibus (GEO) in our study, we selected the bulk RNA sequencing (bulk-seq) data of BC bone metastasis from the GEO database along with the EMT and NM gene lists from the GeneCard database and identified the EMT- and NM-related differentially expressed genes (EMT-NM DEGs) via the Limma package in R9,10,11. Secondly, the Least Absolute Shrinkage and Selection Operator (LASSO) regression was employed to screen out the EMT-NM hub genes of The Cancer Genome Atlas-Breast Cancer (TCGA-BRCA) bone metastasis cohort12,13. Finally, PRDX4 was proven as a key hub gene13,14,15,16. Then, the prognosis model based on PRDX4 and the corresponding immune infiltration pattern and molecular mechanism associated with PRDX4 were investigated and analyzed17,18,19,20.
Recent research21 emphasizes new molecular regulators of breast cancer metastasis. It is identified microRNA-204 as a modulator of the defining hallmark features of breast cancer, including proliferation, invasion, and EMT, which lead to metastatic behavior. The research22 discussed promising molecular and therapeutic strategies that specifically exploit the complexity of the underlying intrinsic mechanisms of bone-metastatic cancers, highlighting the need for precision to move the needle on patient outcomes.
Moreover, we carried out consensus clustering for EMT-NM DEGs of TCGA-BRCA bone metastasis samples to find new molecular subgroups peculiar to BC. Subsequently, we made efforts in drug sensitivity analysis and molecular docking screening to explore potential drugs against bone metastasis of BC, paving the way for future precision medicine applications among BC bone metastasis populations.
Despite the many observations that utilized multi-omics and machine learning (ML) strategies to identify biomarkers of EMT, broadening the horizons of BC and metastatic-based research, focused mainly on single-pathway analyses or transcriptomic-level analysis. In disparity, the research integrates the EMT and NM signaling axes in a multi-omics-ML framework of analysis, which allowed overall interrogation of metabolic-epithelial plasticity that drives metastatic development.
-
The research develops an ML-based multi-omics platform combining EMT and NM signatures for prognostic modeling in bone-metastatic breast cancer.
-
It identified PRDX4 as a major prognostic biomarker, mechanistically related to redox regulation, activation of EMT, and metastatic progression.
-
The research uses LASSO-Cox modeling and consensus clustering to improve patient stratification and prediction of survival.
-
The findings build drug-target networks and molecular docking approaches to propose therapeutics centered on PRDX4, as well as strategies for multi-pathway intervention.
Results
Identification of EMT-NM DEGs in GEO BRCA-BM datasets
GSE39494 and GSE137842 were obtained from the GEO database and then underwent normalization and removal of batch effects, which were illustrated by PCA and a boxplot. Subsequently, DEGs from these 2 datasets were obtained (Fig. 1A, B). Next, the intersection of DEGs from GSE39494 and GSE137842 with NM and EMT was also performed (Fig. 1C). Next, we illustrated 16 NM-EMT DEGs in GSE39494 and GSE137842, respectively (Fig. 1D). Finally, we analyzed the molecular functions of 16 EMT-NM DEGs (Fig. 1E).

A Box, PCA, and Volcano plots of DEGs in GSE39494. B Box, PCA, and Volcano plots of DEGs in GSE137842. C Venn diagram of the intersection of 16 EMT-NM DEGs. D Heatmap illustration of 16 EMT-NM DEGs in GSE39494 and GSE137842, respectively. E KEGG and GO enrichment of EMT-NM DEGs. Abbreviations: EMT, epithelial–mesenchymal transition; NM, nucleotide metabolism; DEG, differentially expressed gene; GEO, Gene Expression Omnibus; PCA, principal component analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology.
Lasso regression for hub gene identification in the TCGA-BRCA-BM cohort
After retrieving EMT-NM DEGs from 2 GEO bulk-seq datasets, we performed Lasso regression analysis to identify hub genes involved in BRCA tumor cells progression (Fig. 2A, B). According the Lasso data, we confirmed that TERF1, YY1, SNRNP70, CREBBP, CYP7B1, LATS1, ENO1, ACO2, LDHA, PRDX4 and E2F1 are associated with poor prognosis of BRCA-BM patients (Fig. 2C–E).

A, B Lasso regression targeting EMT-NM DEGs in TCGA-BRCA-BM cohort. C Risk factors: illustrations of Lasso regression results. D, E KM and time-dependent ROC analysis of Lasso regression results. Abbreviations: LASSO, least absolute shrinkage and selection operator; EMT, epithelial–mesenchymal transition; NM, nucleotide metabolism; DEG, differentially expressed gene; TCGA, The Cancer Genome Atlas; BRCA, breast cancer; BM, bone metastasis; KM, Kaplan–Meier; ROC, receiver operating characteristic.
Table 1 shows that Robustness was demonstrated using 10-fold cross-validation (mean AUC = 0.846, low error = 0.154) and the optimal λ_min = 0.0142 in order to reduce overfitting risk. In addition, we complemented these steps with the use of duplicate GEO-derived EMT-NM DEGs, as well as L1-regularized LASSO and Cox modeling, verified using ROC, calibration, and DCA analyses in order to ensure generalizability even with limited TCGA-BRCA-BM samples.
Prognostic model construction and identification of PRDX4 prognostic efficacy
For the detection of key genes involved in BM progression in the BRCA dataset, we performed uni- and multi-Cox regression (Fig. 3A). We identified PRDX4 as a key gene reflecting the poor prognosis of BRCA-BM patients. Besides, the expression of PRDX4 in BC and BC-BM groups, as well as the diagnostic efficacy of PRDX4 for BC to BC-BM (Fig. 3B–D). Significantly, nomogram, calibration, KM plot, time-dependent ROC, and risk factor plot indicated PRDX4 has excellent prognostic efficacy targeting BC to BC-BM (Fig. 3E, F). The DCA plot also indicates that PRDX4 possesses excellent prognostic accuracy (Fig. 3G).

A Cox regression analysis forest plot. B Bar plot of PRDX4 expression. C Clinical information Sankey plot. D ROC plot of PRDX4. E Prognostic Nomogram and calibration plot of PRDX4. F Risk factor, KM plot, and time-dependent ROC plot of PRDX4. G DCA plot of PRDX4. Abbreviations: PRDX4, peroxiredoxin 4; ROC, receiver operating characteristic; KM, Kaplan–Meier; DCA, decision curve analysis.
Immune and molecular role of PRDX4 in TCGA-BRCA-BM cohort
To comprehensively characterize it employed six complementary approaches were employed to explore PRDX4’s immunological role in the tumor ecosystem: TIMER, CIBERSORT, xCell, EPIC, MCP-counter, and quanTIseq. to systematically evaluate immune cell infiltration patterns in the TCGA-BRCA-BM cohort. This multi-algorithm approach ensured robust cross-validation of results across different computational frameworks. Remarkably, PRDX4 expression showed a strong positive association with CD8⁺ T-cell activation signatures (Fig. 4A, B), suggesting that PRDX4 may participate in shaping the cytotoxic immune landscape of breast cancer with bone metastasis. This finding raises the intriguing possibility that PRDX4 could act as a prognostic biomarker and an immunomodulatory node influencing response to immunotherapy.

A, B Immune infiltration analysis of PRDX4. C Heatmap and KEGG and GO enrichment analysis of PRDX4. Abbreviations: PRDX4, peroxiredoxin 4; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology.
To further elucidate PRDX4’s functional network, we conducted gene–gene correlation analysis, which identified five highly co-expressed genes exhibiting strong statistical association with PRDX4 expression. Functional annotation of these genes revealed enrichment in several biologically significant processes, including chromosome localization, p53-mediated intrinsic apoptotic signaling, mitotic metaphase plate congression, negative regulation of sprouting angiogenesis, and spindle midzone organization. Additionally, these genes are found functionally attached to important cellular components such as the RNA polymerase II transcription regulator complex, microtubule-associated protein complex, and spindle apparatus (Fig. 4C). These results collectively suggest that PRDX4 not only contributes to immune modulation but is also deeply embedded in cell cycle regulation and genome stability maintenance.
Tumor phenotype evaluation of PRDX4 in the TCGA-BRCA-BM cohort
First, the phrase of PRDX4 with microsatellite instability (MSI) as well as TMB scores was carried out, and it was demonstrated that PRDX4 was related to greater MSI propensity and reduced TMB tendency, indicating that BRCA-BM patients are potentially suitable for immunotherapy (Fig. 5A). Besides, the expression of PRDX4 with EMT and ECM degradation score was evaluated, indicating that PRDX4 (Fig. 5B). The expression of PRDX4 with pyrimidine metabolism and purine metabolism is also evaluated (Fig. 5C).

A The relationship between expression of PRDX4 and MSI and TMB score. B The relationship between the expression of PRDX4 and EMT and ECM degradation. C The relationship between the expression of PRDX4 and pyrimidine metabolism and purine metabolism. Abbreviations: PRDX4, peroxiredoxin 4; MSI, microsatellite instability; TMB, tumor mutational burden; EMT, epithelial–mesenchymal transition; ECM, extracellular matrix.
EMT-NM-related molecular subgroups identification via consensus clustering
After identifying 16 robust DEGs from the integrated analysis of two GEO bulk RNA-seq datasets, we performed unsupervised consensus clustering to delineate molecular heterogeneity within the TCGA breast cancer bone metastasis (TCGA-BC-BM) cohort. This approach, which combines hierarchical clustering with resampling-based stability assessment, enabled the recognition of two distinct molecular subgroups, designated C1 and C2 (Fig. 6A–D). The clustering exhibited high consensus scores across multiple iterations, confirming the robustness of subgroup stratification.

A CDF and CDF curve of consensus clustering. B EMT-NM subgroups identification K = 2). C Heatmap illustration of DEGs in C1 and C2 subgroups. D PCA plot of C1 and C2. E KM plot of C1 and C2. Abbreviations: EMT, epithelial–mesenchymal transition; NM, nucleotide metabolism; CDF, cumulative distribution function; DEG, differentially expressed gene; PCA, principal component analysis; KM, Kaplan-Meier.
Across k values from 2 to 6, the empirical cumulative distribution function (CDF) and delta area curves indicated maximal stability at k = 2; the average silhouette width also peaked at k = 2, and the proportion of ambiguous clustering (PAC) was lowest at k = 2 (PAC = 0.085; Supplementary Table S1). Together, these indices support k = 2 as the most stable and least ambiguous clustering solution.
Importantly, survival analysis revealed a striking prognostic divergence between the two clusters, with subgroup C2 showing significantly worse overall survival (OS) compared with subgroup C1 (Fig. 6E). Functional enrichment analysis indicated that C2 was enriched for pathways associated with EMT, oxidative stress response, DNA repair dysregulation, and immune evasion, collectively suggesting a more aggressive and therapy-resistant phenotype. In contrast, subgroup C1 exhibited signatures of immune activation and favorable tumor microenvironmental composition, which may contribute to the improved clinical outcomes observed. These findings underscore the clinical relevance of the identified subgroups and highlight their potential utility for patient risk stratification and the design of precision therapeutic interventions.
Difference in EMT-NM-related molecular subgroups
We firstly compared the DEGs between C1 and C2, and then identified the 16 EMT-NM DEGs expression values (Fig. 7A). Next, we compared the mechanisms and functions between C1 and C2 via gene set enrichment analysis (GSEA) analysis, and discovered that the establishment of sister chromatid cohesion, RORA activates gene expression, Cohesin loading onto chromatin, TGFB pathway, IL5 signaling pathway, pathways in cancer, cytokines, and inflammatory response (Fig. 7B). Besides, the clinical, immune, tumor stemness, and TIDE discrepancy between C1 and C2 were also assessed (Fig. 7C–G).

A Volcano map illustration between C1 and C2. B GSEA analysis between C1 and C2. C Clinical information difference between C1 and C2. D immune checkpoint comparison between C1 and C2. E Immune infiltration difference between C1 and C2. F Tumor stemness between C1 and C2. G TIDE difference between C1 and C2. Abbreviations: EMT, epithelial–mesenchymal transition; NM, nucleotide metabolism; GSEA, gene set enrichment analysis; TIDE, Tumor Immune Dysfunction and Exclusion.
Figure 8A depicts a PRDX4-centric drug-target interaction network, positioning PRDX4 (red square) as the hub connected to 25 candidate drugs color-coded by class; Fig. 8B illustrates the best-scoring docking pose within the PRDX4 pocket, with hydrogen bonds rendered as yellow dashed lines and interatomic distances annotated; Fig. 8C presents a comparative ranking of docking scores for the top 20 candidates, highlighting Docetaxel and Paclitaxel as the strongest predicted binders; and Fig. 8D visualizes a drug-pathway chord map linking top-ranked compounds to five oncogenic pathways, where chord thickness conveys pathway coverage and multitarget potential.

A Drug-target network centered on PRDX4 with 25 candidate drugs. B Top-scoring ligand bound in the PRDX4 pocket with annotated hydrogen bonds. C Docking score comparison for the top 20 drugs, with Docetaxel and Paclitaxel ranking highest. D Drug-pathway chord plot linking top-ranked drugs to five oncogenic pathways, where chord thickness indicates pathway coverage and multitargeting.
Drug targeting PRDX4 prediction and molecular docking validation
We first systematically screened therapeutic agents targeting PRDX4 using the Drug–Gene Interaction Database (DGIdb), which integrates curated drug–gene interactions from multiple pharmacogenomic and cheminformatics resources. This in silico screening yielded several potential candidates, among which Docetaxel Anhydrous emerged as a top-ranked compound based on its strong predicted interaction profile with PRDX4 (Fig. 9A). To further substantiate this prediction, we carried out molecular docking simulations using the high-resolution crystal structure of PRDX4. The docking workflow involved protein preparation, energy minimization, and active-site grid generation, followed by ligand conformational sampling to ensure accurate prediction of binding poses. Our results revealed that Docetaxel Anhydrous formed stable hydrogen bonds and hydrophobic contacts with key catalytic residues within the PRDX4 active pocket, suggesting a favorable and specific binding mode (Fig. 9B). The docking score and calculated binding free energy indicated a high binding affinity, reinforcing the potential of Docetaxel Anhydrous as a PRDX4 modulator. These findings provide a mechanistic rationale for repositioning Docetaxel beyond its established microtubule-stabilizing effects, potentially enabling dual targeting of mitotic regulation and oxidative stress pathways in PRDX4-overexpressing tumors.

A The potential therapeutic agents discovered from the DGIDB database. B Molecular docking validation. Abbreviations: DGIdb, Drug-Gene Interaction Database.
Discussion
Breast cancer remains one of the most common malignancies and a leading cause of cancer-related mortality among women worldwide, particularly when it metastasizes to bone. Bone metastasis is associated with significant morbidity and a poor prognosis, emphasizing the need for improved biomarkers and therapeutic targets.
The research proposed a multi-omics, integrated framework that integrates EMT and NM features to identify PRDX4 as a mechanistic and prognostic biomarker in bone-metastatic breast cancer (BRCA-BM). The originality lies in the combination of LASSO-based ML feature selection, Cox survival modeling, and consensus clustering for a data-driven molecular subgrouping and accurate patient stratification beyond traditional single-pathway analyses. Using a LASSO-based ML approach, PRDX4 was identified as a key prognostic biomarker23. This observation is consistent with previous reports linking EMT-related genes and redox regulators to tumor progression and metastasis24,25,26.
A mechanistic perspective, PRDX4 functions as a redox regulator that links the detoxification of ROS to the activation of the EMT program and colonization of distant tissues. By altering TGF-β/SMAD, PI3K/AKT, and NF-κB signaling, PRDX4 promotes mesenchymal plasticity, ECM remodeling, and invasive potential while preserving redox homeostasis in conditions of oxidative stress. Enrichment analyses additionally associated PRDX4 with p53 signaling, DNA repair, and angiogenesis, which highlights the multifaceted nature of PRDX4’s role in adaptation to a metastatic function.
While PRDX4 has been previously associated with redox homeostasis, regulation of oxidative stress, and metastatic progression, the originality of the research lies in the integrated analytic pipeline and not the gene itself. Using a multi-omics and ML-driven framework, the researchers comprehensively integrated the EMT and NM pathways to identify PRDX4 as a dual-pathway prognostic biomarker. The research also expands previous findings by validating across TCGA and GEO cohorts, and undertakes a drug repurposing therapeutic context using a network pharmacology-based approach that identified Docetaxel as a potential PRDX4-interacting compound. The innovation of the research lies in the integrated methodology, inadequate validation, and therapeutic context of PRDX4, but not just the association of PRDX4 with metastasis.
The LASSO-Cox ML pipeline computationally optimized prognostic capacity (AUC > 0.85) by eliminating redundant genes and highlighting EMT-NM genes with survival information. Consensus clustering identified two EMT-NM molecular subtypes (C1 and C2) that differed in immune profiles, stemness, and outcomes, providing a novel stratification model for bone-metastatic BRCA. Finally, network pharmacology and molecular docking identified Docetaxel as a high-affinity ligand of PRDX4, suggesting new drug repurposing evidence linking redox modulation to interference with the cytoskeleton. The integrative pipeline provides a mechanistically motivated, ML-based approach that goes beyond prior EMT/PRDX4 studies by demonstrating novel molecular subgrouping, cross-validated predictive modeling, and therapeutic targeting potentials in metastatic breast cancer.
The limitations include a relatively small TCGA-BRCA-BM cohort, limited external validation, and use of in silico analyses without experimental or clinical validation of PRDX4’s mechanistic roles. Future studies should include larger multi-cohort studies for validation, functional studies to investigate the regulatory mechanisms of PRDX4, and preclinical studies of therapeutic strategies targeting PRDX4 or docetaxel. Future research should assess PRDX4 knockdown and PRDX4 expression between models of primary and bone-metastatic breast cancer in vitro to confirm its regulatory role in the EMT signaling pathways and redox homeostasis.
This study presents an end-to-end ML-driven framework for biomarker discovery, prognostic modeling, and therapeutic target prioritization in bone-metastatic breast cancer. By integrating EMT and NM gene signatures, we identified PRDX4 as a key prognostic biomarker associated with poor clinical outcomes. Immune infiltration and molecular correlation analyses revealed that PRDX4 expression is linked to an immunosuppressive tumor microenvironment, providing critical insights into its functional relevance in metastatic progression and immune regulation.
Building on this discovery, we applied network pharmacology, molecular docking, and pathway mapping to systematically identify and validate candidate drugs capable of targeting PRDX4. Several top-ranked compounds, including Docetaxel, Paclitaxel, Ixabepilone, Lapatinib, and Curcumin, demonstrated strong binding affinities and collectively covered key pathways such as EMT activation, ROS detoxification, PI3K/AKT signaling, DNA repair, and immune checkpoint regulation. These results establish a rational basis for developing multitargeted combination therapies that address tumor progression, redox imbalance, and immune escape, providing a powerful template for precision oncology and accelerating the transition from computational predictions to clinically actionable interventions.
Methods
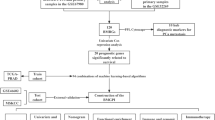
An ML-driven multi-omics pipeline integrating GEO and TCGA transcriptomic data, immune profiles, and clinical annotations to identify prognostic biomarkers and therapeutic targets was implemented in the research. The ML-based research on EMT-associated biomarkers in metastatic breast cancer forward, we applied an integrated EMT-NM network analysis, ML-based feature selection using the LASSO-Cox pipeline, and unsupervised consensus clustering. This approach provided the opportunity to identify robust prognostic gene signatures, novel molecular subgroups, and potentially druggable targets such as PRDX4, validated through cross-dataset discovery and drug-target interaction modeling, all for reproducibility and translational potential.
Source of data and pre-processing of the dataset
The research used bulk-seq data (GSE33494, GSE137842) from the GEO database. In which GSE33494 was from the GPL6480 platform, contained five primary breast tumor samples and five bone metastatic BC tumor samples; GSE137842 was from the GPL570 platform, included three metastatic BC tumor samples and three bone metastatic BC tumor samples27,28,29. The data obtained was in MINiML format30. All existing platforms, sample information, and complete GSE records for each record in this paper were included29. Data that had not been normalized were log2-transformed31. The probe IDs were transformed into gene symbols based on the platform annotation information32. Probes corresponding to more than one gene were removed, and an average value was calculated when multiple probes matched the same gene33. During the removal of the batch effect in the same dataset and platform, to reduce the combined impact, we used the decrease Batching impact function found in the R package limma34,35. In case of the combined analysis of multiple datasets or different platforms within the same dataset (EMT), a total of n genes were extracted from different datasets or platforms. These different datasets or platforms were noted as separate batches, and batch effect-related functions were utilized to delete batch effects. The outcomes of data pre-processing were judged on the boxplot (data normalization status), PCA plot (before/after batch effect removal) (data batch effect status). Gene lists of EMT and NM were obtained from the Genecard database36,37,38,39.
Identification of EMT-NM DEGs
DEGs were identified, accounting for statistical variability with the limma package (v3.40.2) in R after Comprehensive pre-processing and normalization. Raw expression matrices derived from GEO datasets GSE33494 and GSE137842 were log2-transformed and quantile-normalized for sample comparability. A linear model framework that employed empirical Bayes moderation was utilized to accurately estimate gene-wise variances. Statistical significance for DEGs was evaluated in accordance with Benjamini–Hochberg-adjusted P values (false discovery rate [FDR]), where DEGs were deemed statistically significant at an adjusted P < 0.05, and |log₂ fold change| > 0.5. Filtered DEGs were subsequently intersected with curated gene sets of EMT and neuro-mesodermal (NM) genes from the Gene Cards database to yield EMT-NM-specific DEGs. The DEGs were visualized using ggplot2 (v3.3.5) for the volcano plots, and Complex Heatmap (v2.8.0) was used for expression profiling of certain key dysregulated genes.
LASSO regression and feature selection
LASSO regression is a supervised method of ML that uses L1 regularization to find prognostic NM-associated hub genes. It performs feature selection and predictive modeling using the principle of shrinking less informative gene coefficients toward zero, and in practice, it keeps the most relevant variables to optimize the model. The ML workflow, using the glmnet package in R, was trained and validated using training and validation subsets of the full dataset, ensuring that the resulting algorithm was generalizable. The training set underwent a 10-fold cross-validation approach to estimate the appropriate penalty parameter (λ_min) applied to the model to limit overfitting and improve the prediction accuracy of the model. Through this ML-driven feature selection process, we were able to identify EMT-NM hub genes most strongly associated with patient survival outcomes, which formed the basis for the predictive modeling and molecular analyses.
Cox analysis and prognostic model construction
To establish a survival prediction model based on ML by combining the genes identified by LASSO from the EMT-NM gene hub into a Cox proportional hazards regression model. Cox regression is a classic statistical approach to modeling survival data, although when integrated into our adaptation based on LASSO feature selection, it additionally becomes an ML-informed survival model through the data generation of the input variables. A single variable as well as multivariate Cox analysis has been conducted using the survival program in R, which examines hazard ratios, as well as independent prognostic factors. The model assessed robustness and the performance of different models using Kaplan-Meier survival curves and time-dependent ROC curves, decision curves analysis (DCA), and calibration plots to predict patient outcome. This combined LASSO-Cox ML approach allowed for the creation of a high-dimensional, regularized prognostic model of EMT-NM gene signatures predicting OS in bone-metastatic breast cancer.
Immune infiltration analysis
The STAR-counts data and accompanying clinical information were gathered from the TCGA database (BC-BM (idkwh_560, Number of tumors = 64)). Then, data in TPM format were obtained and normalized by log2(TPM + 1). To attain a rigorous immune score, the immunoDeconv R package (version 4.3.1) that integrates six cutting-edge methods: TIMER, xCell, and MCP-counter, and CIBERSORT, is a statistical deconvolution technique that derives the relative ratios of 22 immune cell types in bulk tumor gene expression profiles by comparing those data to a validated leukocyte gene signature matrix (LM22). It uses support vector regression to reduce noise and infer immune cell composition in complex tissue samples, allowing for quantitative characterization of heterogeneity in the tumor immune microenvironment. EPIC (and quantiTiSeq) was employed to calculate the scores as it utilizes all six algorithms and passes systematic benchmark tests with exceptional performance and pros for all six. Analysis and visualization of the outputs were done using the R package ggClusterNet (v1.4.2).
Gene correlation and KEGG and GO enrichment analysis
Downloaded STARCcounts data along with clinical information of 64 BC-BM patients from the TCGA database and obtained them in TPM format. The log2(TPM + 1) transformation was then applied to make the samples univariable and use these as input to generate the gene correlation map in R software (version 4.3.1) (ggstatsplot package)40. To further clarify the biological function of the targets, we conducted functional enrichment analysis on the obtained gene sets.
MSI, TMB, and pathway enrichment analysis
Mutations burden (TMB), in combination with PDL1, has gained increasing interest as two critical predictive markers of patient response to anti-PD1 antibody therapy. MSI plays a key role in predicting patient response to conventional chemotherapy. Furthermore, for the TCGA-BRCA-BM cohort in TPM format, samples were selected only if RNA-seq data and clinical information were both provided, and then the genes involved in each pathway were extracted and processed using the GSVA package of R software, setting the method to’ ssgsea ‘for ssGSEA, and finally Spearman’s correlation was carried out for exploring correlation between the gene expressions and pathway scores.
Consensus clustering
For molecular subtyping, we analyzed the TCGA-BRCA-BM cohort in TPM format, retaining only samples with both RNA-seq data and corresponding clinical information (n = 64). Consensus clustering was accomplished using the Consensus Cluster Plus software (v1.54.0) with the parameters: Minimum number of clusters (k): 6, 100 resampling iterations, 80% item resampling, hierarchical clustering algorithm (“hc”), and Ward.D2 linkage method.
Consensus clustering, a form of unsupervised ML, was used to recognize intrinsic molecular subgroups based on EMT-NM gene expression. The methodology, utilizing a clustering of consensus matrix approach, involved repeatedly clustering resampled data to produce a consensus matrix. This ML-based methodology enables reliable data-driven classification and accurate identification of BC phenotypes.
Heatmaps of gene activity were created using the pheatmap program (v1.0.12). For visualization, only genes with variance >0.1 were included; when more than 1000 genes were available, the top 25% most variable genes were displayed.
Using the longevity package in R, we performed a Kaplan-Meier survival analysis to compare variations in OS amongst biochemical subtypes. Log-rank tests were employed to evaluate statistical significance.
Gene set enrichment analysis (GSEA)
To investigate biological variations between the biochemical subgroups found by consensual clustering, GSEA was conducted using the cluster Profile (v4.6.2) package in R. Gene expression profiles were ranked by signal-to-noise ratio, and enrichment analysis was conducted against the MSigDB c2.cp.all.v2022.1.Hs.symbols.gmt collection includes 3050 canonical pathways. To determine statistical significance, the analysis was conducted with 1000 permutations. Pathways with FDR < 0.05 and a normalized enrichment score (NES)| > 1 have been considered substantially enriched. Results were visualized as enrichment plots and summarized using bar plots for the top positively and negatively enriched pathways.
Clinical values evaluation between molecular subgroups
To determine the clinical significance of the classification defined above, the relationships between molecular subtypes and age, as well as radiotherapy, the researches were carried out using the ggalluvial package of the R software.
Relationship between molecular subgroups and tumor immune microenvironment (TME)
Once the TCGA data in TPM format were retrieved, the CIBERSORT method was used to examine differences in immune cell count between the two separate molecular subgroups. Additionally, ITPRIPL1, SIGLEC15, as well as TIGIT, CD274, HAVCR2, the PDCD1 gene, CTLA4, LAG3, and PDCD1LG2. These are important genes for immune modulation. Analyze the gene expression frequencies of these genes, a deeper understanding of the functional status of immune checkpoints can be obtained.
Tumor stemness analysis and immune response evaluation
In the first place, the TCGA data were retrieved into TPM format. As an ML-based stemness prediction algorithm, the One-Class Logistic Regression (OCLR) model developed by Malta et al. was used. OCLR has been trained on stem cell expression profiles to capture transcriptional features that are specific to pluripotency. The OCLR model predicts a stemness index (mRNAsi) for tumor samples using similarity to the stem cell reference model. A statistical ML method allows quantitative estimation of tumor stemness by linking EMT-NM molecular signatures to dedifferentiation and differentiation potential, which was derived by using the gene activation profile for cancer cells, containing characteristics from 11,774 genes. RNA expression levels were ranked by Spearman correlation, followed by linear transformation to map the stemness index onto [0, 1], which was executed by removing the smallest value and dividing by the largest value.
Drug prediction and molecular docking validation
Candidate drugs targeting PRDX4 were identified using the DGIdb database, and shortlisted compounds were subjected to molecular docking to evaluate their binding affinities. The crystal structure of the PRDX4 protein was retrieved from the Protein Data Bank (PDB), and ligands (including curcumin as a reference compound) were obtained in SDF format from PubChem and the PDB ligand database. Ligand structures were converted to PDB format using PyMOL and then to mol2 format via OpenBabel (v3.1.1). Torsional degrees of freedom were configured in AutoDockTools (v1.5.7), and PDBQT files were generated for docking.
Docking was performed using AutoDock Vina, with a grid box defined around the PRDX4 active site. The best binding positions were graded according to binding energy (kcal/mol), with lower energies indicating higher predicted affinity and greater conformational stability of the receptor–ligand complex. The resulting docked complexes were visualized and analyzed using PyMOL to confirm key hydrogen bonds and hydrophobic interactions. These in silico docking results provided a rational starting point for drug repurposing and precision therapy design targeting PRDX4.
Statistical analysis
All analyses of the related data were finished using the R programming package. First, normality tests were applied to different groups to analyze those groups. If a dataset passes the normality test, a t-test involving paired samples is employed to analyze the difference between matched datasets, while if it fails the test, then the Wilcoxon rank-sum test will be used. In addition, when it comes to a two-tailed P value, statistical significance here refers to P ≤ 0.05; whereas Spearman’s correlation test was used for gene coexpression and immune infiltration correlation analysis.
Data availability
The transcriptomic datasets analyzed in this study are publicly available from the Gene Expression Omnibus (GEO) under accession numbers GSE33494 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33494) and GSE137842 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137842). TCGA-BRCA bone metastasis cohort data were obtained from The Cancer Genome Atlas (TCGA) via the Genomic Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/).
Code availability
The code used in this study is available from the corresponding author upon reasonable request.
References
Dagher, E. et al. Identification of an immune-suppressed subtype of feline triple-negative basal-like invasive mammary carcinomas, spontaneous models of breast cancer. Tumour Biol. 42, 1010428319901052 (2020).
Duderstadt, E. L. et al. Chemical carcinogen-induced rat mammary carcinogenesis is a potential model of p21-activated kinase positive female breast cancer. Physiol. Genom. 53, 61–68 (2021).
Ghosh, A. & Gopinath, S. C. B. Molecular mechanism of breast cancer and predisposition of mouse mammary tumor virus propagation cycle. Curr. Med. Chem. 32, 2330–2348 (2025).
Venetis, K. et al. Breast cancer with bone metastasis: molecular insights and clinical management. Cells 10, 1377 (2021).
Hashemi, M. et al. EMT mechanism in breast cancer metastasis and drug resistance: revisiting molecular interactions and biological functions. Biomed. Pharmacother. 155, 113774 (2022).
Bahreini, F., Rayzan, E. & Rezaei, N. microRNA-related single-nucleotide polymorphisms and breast cancer. J. Cell. Physiol. 236, 1593–1605 (2021).
Mullen, N. J. & Singh, P. K. Nucleotide metabolism: a pan-cancer metabolic dependency. Nat. Rev. Cancer 23, 275–294 (2023).
Xu, Y. et al. CircMMP2(6,7) cooperates with β-catenin and PRMT5 to disrupt bone homeostasis and promote breast cancer bone metastasis. Cancer Res. 84, 328–343 (2024).
Atala, A. Re: Peroxiredoxin 4: a novel secreted mediator of cancer-induced osteoclastogenesis. J. Urol. 195, 220–221 (2016).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Jain, P. et al. NOX4 links metabolic regulation in pancreatic cancer to endoplasmic reticulum redox vulnerability and dependence on PRDX4. Sci. Adv. 7, eabf7114 (2021).
Lai, W. et al. HJURP inhibits sensitivity to ferroptosis inducers in prostate cancer cells by enhancing the peroxidase activity of PRDX1. Redox Biol. 77, 103392 (2024).
Liu, S. et al. Analysis of genomics and immune infiltration patterns of epithelial-mesenchymal transition related to metastatic breast cancer to bone. Transl. Oncol. 14, 100993 (2021).
Ning, Z. K. et al. Single-cell perspective on the Monocyte-to-HDL cholesterol ratio as a metastasis biomarker in papillary thyroid cancer. BMC Cancer 25, 1203 (2025).
Pastushenko, I. & Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 29, 212–226 (2019).
Rafiei, S. et al. Peroxiredoxin 4: a novel secreted mediator of cancer-induced osteoclastogenesis. Cancer Lett. 361, 262–270 (2015).
Ranalli, M. G. et al. M-quantile regression shrinkage and selection via the Lasso and Elastic Net to assess the effect of meteorology and traffic on air quality. Biom. J. 65, e2100355 (2023).
Rhee, S. G. & Kil, I. S. Multiple functions and regulation of mammalian peroxiredoxins. Annu. Rev. Biochem. 86, 749–775 (2017).
Tsuji, M. et al. Design and synthesis of visible light-activatable photocaged peroxides for optical control of ROS-mediated cellular signaling. Bioorg. Med. Chem. 111, 117863 (2024).
Xie, Y., Shi, H. & Han, B. Bioinformatic analysis of underlying mechanisms of Kawasaki disease via Weighted Gene Correlation Network Analysis (WGCNA) and the Least Absolute Shrinkage and Selection Operator method (LASSO) regression model. BMC Pediatr. 23, 90 (2023).
Bermudez, M. et al. Role of microRNA-204 in regulating the hallmarks of breast cancer: an update. Cancers 16, 2814 (2024).
Lan, H., Wu, B., Jin, K. & Chen, Y. Beyond boundaries: unraveling innovative approaches to combat bone-metastatic cancers. Front. Endocrinol. 14, 1260491 (2024).
Yamaguchi, K. et al. Efficacy of pembrolizumab in microsatellite-stable, tumor mutational burden-high metastatic colorectal cancer: genomic signatures and clinical outcomes. ESMO Open 10, 104108 (2025).
Chen, M. et al. ICAM1 promotes bone metastasis via integrin-mediated TGF-β/EMT signaling in triple-negative breast cancer. Cancer Sci. 113, 3751–3765 (2022).
Xu, Y. et al. Single nucleotide polymorphisms within the Wnt pathway predict the risk of bone metastasis in patients with non-small cell lung cancer. Aging 12, 9311–9327 (2020).
Jia, W., Chen, P. & Cheng, Y. PRDX4 and its roles in various cancers. Technol. Cancer Res. Treat. 18, 1533033819864313 (2019).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 41, D991–D995 (2013).
Kang, Y. et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003).
National Center for Biotechnology Information (NCBI). MINiML (MIAME Notation in Markup Language) format description. (2005). https://www.ncbi.nlm.nih.gov/geo/info/MINiML.html.
Brazma, A. et al. Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat. Genet. 29, 365–371 (2001).
Li, W., Suh, Y. J. & Zhang, J. Does logarithm transformation of microarray data affect the ranking order of differentially expressed genes?BMC Bioinform. 6, 187 (2005).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Smyth, G. K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 3 (2004).
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739 (2010).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Wu, Q. et al. Epithelial-mesenchymal transition (EMT) and cancer metastasis. Cancer Lett. 356, 361–373 (2015).
Chaffer, C. L. & Weinberg, R. A. A perspective on cancer cell metastasis. Science 331, 1559–1564 (2011).
Stelzer, G. et al. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 54, 1.30.1–1.30.33 (2016).
Hoadley, K. A. et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 (2014).
Author information
Authors and Affiliations
Contributions
Xiao Zhou and Huawei Yang conceived the study and designed the overall research plan. Longgui Xie and Jianhui Liu collected and curated the data, and together with Geyi Liao performed the analyses and prepared figures. Xiao Zhou and Jianhui Liu drafted the manuscript, and all authors (Xiao Zhou, Longgui Xie, Jianhui Liu, Geyi Liao, and Huawei Yang) reviewed and approved the final version. Huawei Yang supervised the work and is the corresponding author.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, X., Xie, L., Liu, J. et al. Integrative multi-omics and machine learning framework identifies PRDX4 as a redox-EMT regulator and predictive marker in bone-metastatic breast cancer. npj Precis. Onc. 10, 43 (2026). https://doi.org/10.1038/s41698-025-01240-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01240-w
