
Abstract
Extracting structured data from free-text medical records at scale is laborious, and traditional approaches struggle in complex clinical domains. We present a novel, end-to-end pipeline leveraging large language models (LLMs) for highly accurate information extraction and normalization from unstructured pathology reports, focusing initially on kidney tumors. Our innovation combines flexible prompt templates, the direct production of analysis-ready tabular data, and a rigorous, human-in-the-loop iterative refinement process guided by a comprehensive error ontology. Applying the finalized pipeline to 2297 kidney tumor reports with pre-existing templated data available for validation yielded a macro-averaged F1 of 0.99 for six kidney tumor subtypes and 0.97 for detecting kidney metastasis. We further demonstrate flexibility with multiple LLM backbones and adaptability to new domains, utilizing publicly available breast and prostate cancer reports. Beyond performance metrics or pipeline specifics, we emphasize the critical importance of task definition, interdisciplinary collaboration, and complexity management in LLM-based clinical workflows.
Similar content being viewed by others

Introduction
Extracting structured information from free-text electronic medical records (EMR) presents a significant challenge due to their narrative structure, specialized terminology, and inherent variability1. Historically, this process has been labor-intensive and error-prone, requiring manual review by medical professionals2,3,4, thereby hindering large-scale retrospective studies and real-world evidence generation5. Consequently, automated, reliable methods are needed to extract clinically relevant information from unstructured EMR text6.
Natural language processing (NLP) techniques, including rule-based systems and early neural models, have struggled with the nuances of the medical domain7,8. While transformer-based architectures like ClinicalBERT9, GatorTron10, and others11,12,13, furthered the state-of-the art, they often require extensive fine-tuning on large annotated datasets, which are costly and time-consuming to create14,15. The challenge is particularly acute in specialized tasks like the extraction of immunohistochemistry (IHC) results from pathology reports, which requires identifying and mapping tests to the correct results and specimens, resolving synonyms, and navigating diverse terminology.
The rapid emergence of generative large language models (LLMs)16 offers a potentially transformative approach. Their large number of parameters and ability to process extensive context windows enable them to retain and “reason” over substantial amounts of domain-specific knowledge without fine-tuning17,18,19,20. Natural language prompts allow a high degree of flexibility, enabling rapid iteration and adaptation to new entities and instructions21,22.
Recent studies report promising results using LLMs for text-to-text medical information extraction23. Initial efforts have successfully extracted singular/non-compound report-level information, such as patient characteristics from clinical notes24, and tumor descriptors/diagnosis from radiology25 and pathology reports26,27. Studies have also demonstrated the potential for extracting inferred conclusions, such as classifying radiology findings28 and cancer-related symptoms4. However, challenges remain, particularly factually incorrect reasoning29,30, and the potential for information loss when forcing complex medical concepts into discrete categories31.
Evaluating LLM performance is complicated by the lack of standardized error categorization that accounts for clinical significance and the limitations of traditional metrics like exact match accuracy, which are ill-suited for open-ended generation32,33,34,35,36. For example, misclassifying a test result as “negative” versus “positive” is substantially different than minor grammatical discrepancies between labels, e.g., “positive, diffusely” versus “diffuse positive”. This open-ended style thus necessitates methods to constrain generation in order to minimize requisite downstream normalization37,38. Furthermore, many existing clinical NLP datasets utilize BERT-style entity tagging, limiting their use for benchmarking end-to-end information extraction33,39,40. Nonetheless, our lack of pre-annotated data, high degree of entity complexity, and desire for flexibility, coupled with the rapidly improving performance of LLMs41, prompted us to explore their potential.
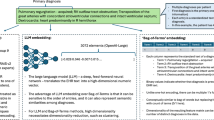
To address these challenges, we developed a novel LLM-based pipeline for end-to-end information extraction from real-world clinical data. We define ‘end-to-end’ here as encompassing: (1) entity identification, (2) clinical question inference (e.g., determining the final diagnosis), (3) terminology normalization, (4) relationship mapping (e.g., linking IHC results to specific specimens), and (5) structured output generation. Our approach leverages three key innovations: flexible prompt templates with a centralized schema (Fig. 1a), multi-step LLM interactions with chain-of-thought reasoning (Fig. 1b), and a comprehensive error ontology. Developed through iterative “human-in-the-loop”42 refinement, this ontology provided a systematic framework not only for classifying discrepancies but also for understanding the diverse contextual challenges inherent in extracting nuanced information from complex medical text, thereby guiding the refinement of our extraction objectives.

a Abbreviated examples of the extraction schema for immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH), histology, and diagnosis, demonstrating the inclusion of entity-specific instructions, standardized labels, and a structured vocabulary for IHC test reporting. An abbreviated report text is included for reference. b Overview of pipeline steps. Each prompt template included base instructions consistent across entities, and placeholders for entity-specific instructions and labels that could be easily “hot-swapped”, with the {{}} indicating where information from the schema is pasted in. All template sets include initial prompts to segment and organize text, a subsequent standardization prompt to normalize labels and produce structured output, and a final Python step for parsing into tabular data. The full output from segmentation steps, both reasoning and the segmented text, is passed to subsequent steps. The resulting data is in tabular format for immediate downstream utility.
We demonstrate this pipeline using renal tumor pathology reports, extracting and normalizing report-level diagnosis, per-subpart/specimen histology, procedure, anatomical site, and detailed multipart IHC results for 30+ assays at both the specimen and tissue block level. We focus primarily on renal cell carcinoma (RCC) given the high volume of RCC patients treated at UT Southwestern, the diversity of RCC subtypes and the wide variety of ancillary studies used for subtyping, and a multidisciplinary UTSW Kidney Cancer Program recognized with a Specialized Program of Research Excellence award from the National Cancer Institute.
This paper details the pipeline’s iterative development utilizing 152 diverse kidney tumor reports, highlighting how the error ontology revealed crucial insights into task specification and report complexity. We subsequently validate the finalized pipeline on 3520 institutional reports and assess its portability using independent, publicly available breast and prostate cancer datasets.
Beyond the technical specifics of our pipeline, we place particular emphasis on the broader considerations that arose during development. Specifically, our focus shifted from engineering prompts that could extract information, to precisely defining what information to extract and why. This experience underscores that, as AI approaches human-level intelligence in many domains41, success may increasingly hinge on the clear articulation of objectives, rather than on singular workflow methodologies. As such, by detailing both our successes and pitfalls, we hope to provide a roadmap of generalizable context and considerations for future AI-powered clinical information extraction workflows.
Results
Workflow refinement and gold-standard set
A development set consisting of 152 reports reflecting diverse clinical contexts was used to guide iterative “human-in-the-loop”42 refinement of our pipeline, ultimately resulting in our error ontology and a set of “gold-standard” annotations reflecting our desired pipeline output; Fig. 2, see Methods for details. In total, 89 reports contained local/regional RCC, 41 contained metastatic RCC, nine contained non-RCC malignancies such as urothelial carcinoma, and 13 contained benign or neoplasms of uncertain behavior of kidney such as renal oncocytoma; Supplementary Fig. 1. A flow chart for defining and documenting discrepancies across iterations as well as an introduction to error contexts, posed as broader questions regarding information extraction aims, is provided in Fig. 3. Several error context examples are provided in Tables 1–5, with the remainder in Supplementary Tables 1–11.

After completing each iteration, the schema, prompts, LLM outputs, and gold-standard are incrementally versioned, e.g., V1, V2, V3, etc. The gold-standard set was structured in the exact format as the table that held the LLM outputs, with columns for report ID, specimen (and block when applicable) name, item name, e.g., histology, and item label, e.g., clear cell RCC, so that items could be programmatically matched for review. The same LLM backbone (GPT-4o 2024-05-13) is utilized through all iterations.

After matching LLM entities to gold-standard entities, e.g., specimen A histology, the labels are checked for discordance (clear cell RCC vs papillary RCC). Discrepancies are then assessed for source and severity. Issue contexts are introduced as questions needing to be asked about both workflow requirements and how certain kinds of deviations from instructions might need to be addressed. For the final two contexts: “Formatting only” refers to discrepancies that are purely due to standardized spellings/punctuation (BAP-1 vs BAP-1), while “Blatantly Incorrect Reasoning” refers to errors not arising from any given nuanced context (e.g., hallucinating a test result not present in the report text).
Following six iterative refinement cycles with GPT-4o 2024-05-1343 as the LLM backbone, our pipeline achieved strong alignment with the gold-standard annotations (1413 total entities: 152 diagnoses, 651 specimen-level labels, 610 IHC/FISH results). The final iteration yielded a major LLM error rate of 0.99% (14/1413) with no major annotation errors identified; Fig. 4a. Schema issues necessitating more than flagging for review were also eliminated; Fig. 4b and Supplementary Figs. 2–4. Supplementary Table 12 details distinct iteration updates with respect to fluctuating error prevalence.

a Error/discrepancy source, severity, and entity type across iterations. “Other Items” includes diagnosis, histology, procedure, and anatomical site. The five “Problematic reports” are consistent across iterations. Counts of 0 are left blank. Column totals are not equal across all iterations due to duplicate IHC/FISH entities and variations in missingness. b Error/discrepancy contexts by source and severity across iterations (IT). Counts of 0 are left blank. Due to the lower number of major annotation errors, they have been grouped with minor annotation discrepancies for ease of visualization. For all panels, the fill color scale is maintained with a maximum of 84 and a minimum of 1.
This iterative refinement process, systematically guided by our error ontology, provided critical insights into the diverse challenges encountered when extracting complex information from pathology reports, primarily falling into categories related to inherent report complexities, difficulties in task specification, normalization hurdles, and the integration of medical nuance.
Report complexities
First, certain inherent report characteristics consistently generated discrepancies. Five complex outside consultations accounted for disproportionately many minor IHC/FISH discrepancies; Fig. 4a. These “problematic reports” all contained a mix of IHC/FISH tests where the associated block or specimen was clearly identified for some tests but not others. In such instances, rather than indicating ambiguity, the LLM would often incorrectly duplicate results across all specimens with similar histology (Table 1). Additionally, outside consultations often contained discordant reporting conventions for specimen names. For example, a specimen designated as “B” by the outside institution, could be referred to internally as specimen “A”; Supplementary Table 1. Despite adding more illustrative examples of correctly mapped output to the prompts, these challenges persisted; Fig. 4a, b. We also observed that major errors in IHC/FISH extraction, primarily missing results, were more prevalent in reports containing a high volume of tests (>10), and ambiguity arose in defining the severity of missing results when tests were mentioned by name but potentially not performed (Supplementary Table 2).
Specification issues
Second, precisely defining the desired information scope and granularity (“specification issues”) was a key refinement focus. This required clarifying relevant entities when multiple labels were possible (e.g., anatomical sites, Table 2) and optimizing the level of detail for IHC results. The latter involved shifting from an exhaustive list of potential test results to a structured vocabulary capturing the distinct dimensions of status, intensity, extent, and pattern, allowing more flexible yet standardized representation; Table 3. Additional instructions were needed to handle nested labels, i.e., one label is more precisely correct than another, and the preferred level of granularity for capturing anatomical sites; Supplementary Tables 3, 4. Ontological nuances also required explicit instructions; for example, clarifying that a “peripancreatic mass” should not be coded as pancreatic metastasis required guidance on interpreting directional terms (Table 444).
We also addressed ontological overlap, i.e., multiple labels could be considered fully correct, by establishing prioritization rules. For example, defining the preferred primary status for BAP-1 when both “Positive” and “Intact” were reported (Supplementary Table 5). Finally, in certain circumstances when individual specimens shared characteristics, we needed to clarify when the label for one entity can affect that of another (e.g., Specimen A and B both originate from a “nephrectomy”, but in the report text specimen A’s procedure is referred to only as a “resection”(Supplementary Table 6).
Normalization difficulties
Third, normalizing terms and handling free-text entries remained challenging. While necessary to avoid information loss31, the inclusion of “Other-<as per report>“ categories consistently generated minor discrepancies due to the difficulty of achieving verbatim matches between the LLM output and gold-standard (Supplementary Table 7).
Specific IHC/FISH term normalization, like standardizing “diffusely” to “diffuse”, also proved problematic. This single normalization challenge accounted for over half of the remaining formatting-only discrepancies in the final iteration (15/28 errors) despite the target term “diffuse” being relatively infrequent overall (46/610 IHC/FISH annotations). Investigating potential causes, we examined the GPT-4o tokenizer’s45 byte pair encoding (BPE)46 behavior. We found “diffusely” tokenization varied with the preceding character: splitting into three tokens (“diff”, “us”, “ely”) after a newline but two (“diffus”, “ely”) after a space. While BPE segmentation is complex, we hypothesize this differential sub-word tokenization may contribute to the inconsistent application of this specific normalization rule. In contrast to these difficulties, the workflow proved highly adept at normalizing historical terminology to updated terms (Fig. 4b and Supplementary Table 8).
Medical nuance
Finally, difficulties in integrating medical history and nuance required clinical domain expertise and corresponding adjustments. For example, the pathologist on our team clarified that in pathology reporting, the terms “consistent with” or “compatible with” often carry more conclusive meaning than in general parlance, leading us to adjust instructions regarding the level of certainty provided by these terms (Supplementary Table 947). Similarly, interpreting complex distinctions, such as delineating local vs. distant lymph node metastases and assessing the relevance of medical history, necessitated providing detailed clinical context within the prompts (Table 5 and Supplementary Tables 10, 11).
Assessing LLM interoperability
LLM backbone interoperability was assessed by comparing pipeline output using GPT-4o 2024-05-13, Llama 3.3 70B Instruct48, and Qwen2.5 72B Instruct49 to the final gold-standard. Exact match accuracies were 84.1% for GPT-4o, 78.1% for Qwen2.5, and 70.1% for Llama 3.3. Applying fuzzy matching for IHC/FISH items (ignoring test name punctuation/capitalization and relaxing specimen/block mapping criteria) improved these respective accuracies to 90.0, 86.1, and 82.0%. See Supplementary Table 13 for concrete examples of matching criteria and Supplementary Table 14 for exact counts of match type for each model. Exact match inter-model agreement between GPT-4o and the open-weight models was high (84.8% with Qwen2.5, 82.2% with Llama 3.3). These results suggest the core prompt and schema logic is transferable, though model choice impacts performance.
Validation against preexisting data
Applying the finalized pipeline (using GPT-4o 2024-08-06) to 2297 internal kidney tumor reports with available structured EMR data for validation yielded high performance for identifying six key kidney tumor histologies (clear cell RCC, chromophobe RCC, papillary RCC, clear cell papillary renal cell tumor/CCPRCT, TFE3-rearranged RCC, and TFEB-altered RCC) with a macro-averaged F1 score of 0.99, and an F1 of 0.97 for metastatic RCC detection (Table 6).
A review of discrepancies for histological subtype showed continued difficulty with integrating medical history; most clear cell false negatives (28/32), for instance, resulted from incomplete use of a patient’s prior history of this subtype. Lower performance in detecting metastatic RCC was primarily attributable to false positives due to misinterpreting medical history (n = 6), local tumor extension (n = 5), and differentiation of regional vs distant lymph nodes (n = 3). Such errors often occurred in complex or ambiguous reports, underscoring the need for mechanisms to flag these cases for human review. Demonstrating clinical utility, the pipeline correctly identified necessary updates or corrections to the preexisting structured data in 27 instances (e.g., reflecting addended results or more current terminology), all subsequently confirmed by a pathologist. Comprehensive details on all discrepancies, both errors and correct updates, are included in Supplementary Table 15.
Comparison to the regex baseline
The LLM pipeline demonstrated distinct advantages over a custom, rule-based regex tool for extracting the six histologies of interest. The regex tool performed reasonably well for common subtypes with fewer historical variations in terminology (F1 scores: 0.96 Clear Cell, 0.91 Chromophobe, 0.89 Papillary RCC; Table 6). However, its performance degraded substantially when encountering wider terminology variation, historical naming conventions, or results reported primarily in comments or addendums—situations common for rarer subtypes. This resulted in much lower F1 scores for CCPRCT (0.71), TFE3-rearranged RCC (0.11), and TFEB-altered RCC (0.35), whereas the LLM pipeline maintained high accuracy and precision. Full results for the regex tool are available in Supplementary Table 16.
Gauging internal consistency
To assess the internal consistency of extracted data across a broader cohort, we analyzed the full set of 3520 internal reports (see Methods/Supplementary Fig. 1 for cohort criteria). This group included the aforementioned 2297 reports, plus an additional 1223 reports for which no structured EMR data was available. For this analysis, we examined the concordance between the pipeline’s extracted histology and associated IHC results within the same specimen/subpart. Out of all available reports, 2464 subparts/specimens were identified as containing any of the histologies of interest. Of these, 1906 were identified to have only a single histology and corresponding IHC results for the same subpart; Supplementary Fig. 1.
The pipeline showed a high degree of consistency, for example, 87/87 CD117 tests on specimens with chromophobe RCC were positive, and accurate extraction of the CA-IX “cup-like” expected staining pattern for CCPRCT was demonstrated (Table 7). The two “box-like” results found for CCPRCT corresponded to two tumors in a single report, wherein the LLM was consistent with the report text. The case was subsequently reviewed and found to have a “cup-like” pattern and a correction was issued; Details on all unexpected findings are documented in Supplementary Table 17.
Assessing clinical domain interoperability
Portability of the workflow and final prompt templates was assessed using publicly available data from The Cancer Genome Atlas (TCGA)50,51,52,53. To gauge generalization, prompt templates were not modified, and improvements were performed on only the schema, with iterations concluding once the schema adequately covered essential domain concepts and terminology. Of the 757 available TCGA Breast Invasive Carcinoma54 (BRCA) reports, 53 contained the words “immunohistochemistry” and “HER2” in the processed text. Three schema-only iterations were required to incorporate domain-specific terminology and reporting conventions for extracting HER2 (IHC and FISH), estrogen receptor (ER), and progesterone receptor (PR) status. The pipeline (using GPT-4o 2024-08-06) subsequently achieved 89% agreement with the curated tabular data; Supplementary Table 18.
Manual review of discrepancies suggested many were not LLM errors: nine cases involved results present in the curated data but absent in the scanned PDF, potentially indicating an alternate source for the curated result. These nine specific data points, where required information was not in the source text, were excluded from agreement calculations. Another result that first appeared to be an LLM false positive was subsequently found to reflect known clinical ambiguity regarding the classification of ER low-positive results (1–9% staining)55.
Of the 333 available TCGA Prostate Adenocarcinoma56 (PRAD) reports, 253 had corresponding Gleason Scores in the structured clinical data and available report text. For this task, 98% agreement was achieved on the first run (Supplementary Table 19). The difference in convergence speed (one run for PRAD vs. three for BRCA) highlights the impact of task and report complexity: extracting multiple, detailed IHC results from longer, more variable BRCA reports (average 9250 characters) proved more challenging than extracting a single report-level Gleason Score from shorter PRAD reports (average 3440 characters).
Discussion
This study demonstrates that high accuracy in automated pathology report information extraction with large language models (LLMs) is possible but hinges on careful task definition and refinement. Although our pipeline yielded strong performance—for instance, a macro-averaged F1 score of 0.99 on identifying important RCC histological subtypes and adaptability to new entities such as Gleason Scores from prostate adenocarcinoma reports—our experience underscores that the process of defining information extraction goals may hold broader relevance than the performance metrics or workflow technicalities alone.
It became evident that the model’s success depended heavily on the clarity and depth of instructions. Consequently, a multidisciplinary team with domain expertise in NLP and LLMs, downstream statistical analysis, and clinical pathology became instrumental for success. Additional collaboration came from the LLM itself—particularly through review of the “reasoning” output. We found that well-meaning instructions like “focus on the current specimen…, not past medical history” led to instances of ‘malicious compliance’ where the LLM followed instructions too literally, discarding important contextual information. Rectifying this required careful consideration of how instructions might be interpreted by the model, leading to increased specificity (Table 5). Underpinning this iterative process, the systematic application of our error ontology proved invaluable, not just for classifying discrepancies, but for prompting essential questions (as per Fig. 3) and actively guiding the refinement of our extraction goals.
Cross-specialty collaboration was particularly vital for developing the error ontology. While developed with expert clinical input, we do acknowledge that our distinction between schema issues, major, and minor errors relied on contextual interpretation and our specific downstream use case. We thus caution against interpreting raw performance numbers in isolation. Instead, we advocate for a holistic interpretation considering the clinical significance of errors and their potential downstream impact, informed directly by stake-holding researchers57,58. Interpretations could therefore differ between groups. For example, in broader kidney cancer research, deviations in the detailed staining pattern modifiers for CA-IX could be considered ‘major’ errors, as the distinction between “box-like” vs “cup-like” is decisive for RCC subtyping. However, if one were exclusively studying oncocytoma, where CA-IX is canonically negative59, variations in the details of a “Positive” result might sufficiently exclude this histology and be considered ‘minor’.
We note several limitations of our study and workflow. The iterative refinement process risked adding overly specific ‘one-off’ rules; we mitigated this by striving for generalizable instructions, using specific cases as examples rather than distinct rules (Table 4). Each refinement cycle also required highly detailed and time-intensive human review. One might ask whether an end-to-end manual annotation followed by traditional fine-tuning would have been easier. However, such an approach would have still demanded extensive manual review4, offered no guarantee of easily handling the diverse and complex “error-inducing” contexts that we encountered, and might not have spurred the same level of insight into refining our information extraction goals. Moreover, generative LLM technology is evolving rapidly41; our prompt-based pipeline is more adaptable to both changes in clinical entities and new model upgrades than static, fine-tuned architectures.
Furthermore, this approach currently produces only semi-structured data that, in some instances, requires further downstream normalization. While our research team possessed this domain-specific skillset, allowing us to prioritize granular detail over strict upfront standardization, this trade-off may not be acceptable for all teams and situations. Future work could leverage guided-decoding backends37,38 that can help constrain LLM generation to a predefined JSON schema. Such tools, along with increasing baseline AI performance and methods to flag problematic reports for human review, could further shift workflow efforts from managing formatting and data structuring requirements to well-informed task specification.
Limitations related to gold-standard development and validation also warrant mention. The standard was developed iteratively by the core team to refine complex task specifications, precluding formal inter-reviewer agreement metrics and limiting assessment of absolute annotation reliability. Our validation strategy—large-scale internal checks on core entities and external portability tests—aligned with the study’s focus on the refinement methodology and task specification challenges, rather than exhaustive benchmarking across every field against a static, held-out test set. As such, we posit that as LLMs are utilized to perform increasingly complex tasks, particularly those involving ambiguity and advancing medical knowledge, it may be useful to conceptualize “gold-standard” not merely as static ground truth, but as a dynamic reflection of evolving research goals and domain understanding40,57,58.
In summary, our LLM-based pipeline for pathology report information extraction highlights both strong performance metrics and the intricate processes required to achieve them. Our experience illustrates the importance of thoroughly understanding extraction intentions and goals, and how collaboration between domain experts—and even insights derived from the LLMs themselves—are crucial to this process. By documenting these complexities, we aim to provide a set of generalizable considerations that can inform future LLM-based clinical information extraction pipelines. As generative AI matures, flexible, human-in-the-loop strategies may prove essential to ensuring workflows remain grounded in real-world clinical objectives.
Methods
Defining the task
As detailed in the Introduction, we defined “end-to-end” information extraction for this study to encompass entity identification, clinical question inference, terminology normalization, relationship mapping, and structured output generation suitable for downstream analysis (e.g., tabular datasets).
Initial entities to extract and normalize from reports included: (1) report-level diagnosis; (2) per-subpart/specimen histology, procedure type, and anatomical site; and (3) detailed specimen and tissue block-level IHC/FISH (fluorescence in situ hybridization) test names and results. We first defined an ‘extraction schema’, outlining standardized labels, a structured vocabulary of terms and preferred synonyms for IHC results, and unique entity-specific instructions; see Fig. 1a. Unique instructions provided entity-specific context and logic, like differentiating regional vs. distant metastasis based on lymph node involvement for diagnosis.
Labels for diagnosis were primarily derived from ICD-10 descriptors, with procedure and histology labels primarily sourced from the contemporary College of American Pathologists (CAP) Cancer Protocol Templates for kidney resection and biopsy60,61. Labels for anatomical site and IHC/FISH, along with specialized labels such as the diagnosis “Metastatic RCC,” were developed with guidance from pathology experts. While the normalized lists covered common entities, free-text options (e.g., “Other- <fill in as per report>”) were essential for capturing less frequent findings or specific nuances not fully represented, thus preventing information loss.
Prompt templates
We used Microsoft Prompt flow62 to organize the workflow as a directed acyclic graph, with nodes for Python code execution or LLM requests via specific prompt templates. Reusable, modular prompt templates were designed for portability. We developed three distinct template sets, each optimized for a specific class of entity: The ‘feature report’ set for entities with a single label per report, such as diagnosis; The “feature specimen” set for entities with one label per specimen/subpart; And an IHC/FISH specific set as it uniquely requires matching any number of specimens, blocks, test names, and test results; see Fig. 1b. Initial prompts segmented and organized relevant text, while final prompts generated schema-normalized, parsable JSON output.
All prompts required initial “reasoning” generation, passed to subsequent prompts to create a “chain-of-thought”63. This has been shown to enhance LLM performance across a wide range of tasks64,65, and provides insight into specific limitations and usage of our instructions66. The reasoning instructions prompted the LLM to consider uncertainties and articulate its decision-making process for text segmentation or standardization. Prompt templates were written in markdown format for organization and included examples of properly structured JSON responses. Full schema and templates are included in the Supplementary Information and a GitHub repository for implementation can be found at github.com/DavidHein96/prompts_to_table.
Workflow refinement and gold-standard set
We selected 152 reports reflecting diverse clinical contexts from a set of patients with kidney tumor related ICD-10 codes in their EMR history; Supplementary Fig. 1. This included reports with multiple specimens, complex cases that required expert consultation, biopsies and surgical resections, various anatomical sites such as primary kidney and suspected metastases, and both RCC and non-RCC histologies. All data was collected under the purview of our institutional IRB STU 022015-015.
These reports were used to iteratively develop the pipeline, error ontology, and a corresponding “gold-standard” annotation set that could be used for benchmarking. First, the workflow was executed with preliminary prompts and schema to generate rough tabular outputs, allowing for expedited manual review and reducing annotation burden for creating the initial gold-standard set. Subsequently, the prompts and schema were updated to reflect desired changes, and the workflow was executed again. The output was then matched to gold-standard annotations and discordant results were manually reviewed, with source and severity of the discrepancy documented and used to inform subsequent adjustments to the workflow and gold-standard. This process was then repeated iteratively, as outlined in Fig. 2.
A data scientist (5 years clinical oncology experience) and a statistician (13 years kidney cancer research experience) reviewed annotations and discrepancies with all uncertainties or ambiguities deferred to a board-certified pathologist specializing in genitourinary pathology (23 years’ experience). All development used GPT-4o (2024-05-13, temperature 0) via a secure HIPAA-compliant Azure API43.
Creating an error ontology
A structured error ontology was developed to both provide a framework for classifying the source and severity of discrepancies between the LLM outputs and the gold-standard, and to highlight generalizable contexts in which discrepancies arose. The ontology comprises three sources of discrepancy: LLM, manual annotations (errors introduced to the gold-standard set by incorrect or insufficient annotation in a prior step), and what we term “schema issues”. Schema issues represent instances where the LLM and gold-standard were discordant, yet both appeared to have adhered to the provided instructions. In these cases, the instructions themselves were found to be insufficient or ambiguous. Both LLM and manual annotation discrepancies were further subclassified as “major” or “minor” severity based on their potential impact on clinical interpretation or downstream analysis.
A flow chart for defining and documenting discrepancies as well as an introduction to the error contexts, presented as broad questions regarding information extraction aims, is provided in Fig. 3. Detailed examples for a subset of contexts are given in Tables 1–5, with the remainder plus additional examples in Supplementary Tables 1–11. For each context, we first provide two potential labels for an entity arising from insufficient instructions in the given context. This is followed by our addressing methodology, and further examples of LLM or annotation error severity in similar contexts (provided that we found our instructions to be sufficient).
Stopping criteria
Refinement concluded upon reaching zero major manual annotation errors, a near-zero rate of minor annotation discrepancies, a major LLM error rate near or below 1%, and an elimination of most schema errors, except those arising from complex cases deemed requiring human review. Upon reaching these criteria, we retrospectively reviewed the error documentation for each iteration to ensure consistency in our categorization (e.g., refining initial broad specification issues into more granular categories).
Assessing LLM interoperability
LLM backbone interoperability was assessed by comparing GPT-4o 2024-05-13, Llama 3.3 70B Instruct48, and Qwen2.5 72B Instruct49 outputs to the final gold-standard: First by exact match using the full specimen, block and test name when applicable, then by using an additional “fuzzy match” for IHC/FISH items that ignored punctuation and capitalization in the test name and did not require the specimen or tissue block identifiers to match the gold-standard. See Supplementary Table 13 for examples of match criteria. Exact match inter-model agreement between GPT-4o and the two open-weight models was also calculated. Both open-weight models were run on local compute infrastructure, with generations using a temperature of 0.
Internal application and validation
Our finalized pipeline was run using GPT-4o 2024-08-06 on the free text portion (final diagnosis, ancillary studies, comments, addendums) of 3520 internal pathology reports containing evidence of renal tumors spanning April 2019–April 2024; see Supplementary Fig. 1 for additional details on inclusion criteria. Of these reports, 2297 utilized additional discrete EMR fields corresponding to CAP kidney resection/biopsy and internal metastatic RCC pathology templates; This structured data could be pulled separately from the report text and was used for cross-referencing LLM output regarding metastatic RCC status and the presence or absence of six kidney tumor subtypes: clear cell RCC, chromophobe RCC, papillary RCC, clear cell papillary renal cell tumor (CCPRCT), TFE3-rearranged RCC, and TFEB-altered RCC. Discrepancies were manually reviewed using the free-text report as ground truth. Notably, the LLM needed to infer updated TFE3/TFEB subtypes from older ‘MiT family translocation RCC’ terms present in some structured data (per CAP Kidney 4.1.0.0)60,61.
To provide a baseline for histology extraction and mapping to updated terminology, we developed a custom rule-based regular expression (regex) tool targeting predictable structural and lexical patterns commonly observed in the “final diagnosis” section of reports, where primary histology is typically stated. This represented a pragmatic, non-machine learning approach using domain heuristics for comparison.
To attempt scalable validation of LLM extracted histology and IHC/FISH results across all 3520 reports, including those with no available templated data, we selected all extracted subparts/specimens with a single histology of the above six for which IHC/FISH results were also extracted. We then assessed the consistency of the histological subtype with the expected IHC/FISH pattern for five common markers used to differentiate renal tumor subtypes; CA-IX, CD117, Racemase, TFE3, and TFEB67. Unexpected findings were subject to manual review of the report text.
Assessing clinical domain interoperability
Adaptability to different clinical domains was evaluated using The Cancer Genome Atlas (TCGA)50,51,52 breast invasive carcinoma (BRCA)54 and prostate adenocarcinoma (PRAD)56 pathology reports that had undergone scanned PDF to text optical character recognition (OCR) processing and had corresponding tabular clinical data available53. For BRCA reports, we extracted results for HER2 (both FISH and IHC separately), progesterone receptor (PR), and estrogen receptor (ER), utilizing only modified IHC/FISH schema labels and instructions. We restricted the reports to those containing the words “immunohistochemistry” and “HER2” to ensure IHC results were present in the OCR processed text.
For PRAD reports, we extracted Gleason Scores using the ‘feature report’ flow with only modifications to the schema instructions and labels. All external validation was done using GPT-4o (2024-08-06 via Azure, temperature of 0).
Data availability
The data used in this study contains patient identifiers and cannot be shared publicly due to privacy regulations and institutional policies. The publicly available breast cancer reports and clinical data can be found at https://www.cbioportal.org/study/clinicalData?id=brca_tcga_pub2015. The publicly available prostate cancer reports and clinical data can be found at https://www.cbioportal.org/study/summary?id=prad_tcga_pub. The optical character recognition processed reports are available at https://data.mendeley.com/datasets/hyg5xkznpx/1.
Code availability
Code for implementing the workflow described in this paper is available at https://github.com/DavidHein96/prompts_to_table.
References
Li, I. et al. Neural natural language processing for unstructured data in electronic health records: a review. Comput. Sci. Rev. 46, 100511 (2022).
Zozus, M. N. et al. Factors affecting accuracy of data abstracted from medical records. PLoS ONE 10, e0138649 (2015).
Brundin-Mather, R. et al. Secondary EMR data for quality improvement and research: a comparison of manual and electronic data collection from an integrated critical care electronic medical record system. J. Crit. Care 47, 295–301 (2018).
Sushil, M. et al. CORAL: expert-curated oncology reports to advance language model inference. NEJM AI 1 (2024).
Jee, J. et al. Automated real-world data integration improves cancer outcome prediction. Nature 636, 728–736 (2024).
Sedlakova, J. et al. Challenges and best practices for digital unstructured data enrichment in health research: a systematic narrative review. PLoS Digit. Health 2, e0000347 (2023).
Xu, H., Anderson, K., Grann, V. R. & Friedman, C. Facilitating cancer research using natural language processing of pathology reports. Stud. Health Technol. Inform. 107, 565–572 (2004).
Hripcsak, G. et al. Unlocking clinical data from narrative reports: a study of natural language processing. Ann. Intern. Med. 122, 681–688 (1995).
Alsentzer, E. et al. Publicly available clinical BERT embeddings. Preprint at https://doi.org/10.48550/ARXIV.1904.03323 (2019).
Yang, X. et al. A large language model for electronic health records. Npj Digit. Med. 5, 194 (2022).
Rasmy, L., Xiang, Y., Xie, Z., Tao, C. & Zhi, D. Med-BERT: pretrained contextualized embeddings on large-scale structured electronic health records for disease prediction. Npj Digit. Med. 4, 86 (2021).
Lee, J. et al. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 36, 1234–1240 (2020).
Peng, Y., Yan, S. & Lu, Z. Transfer learning in biomedical natural language processing: an evaluation of BERT and ELMo on ten benchmarking datasets. Preprint at https://doi.org/10.48550/ARXIV.1906.05474 (2019).
Su, P. & Vijay-Shanker, K. Investigation of improving the pre-training and fine-tuning of BERT model for biomedical relation extraction. BMC Bioinformatics 23, 120 (2022).
Li, Y., Wehbe, R. M., Ahmad, F. S., Wang, H. & Luo, Y. A comparative study of pretrained language models for long clinical text. J. Am. Med. Inform. Assoc. 30, 340–347 (2023).
Brown, T. et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 33, 1877–1901 (2020).
Liu, H. et al. Evaluating the logical reasoning ability of ChatGPT and GPT-4. Preprint at https://doi.org/10.48550/ARXIV.2304.03439 (2023).
Nori, H., King, N., McKinney, S. M., Carignan, D. & Horvitz, E. Capabilities of GPT-4 on medical challenge problems. Preprint at https://doi.org/10.48550/ARXIV.2303.13375 (2023).
Singhal, K. et al. Large language models encode clinical knowledge. Nature 620, 172–180 (2023).
Agrawal, M., Hegselmann, S., Lang, H., Kim, Y. & Sontag, D. Large language models are few-shot clinical information extractors. Preprint at https://doi.org/10.48550/ARXIV.2205.12689 (2022).
Hu, Y. et al. Improving large language models for clinical named entity recognition via prompt engineering. J. Am. Med. Inform. Assoc. 31, 1812–1820 (2024).
Sivarajkumar, S., Kelley, M., Samolyk-Mazzanti, A., Visweswaran, S. & Wang, Y. An empirical evaluation of prompting strategies for large language models in zero-shot clinical natural language processing: algorithm development and validation study. JMIR Med. Inform. 12, e55318 (2024).
Peng, C. et al. Generative large language models are all-purpose text analytics engines: text-to-text learning is all your need. J. Am. Med. Inform. Assoc. 31, 1892–1903 (2024).
Burford, K. G., Itzkowitz, N. G., Ortega, A. G., Teitler, J. O. & Rundle, A. G. Use of generative AI to identify helmet status among patients with micromobility-related injuries from unstructured clinical notes. JAMA Netw. Open 7, e2425981 (2024).
Hu, D., Liu, B., Zhu, X., Lu, X. & Wu, N. Zero-shot information extraction from radiological reports using ChatGPT. Int. J. Med. Inf. 183, 105321 (2024).
Huang, J. et al. A critical assessment of using ChatGPT for extracting structured data from clinical notes. Npj Digit. Med. 7, 106 (2024).
Johnson, B. et al. Large language models for extracting histopathologic diagnoses from electronic health records. Preprint at https://doi.org/10.1101/2024.11.27.24318083 (2024).
Le Guellec, B. et al. Performance of an open-source large language model in extracting information from free-text radiology reports. Radiol. Artif. Intell. 6, e230364 (2024).
Liu, F. et al. Large language models are poor clinical decision-makers: a comprehensive benchmark. Preprint at https://doi.org/10.1101/2024.04.24.24306315 (2024).
Omiye, J. A., Gui, H., Rezaei, S. J., Zou, J. & Daneshjou, R. Large language models in medicine: the potentials and pitfalls: a narrative review. Ann. Intern. Med. 177, 210–220 (2024).
Sushil, M. et al. A comparative study of large language model-based zero-shot inference and task-specific supervised classification of breast cancer pathology reports. J. Am. Med. Inform. Assoc. 31, 2315–2327 (2024).
Wang, L. L. et al. Automated metrics for medical multi-document summarization disagree with human evaluations. Proc. Conf. Assoc. Comput. Linguist. Meet. 2023, 9871–9889 (2023).
Wornow, M. et al. The shaky foundations of large language models and foundation models for electronic health records. Npj Digit. Med. 6, 135 (2023).
Tang, L. et al. Evaluating large language models on medical evidence summarization. Npj Digit. Med. 6, 158 (2023).
Reichenpfader, D., Müller, H. & Denecke, K. A scoping review of large language model based approaches for information extraction from radiology reports. Npj Digit. Med. 7, 222 (2024).
Tian, S. et al. Opportunities and challenges for ChatGPT and large language models in biomedicine and health. Brief. Bioinform. 25, bbad493 (2023).
Willard, B. T. & Louf, R. Efficient guided generation for large language models. Preprint at https://doi.org/10.48550/ARXIV.2307.09702 (2023).
Dong, Y. et al. XGrammar: flexible and efficient structured generation engine for large language models. Preprint at https://doi.org/10.48550/ARXIV.2411.15100 (2024).
Fleming, S. L. et al. Medalign: a clinician-generated dataset for instruction following with electronic medical records. in Proceedings of the AAAI Conference on Artificial Intelligence vol. 38 22021–22030 (2024).
McIntosh, T. R. et al. Inadequacies of large language model benchmarks in the era of generative artificial intelligence. Preprint at https://doi.org/10.48550/ARXIV.2402.09880 (2024).
Zhong, T. et al. Evaluation of OpenAI o1: opportunities and challenges of AGI. Preprint at https://doi.org/10.48550/ARXIV.2409.18486 (2024).
Goel, A. et al. Llms accelerate annotation for medical information extraction. in Machine Learning for Health (ML4H) 82–100 (PMLR, 2023).
OpenAI et al. GPT-4o system card. Preprint at https://doi.org/10.48550/ARXIV.2410.21276 (2024).
Lavu, H. & Yeo, C. J. Metastatic renal cell carcinoma to the pancreas. Gastroenterol. Hepatol. 7, 699–700 (2011).
OpenAI. tiktoken. OpenAI (2025).
Gage, P. A new algorithm for data compression. C. Users J. 12, 23–38 (1994).
Oien, K. A. & Dennis, J. L. Diagnostic work-up of carcinoma of unknown primary: from immunohistochemistry to molecular profiling. Ann. Oncol. 23, x271–x277 (2012).
Grattafiori, A. et al. The Llama 3 Herd of models. Preprint at https://doi.org/10.48550/ARXIV.2407.21783 (2024).
Qwen et al. Qwen2.5 technical report. Preprint at https://doi.org/10.48550/arXiv.2412.15115 (2025).
De Bruijn, I. et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 83, 3861–3867 (2023).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Kefeli, J. & Tatonetti, N. TCGA-Reports: a machine-readable pathology report resource for benchmarking text-based AI models. Patterns 5, 100933 (2024).
Ciriello, G. et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163, 506–519 (2015).
Makhlouf, S. et al. The clinical and biological significance of estrogen receptor-low positive breast cancer. Mod. Pathol. 36, 100284 (2023).
Abeshouse, A. et al. The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015).
Shool, S. et al. A systematic review of large language model (LLM) evaluations in clinical medicine. BMC Med. Inform. Decis. Mak. 25, 117 (2025).
Ho, C. N. et al. Qualitative metrics from the biomedical literature for evaluating large language models in clinical decision-making: a narrative review. BMC Med. Inform. Decis. Mak. 24, 357 (2024).
Büscheck, F. et al. Aberrant expression of membranous carbonic anhydrase IX (CAIX) is associated with unfavorable disease course in papillary and clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 36, 531.e19–531.e25 (2018).
Murugan, P. Protocol for the examination of resection specimens from patients with renal cell carcinoma. (2024).
Murugan, P. Protocol for the examination of biopsy specimens from patients with renal cell carcinoma. (2024).
Microsoft Corporation. Prompt flow. Microsoft (2024).
Wei, J. et al. Chain-of-thought prompting elicits reasoning in large language models. Adv. Neural Inf. Process. Syst. 35, 24824–24837 (2022).
Yu, Z., He, L., Wu, Z., Dai, X. & Chen, J. Towards better chain-of-thought prompting strategies: a survey. Preprint at https://doi.org/10.48550/ARXIV.2310.04959 (2023).
Chen, Q. et al. Towards reasoning era: a survey of long chain-of-thought for reasoning large language models. Preprint at https://doi.org/10.48550/ARXIV.2503.09567 (2025).
Yeo, W. J., Satapathy, R., Goh, R. S. M. & Cambria, E. How interpretable are reasoning explanations from prompting large language models? Preprint at https://doi.org/10.48550/ARXIV.2402.11863. (2024)
Kim, M. et al. Comprehensive immunoprofiles of renal cell carcinoma subtypes. Cancers 12, 602 (2020).
Acknowledgements
This work was supported by the NIH-sponsored Kidney Cancer SPORE grant (P50CA196516) and endowment from the Jan and Bob Pickens Distinguished Professorship in Medical Science and Brock Fund for Medical Science Chair in Pathology. The authors thank the UTSW data warehouse team, who are supported by UL1TR003163, for their assistance in retrieving the data used in this study.
Author information
Authors and Affiliations
Contributions
D.H.: Conceptualization, methodology, data collection, data analysis, software, visualization, writing original draft, writing review and editing. A.C.: Conceptualization, data collection, data analysis, writing review and editing. M.H.: Software, methodology, writing review and editing. B.X.: Conceptualization, data collection, writing review, and editing. A.J.J.: Writing review and editing. J.V.: Data collection, clinical expertise, writing review and editing. N.R.: Data collection. A.H.S.: Software. S.C.: Data collection, software. L.G.C: Conceptualization, writing review and editing. J.B.: Writing review and editing, project administration and supervision, clinical expertise. A.R.J.: Writing review and editing, project administration and supervision. P.K.: Conceptualization, data collection, writing review and editing, project administration and supervision, clinical expertise.
Corresponding author
Ethics declarations
Competing interests
[D.H., M.H, A.H.S, A.R.J.] Azure compute credits were provided to Dr. Jamieson [A.R.J.] and lab members [D.H., M.H., A.H.S.] by Microsoft as part of the Accelerating Foundation Models Research initiative. No otherwise competing interests are declared for these authors. Remaining authors, [A.C., B.X, A.J.J., J.V., N.R., S.C., L.G.C., and P.K.] declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hein, D., Christie, A., Holcomb, M. et al. Iterative refinement and goal articulation to optimize large language models for clinical information extraction. npj Digit. Med. 8, 301 (2025). https://doi.org/10.1038/s41746-025-01686-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41746-025-01686-z
