
Abstract
Mitochondrial genome is an essential resource to investigate the dispersion and diversification of human populations. However, the lack of genomic data from non-European ancestry hinders demographic, anthropologic and genetic studies. Indigenous people from the Brazilian Amazon are especially underrepresented among large genetic datasets, despite their significative contribution to the Brazilian genetic pool. We investigated 157 mitochondrial genomes from the Brazilian Amazon using bioinformatics tools for ancestry inference and phylogenetics. Results were compared with 2578 global mitogenomes to assess haplogroup frequencies and genetic distances. Thirteen macrohaplogroups were identified from investigated samples, including Indigenous, European and African ancestries. Despite being prominent in our sample, Indigenous haplogroup assignment confidence by prediction tools was lower and many samples lacked key-defining variants, suggesting misassignment due to low representation in reference datasets. We also identified potential new groups sharing specific variants. Thus, underrepresentation of Indigenous haplogroups contributes to phylogenetic inconsistencies and needs future investigations.
Similar content being viewed by others


Introduction
Data produced by whole mitochondrial genome (mitogenome) sequencing can substantially increase the discriminatory power of mitochondrial DNA (mtDNA), revealing ancestry-specific variants1. Due to its maternal inheritance, mtDNA can be used to obtain information about genetic ancestry through the identification of mitochondrial haplogroups, defined by the sequential accumulation of alterations inherited from a common ancestor1,2. This information can be used to trace the phylogeographic distribution of mtDNA in different populations and to identify lineages of specific geographic regions. Further, mtDNA variation may contribute to differences in the prevalence of diseases observed among different ethnic groups1,2.
However, current mtDNA haplogroup identification methods face certain difficulties, such as the bias of classification based on consensus haplotypes of the revised Cambridge Reference Sequence (rCRS)3,4. Additionally, admixed populations, such as those in the Americas, represent a peculiar case for genetic ancestry inference because they originated through a complex process of admixture from population of different continents: Indigenous, Europeans, Africans, and more recently East Asians5,6.
In this context, the Brazilian population is one of the most ethnically diverse in the world and is currently divided into five main macro-regions (North, Northeast, Central-West, Southeast, and South), each with distinct histories of colonization and settlement. This diversity is reflected in the genetic structure of the country7,8. Thus, characterization of mitochondrial ancestry markers can elucidate past and current patterns of admixture, complementing prior knowledge about the biological composition of the Brazilian population7,8,9,10.
Among the Brazilian macro-regions, the Amazonian (or North) region stands out for having a larger contribution of Indigenous mitochondrial haplogroups (A–D and X)11,12,13 in its population compared to other regions of the country7,14,15,16. However, there is still a lack of complete mitogenome data for this territory, which hinders demographic genetic studies and limits our understanding of individuals health-disease status, especially those of Indigenous ancestry.
Here, we investigated the mitochondrial haplogroups of the Amazonian region of Brazil using mitogenomes previously analyzed and published by our research group. We assessed the contribution of Indigenous lineages to the formation of this highly admixed population and evaluated the reliability of maternal ancestry predictors in defining Indigenous haplogroups.
Results and discussion
Characterization and genetic composition of the cohort
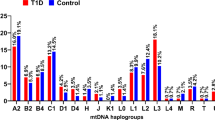
We investigated the mitogenome of 157 admixed individuals from the North region. Although there were more male than females, they both presented similar average age (Table 1). Thirteen macrohaplogroups were identified using HaploGrep3, which can be divided among those of Indigenous (macrohaplogroups A, B, C, and D), European (macrohaplogroups H, J, N, T, and U), and African (macrohaplogroups L0, L1, L2, and L3) ancestry (Fig. 1A). As expected, Indigenous haplogroups represented the majority (64.97%), followed by African (28.03%) and European haplogroups (7%). This haplotypic distribution is consistent with the literature and reflects the processes of occupation and colonization of the Brazilian territory, especially of the Amazon region7,17,18,19,20. When sub-structuring the Indigenous macrohaplogroups, haplogroups C, A2, and B2 were the most frequent in the cohort (Fig. 1B), consistent with previous description of Indigenous population from the region7,19,20,21. Although haplogroups A–D are also present in East Asia, fact that supports the East Asian origin of Indigenous lineages21, we find unlikely that all of the individuals here with these haplogroups are indeed of genetic East Asian origin.

A It shows the frequency of all macrohaplogroups found in our cohort and B It shows only Indigenous haplogroups in detail. Bars were colored according to haplogroup ancestry.
Evaluation of indigenous haplogroup identification
While evaluating the mitochondrial haplogroup assignments of HaploGrep3 tool, we observed that those belonging to European and African ancestries had an average prediction confidence around 0.95 while Indigenous haplogroups was around 0.89 (Fig. 2A). The lower assignment confidence of Indigenous haplogroups reflects the limited data available for such ethnic background in global public databases22,23. Admixed Americans comprise only 3.72% gnomAD24, currently one of the largest genomic databases. The mtDNA-specialized database EMPOP25 has 17% of their samples attributed to Indigenous origin and 6% to admixed Americans, among which only eight Indigenous samples are from Brazil and none are from the Amazon region. Such restrictions interferes with the correct identification of mitochondrial lineages that are still represented today in the Amazon region.

Each point represents a mitogenome haplogroup assignment confidence. Samples were grouped according to their geographic region and their average ±1 SD confidence indicated by black point and line. A presents mitogenomes from this study grouped according to their haplogroup ancestry in African (BRA [AFR]), European (BRA [EUR]), and Indigenous (BRA [IND]). B presents mitogenomes from 1000 Genomes Project grouped according to their geographic groups in African (AFR), East Asian (EAS), European (EUR), South Asia (SAS), and Admixed American (AMR) and those from this study (BRA [ALL]).
Further, European, African and East Asian populations from the 1000 Genomes Project (1000G) have an average prediction confidence greater than 0.95 when carrying out the same analysis, while our sample presented an average confidence of 0.9 (Fig. 2B). This result is not surprising given that the populations included in 1000G were more frequently investigated and have more information available in public databases, unlike of the Amazonian populations from Brazil21.
Underrepresentation of ethnically diverse populations, such as those from the Amazon, in global genetic datasets can impact ancestry inference, impair discovery of population-specific variants, hinder identification of disease causal variants, reduce accuracy of genetic risk predictions, and limit our understanding of shared genetic risk factors affecting health outcomes22,26,27.
Distribution of indigenous mitochondrial variants
We investigated all the variants highlighted by HaploGrep3, as well as all the expected variants for the assigned haplogroup that were absent in the sequence of 102 mitogenomes of Indigenous ancestry. Most ancestry-defining haplotypes were identified. However, the classification of some individuals presented a significant margin of error, attributed to the absence of certain ancestry-defining variants (Table 2).
Although most of A2, B2, and C individuals presented all their ancestry-defining variants, not all of them possess every required mutation, which suggests that the missing variants may have undergone reverse selection. Eight individuals from haplogroup A2 lacked the 16111T and 1736G variants and 14 individuals did not present 8027A, all haplogroup-defining variants, thus being uncertain if they were correctly classified. Unlike those, an A8 individual did not present the variant 16242T, essential for the characterization of such haplogroup, so this individual should have been assigned to the haplogroup A, due to the presence of 16223T, 16290T and 16319A and absence of 16242T28,29.
Regarding haplogroups B2 and B4, all B2 individuals lacked the 15535T mutation, and seven lacked the 3547G mutation. Among the C individuals, five lacked the 10873C mutation, and one individual did not present the 16327 T mutation, which is crucial for the characterization of this haplogroup. This suggests that this individual might be an intersection between haplogroups C and CZ28,29. In our whole cohort, only 23 individuals had the 7028T mutation (A2 = 4, B2 = 6, C = 4, C1 = 1, D1 = 1, L1 = 1, L2 = 3, L3 = 2, and N = 1). This is curious since this variant is highly frequent in most populations, especially in Asian, African, and Latin American/admixed populations, except for the European haplogroup H24,30.
Phylogenetic analyses
Aiming to explore the relationship between our samples, we reconstructed a phylogenetic tree using the maximum parsimony method and rooted the resulting tree on two L0 individuals (Methods; Figs. 3, S1). The resulting tree reflects the major human mitochondrial divisions presented in PhyloTree28,31, where all L samples were outgroups and meta-haplogroups M (composed by haplogroups C and D), N (A and N), and R (H, J, T, U, and B) grouped as expected. Overall, samples were clustered according to their assigned macrohaplogroups, thus reinforcing the robustness of this analysis.

The tree was constructed using maximum parsimony method with 1500 bootstrap samples and rooted on two L0 individuals. Represented using a circular layout that preserves the topological information but not the branches length. Tree tips and branches shared by same haplogroup were colored according to their haplogroup assignment. Branches shared by multiple haplogroups remained gray. Internal clades with more than 50% of bootstrap support are indicated by a dot. Tip shapes indicate haplogroups ancestry.
However, we observed unexpected behaviors when analyzing within Indigenous haplogroups. Individuals classified as C and C1 constitute a single indistinguishable clade from each other. Haplogroup C1 is mainly distinguished from C by the presence of 16325C, but almost all samples classified as C present this variant. Individuals classified as D1 and D4 presented a similar pattern with all grouped under a single clade. Haplogroup D1 is distinguished from D4 by the presence of 16325C and 2092T, here we found that most D1 individuals lack 16325C while all D4 individuals presented the variant. These haplogroups are distinguished only by the presence of 2092T. Underrepresentation of samples of Indigenous ancestry in the PhyloTree construction, especially those from the Amazon region, may be impairing the HaploGrep3 classification.
Representativeness of cohort haplogroups in other populations
We constructed a PCA based on mitochondrial macrohaplogroup frequencies to explore the genetic relationship between Amazon population (BRA) and 1000G populations (Fig. 4A), which includes African, European, East Asian, South Asian and Admixed American populations32. PC1 explained 29.3% of the dataset variation and PC2 21.3%. PCA indicated the existence of five distinct clusters, corresponding to the global African, American, European, South and East Asian populations. Notably, our Amazonian sample was in an intermediate position between all clusters, which is in agreement with the literature and demonstrates the historical processes of migration, admixture and colonization of the Brazilian territory6,33,34.

A PCA based on the frequency of mitochondrial macrohaplogroups among the Amazonian population (BRA) and 26 world populations from 1000G, colored according to their geographic region. B Absolute frequency of haplogroups found in this study among our cohort (BRA) and 1000 G populations. Only 1000G populations presenting an haplogroup identified in this study were included. Dots indicate a haplogroup presence with size and color indicating their absolute frequency. Selected populations (y-axis) are grouped according to their geographic regions.1000G population labels can be found at Supplementary Table 1.
Next, we compare the frequency of haplogroups from our cohort to selected populations from 1000G that presented haplogroups found among our samples (Fig. 4B). The African haplogroups from our cohort (L0, L1, L2 and L3) had a relevant presence only in populations from Nigeria, Sierra Leone, Kenya, Gambia, Barbados and among Afro-Americans from the US.
Indigenous haplogroups were only prevalent among American populations that underwent similar admixture processes as Brazil (Fig. 4B). Haplogroup A2 was found mostly in populations from Puerto Rico, Peru, Mexicans from California and Colombia. Peruvian population presented a significative presence of B2, C and D1 with a similar distribution as our sample. Although Indigenous haplogroups are found among Admixed American populations, they remain under-represented in general and misses specific haplogroups such as A8 and D4. Thus, this evaluation demonstrates the lack of Indigenous ancestry data in public datasets.
We further explored the genetic similarities and divergences between our sample and 1000 G populations by calculating their pairwise Fst genetic distance. For this analysis, we divided our cohort according to their haplogroup ancestry (European: BRA-EUR, African: BRA-AFR, and Indigenous: BRA-IND). These populations grouped with themselves and distinguished from all other populations (Figure S2A). BRA-EUR presented the largest distance to all other populations, with highest value against IBS (Fst=0.13). Our cohort presented the smallest distance to Afro-American populations, such as ASW and BRA-IND (Fst=0.08). These results were reinforced when analyzing pairwise Fst genetic distance of this study groups and 1000 G populations grouped by their geographic region in American, European, South Asian, East Asian, African and African-American (Figure S2B). Our cohort presented the largest distance to European populations (Fst = 0.10), and greater proximity to African-American (Fst = 0.08) and African populations (Fst = 0.09).
In one of the few studies about the complete mitogenome in Brazil, Avila et al.35 produced similar findings when analyzing 96 individuals from South, Southeast and Central-West regions of Brazil. They indicated that the Brazilian population had a high proximity to the African-American population, a low proximity to European populations, and an intermediate proximity to South Asian populations. These findings are supported by genetic, historical and ethnogeographic information previously reported in the Brazilian territory 5,7,33,35.
Haplogroups representation among indigenous populations
Next, we compared the frequency of Indigenous haplogroups (Fig. 5) on our Admixed Amazonian cohort to 74 Indigenous mitogenomes included in the construction of PhyloTree21,31,36 (Supplementary Table 2). Our cohort presented a greater haplogroup frequency similarity to Amazon and South American Indigenous population. Amazonian Indigenous and our cohort presented a greater predominance of haplogroups A2, B2, and C1 than other groups, as previously described1,11,13,18,37,38, while other Brazilian Indigenous populations lack A2 and C. Further, the X2 haplogroup specificity to North American Indigenous is in agreement with the literature21,39 and reflects the limited expansion of this group to the rest of the continent.

Each point size and color indicate the relative frequency of Indigenous haplogroups (x-axis) among our cohort (Admixed Amazon) and Indigenous Populations along the American continent. Populations’ sample size is indicated next to their label.
We further explored the relationship between these samples by constructing a heatmap for mutation presence for haplogroups A to D (Supplementary Figs. 3–6). All samples presented a low haplogroup assignment confidence by HaploGrep3 (0.89 ± 0.06). In fact, samples assigned to different haplogroups presented a high genetic similarity. Two A2 samples clustered with A2 + (64), despite lacking 64 T, due to their shared 310C (Supplementary Fig. 3). Haplogroups B2, B2b, and B2h did not cluster together (Supplementary Fig. 4). One B2e and two B2 clustered due to their exclusive 8281–8289 deletion. B2 and B2*2 clustered due to three variants (103A, 14470C, and 16241G).
Among macrohaplogroup C, most C1b and C1c samples grouped according to their haplogroup assignments (Supplementary Fig. 5). Most samples assigned as C presented 16325C, which defines the C1 clade, and at least half of these also presented 493G, which defines C1b. C1d1 cluster included two C assigned samples, which presented C1d defining variants (16325C and 16051G). Another cluster of three C1c and six C samples presented all C1c defining variants (1888A and 15930A). These examples present strong indication of misassignment of those C samples.
Macrohaplogroup D presented the highest average assignment confidence (0.95 ± 0.02) compared to the others (Supplementary Fig. 6). All D4 samples lacked 2092T but presented 16325C, which defines haplogroup D1. They all clustered with other D1 samples and differed from them by a single mutation.
Among macrohaplogroups A, B, and C, potential new clusters can be identified (Supplementary Figs. 3–5). Three A2 samples clustered due to five exclusive variants (9096C, 10463G, 15951G, 16126C and 16259T). Three B2 samples shared four variants (2392C, 5150T, 15936G, 16178C) and other three B samples shared 16 variants (top of Supplementary Fig. 6). One C sample clustered with C1b*3 due to three exclusive variants (8383C, 10586A, and 16093C), a cluster of six C samples presented C1d defining variants and shared 7697A and 15004T, and other three C samples shared six variants that were almost exclusive (top of Supplementary Fig. 5).
Final remarks
Representation of ancestries and haplogroups from different continents or geographic regions varies widely between genetic panels. Panels more representative of ethnically diverse populations lead to better genomic imputation compared to others40. Previous studies have reported that under-representation of non-European populations influence the accuracy of genotype imputation27,41. Worldwide GWAS are mainly concentrated in USA and European countries, which contribute to underrepresentation of the worldwide genomic diversity, and leads to a low prediction quality among underrepresented populations22,26,27,42. As of November 2024, the majority of genomic studies were conducted on individuals of European ancestry, which represents 90% of all investigated individuals, while admixed individuals from Latin America accounted for only 0.34%42. Previous studies have also shown that Hispanic/Latino and African American genomic studies contribute with a disproportionate larger number of associations compared to a similarly sized study with European ancestry42,43. Therefore, increasing representation of samples of non-European ancestry in genomic datasets is critical to discover new variants associated with diseases and explain complex phenotypes.
Various Indigenous haplogroups are present among the Brazilian Amazon populations and their haplotype are underrepresented among large genomic datasets such as 1000 G and PhyloTree. This lack of data produces poor assignment confidence to Indigenous haplogroups tools such as HaploGrep3. Although there are efforts to increase the diversity of genomic information from Latin America countries, such as PoblAr44, Urugenomes45 and LatinGen46, there is still a scarcity of data from Brazil and Brazilian Amazon, especially regarding the mitochondrial genome. In this sense, this work complements efforts to improve mitochondrial reference data and increase our understanding of underrepresented populations phylogenetic distribution worldwide. However, due to limitations of the current study, further studies with larger cohorts should be carried out to characterize the genetic diversity of Amazonian populations.
Methodology
Sample characterization
A total of 157 mitogenomes from Brazilian Amazon were analyzed in this study. Data was gathered from two previous studies from our research group:
-
De Souza et al.47 analyzed the mitogenome of 33 individuals diagnosed with leprosy and 37 control individuals.
-
Matos et al.48 analyzed the mitogenome of 45 individuals diagnosed with Parkinson disease and 42 control individuals.
DNA extraction, quantification, amplification, and sequencing are described in their respective studies. This work adhered to the Declaration of Helsinki.
To expand our analysis, we also integrated to our dataset the mitogenome of 2504 individuals from the 1000 G phase 332 and 74 Indigenous mitogenome from various regions the Americas21,36. Their detailed population description can be found on Table S1 and Table S2, respectively.
Sequencing pre-processing and bioinformatics analysis
To assess sequencing quality, we used FastQC v0.12.149 and MultiQC v1.1950 before and after data pre-processing. We used FastP (v0.23.4)51 to remove low-quality bases, sequencing adapters, and reads with less than 36 bp. After treatment and quality assessment, reads were aligned to the mtDNA reference sequence (rCRS) using the Burrows-Wheeler alignment tool (BWA) v0.7.1752. The resulting alignments were sorted using SAMTools v1.15.153, and duplicate reads were removed using Picard v2.27.554. To identify and remove samples with contamination issues, we employed Haplocheck v1.3.355 tolerating up to 10% of contamination and excluding samples exceeding this threshold.
Detection and analysis of variants
After pre-processing and aligning the sequences, BAM files were submitted to mutserve v2.0.1 for single nucleotide polymorphisms (SNPs) calling, annotation and heteroplasmy detection (https://mitoverse.readthedocs.io/mutserve/mutserve/)56. Functional annotation of each variant, locus and mtDNA complex was then summarized through custom scripts developed with R programming language57 and RStudio environment58. To improve the reliability of the variant analyses, we required an overall average depth > 550x and heteroplasmy levels below < 0.10.
Inference of mitochondrial haplogroups
Inference of mitochondrial haplogroups was conducted using HaploGrep3 v3.2.159 (available at https://haplogrep.i-med.ac.at/), which performs automatic assignment of mtDNA haplogroups based on PhyloTree 17—Forensic Update 1.231. The inference is performed using the Kulczynski calculation between the sample and presumed haplogroup based on sets of expected and observed polymorphisms, weighted by the relative recurrence of each polymorphism in the phylogenetic tree.
Phylogenetic analysis
Mitogenome sequences were generated using HaploGrep3 with -write-fasta-msa option. The resulting.fasta files were aligned using MAFFT v7.526 software60,61 and later loaded into MEGA v11.0.10 software62,63. We opted to build a Maximum Parsimony tree with 1500x bootstrap64 and exported it in Newick format (.nwk). The resulting tree was rooted in two L0 individuals, to reflect our current understanding of human origin13,31. This strategy was developed following the recommendations from Behar et al.65. The resulting tree was visualized in R programming language using the treeio66 and ggtree67 packages.
Worldwide mitogenome diversity
We performed a Principal Component Analysis (PCA) based on the frequency of all Macrohaplogroups with 1000 G populations and this study cohort. To calculate the Fst pairwise distance between populations, we aggregated our genotype data to mitochondrial data from 1000 G to create a genotype matrix in the R environment using the vcfR package68. The resulting matrix was converted to a hierfstat dataframe used to calculate Fst according to Weir and Cockerham69 equation using hierfstat package70. The results were visualized using pheatmap package71, factoextra72, FactoMineR73 and plotly74.
Then, we combined data from this study with a 74 Indigenous mitogenome from various regions of the Americas21,36. We compared the relative haplogroup frequency among our cohort and different Indigenous populations. Next, we compared these samples mutation profile and presented it using a presence heatmap. The heatmap was visualized using ComplexHeatmap package75.
Statistics and reproducibility
Statistical analyses and visualizations were conducted using R (v. 4.3.2). The statistical methods and tests employed for each analysis are explained in the text and figure legends, when applicable. A comprehensive description of the statistical methodology applied for each of haplogroups assignment, phylogenetics and worldwide mitogenome diversity analysis, respectively, is available in the “Methods” section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
The computer code and scripts used in this study are available at the following Zenodo76.
References
Wood, M. R. et al. Resolving mitochondrial haplogroups B2 and B4 with next-generation mitogenome sequencing to distinguish Native American from Asian haplotypes. Forsenic Sci. Int. Genet. 43, 102143 (2019).
Mitchell, S. L. et al. Characterization of mitochondrial haplogroups in a large population-based sample from the United States. Hum. Genet. 133, 861–868 (2014).
Andrews, R. M. et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 (1999).
Rubin, J. D., Vogel, N. A., Gopalakrishnan, S., Sackett, P. W. & Renaud, G. HaploCart: Human mtDNA haplogroup classification using a pangenomic reference graph. PLOS Comput. Biol. 19, e1011148 (2023).
Escher, L. M. et al. Challenges in selecting admixture models and marker sets to infer genetic ancestry in a Brazilian admixed population. Sci. Rep. 12, 21240 (2022).
Schaan, A. P. et al. New insights on intercontinental origins of paternal lineages in Northeast Brazil. BMC Evol. Biol. 20, 15 (2020).
Schaan, A. P. et al. mtDNA structure: the women who formed the Brazilian Northeast. BMC Evol. Biol. 17, 185 (2017).
Resque, R. et al. Male Lineages in Brazil: intercontinental admixture and stratification of the European Background. PLoS ONE 11, e0152573 (2016).
Hernández, C. Mitochondrial DNA in human diversity and health: from the Golden Age to the Omics Era. Genes 14, 1534 (2023).
Criollo-Rayo, A. A. et al. Native American gene continuity to the modern admixed population from the Colombian Andes: implication for biomedical, population and forensic studies. Forensic Sci. Int. Genet. 36, e1–e7 (2018).
Martínez-Cortés, G. et al. Maternal admixture and population structure in Mexican–Mestizos based on mtDNA haplogroups. Am. J. Phys. Anthropol. 151, 526–537 (2013).
Torroni, A. et al. Native American mitochondrial DNA analysis indicates that the Amerind and the Nadene populations were founded by two independent migrations. Genetics 130, 153–162 (1992).
Badano, I. et al. Mitochondrial DNA ancestry, HPV infection and the risk of cervical cancer in a multiethnic population of northeastern Argentina. PLoS ONE 13, e0190966 (2018).
Poletto, M. M., Malaghini, M., Silva, J. S., Bicalho, M. G. & Braun-Prado, K. Mitochondrial DNA control region diversity in a population from Parana state—increasing the Brazilian forensic database. Int. J. Leg. Med. 133, 347–351 (2019).
Freitas, J. M., Fassio, L. H., Braganholi, D. F. & Chemale, G. Mitochondrial DNA control region haplotypes and haplogroup diversity in a sample from Brasília, Federal District, Brazil. Forensic Sci. Int. Genet. 40, e228–e230 (2019).
Sanches, N. M., Paneto, G. G., Figueiredo, R. F., De Mello, A. O. & Cicarelli, R. M. B. Mitochondrial DNA control region diversity in a population from Espirito Santo state, Brazil. Mol. Biol. Rep. 41, 6645–6648 (2014).
Carvalho, B. M., Bortolini, M. C., Santos, S. E. B. D. & Ribeiro-dos-Santos, ÂK. C. Mitochondrial DNA mapping of social-biological interactions in Brazilian Amazonian African-descendant populations. Genet. Mol. Biol. 31, 12–22 (2008).
Pena, S. D. J. et al. The genomic ancestry of individuals from different geographical regions of Brazil is more uniform than expected. PLoS ONE 6, e17063 (2011).
dos Santos, A. K. C. R. & Zago, M. A. Diversidade de linhagens do DNA mitocondrial de Ameríndios da Amazônia:populações contemporâneas e ancestrais (1996).
Freires Junior, A. A. Diversidade das regiões hipervariáveis (HVS-I, HVS-II, HVS-III) do DNA mitocondrial em tribos indigenas da Amazônia Brasileira (Universidade Federal do Pará, 2008).
Fagundes, N. J. R. et al. Mitochondrial population genomics supports a single pre-clovis origin with a coastal route for the peopling of the Americas. Am. J. Hum. Genet. 82, 583–592 (2008).
Fatumo, S. et al. A roadmap to increase diversity in genomic studies. Nat. Med. 28, 243–250 (2022).
Aquilano, E., De La Fuente, C., Rodríguez Golpe, D., Motti, J. M. B. & Bravi, C. M. Sequencing errors in Native American mitogenomes: Impact on clade definitions, haplogroup assignation, and beyond. Mitochondrion 70, 54–58 (2023).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Parson, W. & Dür, A. EMPOP—a forensic mtDNA database. Forensic Sci. Int. Genet. 1, 88–92 (2007).
Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell 177, 26–31 (2019).
O’Connell, J. et al. A population-specific reference panel for improved genotype imputation in African Americans. Commun. Biol. 4, 1269 (2021).
Dür, A., Huber, N. & Parson, W. Fine-tuning phylogenetic alignment and haplogrouping of mtDNA sequences. Int. J. Mol. Sci. 22, 5747 (2021).
Weissensteiner, H. et al. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 44, W58–W63 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Van Oven, M. & Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 30, E386–E394 (2009).
The 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Pena, S. D. J., Santos, F. R. & Tarazona-Santos, E. Genetic admixture in Brazil. Am. J. Med. Genet. C Semin. Med. Genet. 184, 928–938 (2020).
Simão, F., Ferreira, A. P., De Carvalho, E. F., Parson, W. & Gusmão, L. Defining mtDNA origins and population stratification in Rio de Janeiro. Forensic Sci. Int. Genet. 34, 97–104 (2018).
Avila, E. et al. Full mtDNA genome sequencing of Brazilian admixed populations: a forensic-focused evaluation of a MPS application as an alternative to Sanger sequencing methods. Forensic Sci. Int. Genet. 42, 154–164 (2019).
Kivisild, T. et al. The role of selection in the evolution of human mitochondrial genomes. Genetics 172, 373–387 (2006).
Alves-Silva, J. et al. The ancestry of Brazilian mtDNA lineages. Am. J. Hum. Genet. 67, 444–461 (2000).
Batista Dos Santos, S. E., Rodrigues, J. D., Ribeiro-Dos-Santos, A. K. C. & Zago, M. A. Differential contribution of indigenous men and women to the formation of an urban population in the Amazon region as revealed by mtDNA and Y-dNA. Am. J. Phys. Anthropol. 109, 175–180 (1999).
Havaš Auguštin, D. et al. Refining the global phylogeny of mitochondrial N1a, X, and HV2 haplogroups based on rare mitogenomes from Croatian isolates. Genes 14, 1614 (2023).
Sengupta, D. et al. Performance and accuracy evaluation of reference panels for genotype imputation in sub-Saharan African populations. Cell Genom. 3, 100332 (2023).
Cahoon, J. L. et al. Imputation accuracy across global human populations. Am. J. Hum. Genet. 111, 979–989 (2024).
Mills, M. C. & Rahal, C. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat. Genet. 52, 242–243 (2020).
Martin, A. R. et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019).
Dopazo, H., Llera, A. S., Berenstein, M. & Gonzáles-José, R. Genomas, enfermedades y medicina de precisión: un Proyecto Nacional. Cienc. Tecnol. Política 2, 019 (2019).
Wire, B. Institut Pasteur de Montevideo, Genomic Medicine Institute-Seoul National University, and Macrogen Launch Urugenomes Project to Decode the Genome of Uruguyan Population (2014).
Giugliani, R. & Matte, U. The Latin-American network of human genetics. Public Health Genom. 7, 74–75 (2004).
De Souza, F. G. et al. Whole mitogenome sequencing uncovers a relation between mitochondrial heteroplasmy and leprosy severity. Hum. Genom. 17, 110 (2023).
Matos, G. B. et al. The mitogenome mutation repertoire affects progression of Parkinson´s Disease. Preprint at https://doi.org/10.21203/rs.3.rs-5411701/v1 (2024).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. Babraham Bioinform. (2010).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107 (2023).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformation 26, 589–595 (2010).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Toolkit, P. Broad Institute, GitHub Repository. Broad Inst. (2019).
Weissensteiner, H. et al. Contamination detection in sequencing studies using the mitochondrial phylogeny. Genome Res. 31, 309–316 (2021).
Weissensteiner, H. et al. mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 44, W64–W69 (2016).
Core Team, R. R: A Language and Environment for Statistical Computing (R Foundation of Statistical Computing, 2014).
Team, R. Studio R Studio: Integrated Development Environment for R (R Foundation of Statistical Computing, 2021).
Schönherr, S., Weissensteiner, H., Kronenberg, F. & Forer, L. Haplogrep 3 - an interactive haplogroup classification and analysis platform. Nucleic Acids Res. 51, W263–W268 (2023).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166 (2019).
Kuraku, S., Zmasek, C. M., Nishimura, O. & Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 41, W22–W28 (2013).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Stecher, G., Tamura, K. & Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 37, 1237–1239 (2020).
Felsenstein, J. Confidence limits on phylogenies: an approach using the Bootstrap. Evolution 39, 783–791 (1985).
Behar, D. M. et al. A Copernican reassessment of the human mitochondrial DNA tree from its root. Am. J. Hum. Genet. 90, 675–684 (2012).
Yu G. [Aut, C. treeio. Bioconductor https://doi.org/10.18129/B9.BIOC.TREEIO (2017).
Yu G. [Aut, C. ggtree. Bioconductor https://doi.org/10.18129/B9.BIOC.GGTREE (2017).
Knaus, B. J. & Grünwald, N. J. vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358 (1984).
Goudet, J. Hierfstat, a package for R to compute and test hierarchical F -statistics. Mol. Ecol. Notes 5, 184–186 (2005).
Kolde, R. & Kolde, M. R. Package ‘pheatmap’. R. Package 1, 790 (2015).
Kassambara, A. & Mundt, F. factoextra: extract and visualize the results of multivariate. Data Analyses 1, 0–7 (2016).
F. Husson, J. Josse, S. Le, J. Mazet. FactoMineR: multivariate exploratory data analysis and data mining. 2. 11 https://doi.org/10.32614/CRAN.package.FactoMineR (2006).
Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny. CRC Press, Taylor and Francis Group2020.
Zuguang Gu. ComplexHeatmap. Bioconductor https://doi.org/10.18129/B9.BIOC.COMPLEXHEATMAP (2017).
Gouvea de Souza, F., Ribeiro-dos-Santos, A. M. & Araújo, G. Mitochondrial Ancestry from Complete Mitogenomes Highlights a Lack of Characterization of Indigenous Haplogroups in Brazilian Amazon Population. Zenodo https://doi.org/10.5281/ZENODO.15120168 (2025).
Barra Matos, G. & Araújo, G. mtDNA-network data - Parkinson’s Disease: mtDNA sequencing. Zenodo https://doi.org/10.5281/ZENODO.15150211 (2025).
Acknowledgements
This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-620139/2006-4), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Biocomputacional-Protocol no. 3381/2013/CAPES; Grant-Protocol no. 88887.912165/2023-00/CAPES); A.R.S is supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq/Brazil) with Produtividade (304413/2015–1) and Pró-Reitoria de Pesquisa e Pós-Graduação da Universidade Federal do Pará (PROPESP/UFPA).
Author information
Authors and Affiliations
Contributions
Â.R.S. and G.S.A. designed the study; C.G.S., A.M.R.S., C.S.S., T.P.S., A.R.G., P.F.C., G.L.E., A.V.S.F., G.C.C. and B.L.S.L. collected the data and revised the manuscript; F.G.S., G.B.M. and G.S.A. performed the analyses; C.G.S., B.L.S.L., S.S. and Â.R.S., acquired the resources; F.G.S., G.B.M. and A.M.R.S. wrote and revised the manuscript; Â.R.S., B.L.S.L., C.G.S., and G.S.A. supervised the project. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Aylin Bircan. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
de Souza, F.G., Matos, G.B., Sena Santos, C. et al. Mitochondrial ancestry from complete mitogenomes highlights a lack of characterization of indigenous haplogroups in Brazilian Amazon population. Commun Biol 8, 835 (2025). https://doi.org/10.1038/s42003-025-08126-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08126-4
