
Abstract
Non-small cell lung cancer (NSCLC) is a leading cause of cancer mortality, and therapies utilizing tumor-infiltrating lymphocytes (TILs) show significant promise. However, molecular signatures defining a productive TIL-mediated response remain poorly characterized. Here, we establish a patient-derived organoid and autologous TIL co-culture platform to show that expanded TILs mediate potent, specific cytotoxicity against NSCLC organoids. This functional response is associated with a crucial shift in T-cell states from proliferative towards effector memory phenotypes and involves activating key signaling networks, including the TNF and IL-17 pathways. Furthermore, T-cell receptor (TCR) analysis confirms the expansion process selectively enriches tumor-associated clonotypes, resulting in a more focused repertoire. This work delineates the transcriptional and clonal signatures of an effective anti-tumor immune response, providing a robust framework to guide next-generation personalized TIL therapies.
Similar content being viewed by others
Introduction
Lung cancer remains a leading cause of cancer-related deaths globally, with non-small cell lung cancer (NSCLC) accounting for ~85% of all the cases1. Despite advancements in treatment strategies, including targeted therapies and immunotherapies, the overall survival for NSCLC patients remains low2. Tumor microenvironment, particularly tumor-infiltrating lymphocytes (TILs), plays a crucial role in cancer progression and treatment efficacy3,4. Recent studies have highlighted the potential of TILs in predicting treatment outcomes and as effectors for immunotherapy in NSCLC5,6,7. However, the function and heterogeneity of TILs in NSCLC remain unclear due to the lack of physiologically relevant models that could recapitulate the complex tumor-immune cell interactions.
While patient-derived cell lines and xenograft models failed to fully capture the cellular diversity and heterogeneity of TILs from the original tumors8, organoid technology has emerged as a promising approach to address these limitations. Organoids are three-dimensional cellular structures derived from stem cells or tissue samples that can organize to form structures and functions like the original tissue9. Organoid models effectively preserve the histological, genetic, and functional characteristics of primary tumors while reflecting intertumoral heterogeneity, providing an experimental platform relevant to the in vivo microenvironment for studying tumor development mechanisms, drug screening, and personalized treatment10,11,12,13. Recently, this approach has been extended to model the tumor-immune microenvironment. Foundational studies have established co-culture systems to generate tumor-reactive T cells from peripheral blood and to explore combination immunotherapies using autologous TILs in NSCLC14,15. However, while these works have established the principle of modeling anti-tumor immunity with organoids, the specific molecular programs, cellular state transitions, and clonal dynamics that define a productive TIL response against NSCLC remain to be fully characterized at high resolution.
The advent of single-cell RNA sequencing (scRNA-seq) has significantly advanced our understanding of cellular heterogeneity within tumors and their microenvironment. This technology provides promising resolution for dissecting diverse cell populations in complex tissues, revealing intricate details of tumor evolution and immune cell dynamics16,17. In this study, we use a co-culture system of patient-derived organoids and autologous TILs to demonstrate that expanded TILs exhibit potent and specific cytotoxicity against NSCLC organoids. This functional response is associated with a shift from proliferative to effector memory T-cell phenotypes and the activation of key signaling networks, including the TNF and IL-17 pathways. Furthermore, the expansion process enriches for tumor-associated T-cell receptor clonotypes. Together, these findings delineate the key signatures of a productive anti-tumor response and provide a framework for developing personalized cell therapies.
Results
Establishment and characterization of non-small cell lung cancer organoids
The workflow for this study is illustrated in Fig. 1A. Tumor tissues were minced and digested into single-cell suspensions, then cultured in Matrigel, which is widely used in in vitro and in vivo models18. Simultaneously, we isolated and cultured TILs from the same patient sample.

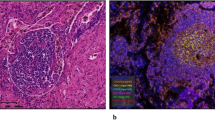
A Schematic of the experimental workflow for generating non-small cell lung cancer (NSCLC) organoids and autologous tumor-infiltrating lymphocytes (TILs) from patient tumor tissue. B Representative bright-field microscopy and immunohistochemistry (IHC) images comparing a primary lung adenocarcinoma (LUAD) tumor with its derived organoid. Staining for the lineage-specific markers TTF-1 and NAPSIN A shows conserved expression. C Representative bright-field and IHC images comparing a primary lung squamous cell carcinoma (LUSC) tumor with its derived organoid. Staining for the lineage-specific markers p63 and CK5/6 shows conserved expression. D A table summarizing the clinical characteristics of the 12 patients included in the study. Scale bars, 200 µm for all primary tissue IHC images; 100 µm for all H&E stains, bright-field, and organoid images.
The established organoids exhibited typical three-dimensional spherical structures. To validate these models, we used immunohistochemistry (IHC) to compare the expression of key lineage markers between the organoids and their corresponding primary tissues. We assessed established markers for lung adenocarcinoma (LUAD), TTF-1 and NAPSIN A, and for lung squamous cell carcinoma (LUSC), p63 and CK5/619,20. As shown in Fig. 1B, C, the organoids were positive for their respective lineage-specific markers, and their expression patterns were consistent with the primary tumors. These findings indicate that our organoid models show consistency in the expression of key histopathological markers with their tumors of origin. These findings indicate that our organoid models faithfully preserve the key histopathological markers of their tumors of origin. We successfully constructed these organoid models from all 12 patient samples (8 LUAD and 4 LUSC), highlighting the stability and reproducibility of this method (Fig. 1D).
We performed IHC, TIL culture, and co-culture experiments on most samples, and selected 5 samples for single-cell RNA sequencing analysis, which is conducive to further revealing complex cell-cell interactions in the tumor microenvironment21. Supplementary Fig. 1 shows the CT imaging data of patients, displaying the location and morphological characteristics of lung tumors in these 12 patients.
In vitro expansion focuses the TCR repertoire on tumor-associated clones
To investigate whether our in vitro expansion protocol enriched for tumor-specific T cell populations, we performed TCR sequencing on TILs from two representative patients (LCP5 and LCP8) at Day 0 and Day 14 of culture. The results, now presented in Fig. 2, indicate that the expansion process reshaped and focused the TCR repertoire. From Day 0 to Day 14, we observed a marked decrease in the total number of unique TCR clones (Fig. 2A, E), which was also reflected in a lower Shannon index (Fig. 2B, F). This contraction in diversity was accompanied by an increase in a “Lung Cancer TCR Score,” suggesting an enrichment of clones functionally relevant to the tumor (Fig. 2C, G). Furthermore, we found that the cumulative frequency of the dominant shared clones across all tiers analyzed (Top 10, 50, 100, and 500) increased after 14 days of culture (Fig. 2D, H).

A The total number of unique T-cell receptor (TCR) clonotypes for patient LCP5 at Day 0 (pre-expansion) and Day 14 (post-expansion), showing a marked decrease after expansion. B The Shannon index for patient LCP5, a measure of clonal diversity and evenness, illustrating a reduction in repertoire diversity post-expansion. C The Lung Cancer TCR Score for patient LCP5, indicating an increased enrichment of tumor-relevant clonotypes after 14 days of culture. D The cumulative frequency of the top shared clonotypes for patient LCP5, demonstrating the increased dominance of a few key clones post-expansion. E–H Corresponding analyses for patient LCP8, confirming a similar trend of TCR repertoire focusing.
Taken together, these TCR sequencing results suggest that the expansion protocol induces a selective process, leading to a less diverse but more focused repertoire enriched with tumor-associated T cell clones.
Assessment of killing effect of TILs on lung cancer organoids in vitro
To gain an in-depth understanding of the killing effect of TILs on NSCLC organoids, we established an organoid-TIL co-culture system and conducted quantitative and qualitative analyses through various methods. We selected two representative patient samples (LCP76 and LCP81) for these assessments (Fig. 3).

A Fluorescence microscopy images of LCP76 organoids, labeled with the green fluorescent dye CFSE, taken at the indicated time points. Images compare organoids co-cultured with TILs to organoids cultured alone over a 48-h period. Scale bars, 100 μm. B A high-magnification image showing TILs (indicated by black arrows) aggregating around a single LCP76 organoid during co-culture. Scale bars, 25 μm. C Microscopy images of LCP81 organoids co-cultured with TILs in the presence of a Caspase 3/7 reagent, taken at the indicated time points. The reagent emits a green fluorescent signal upon apoptosis induction. Scale bars, 100 μm. D Quantification of the mean fluorescence intensity (MFI) over 48 h from the experiment shown in panel (C). Data are shown as mean ± standard error of the mean (SEM) from three technical replicates. E Representative flow cytometry plots quantifying viable LCP81 organoid cells (CFSE positive, DAPI negative) after 48 h of culture alone, co-culture with peripheral blood mononuclear cells (PBMCs), or co-culture with TILs. F Statistical quantification of the percentage of viable organoid cells from the experiment shown in panel (E). Individual data points represent three technical replicates. For panels (D) and (F), statistical significance was determined by Welch’s t test.
For LCP76, we utilized CFSE labeling to track the dynamics of the organoids during the co-culture process with TILs22. Figure 3A showed that the organoids gradually transitioned from the initial intact spherical structure (at 0 h) to a state of decreased number, reduced volume, and partial disintegration (at 36 h), and finally almost destroyed at 48 h, leaving only small residual structures. In comparison, the organoids cultured alone maintained their intact morphology and quantity within 48 h. High-magnification image of LCP76 organoid co-cultured with TILs demonstrated TIL aggregation (arrows) around the organoid (Fig. 3B). For sample LCP81, we employed Caspase 3/7 fluorescent dyes to scrutinize the apoptotic process (Fig. 3C)23. Compared with the control group, the organoids in the experimental group exhibited significant dynamic changes. After 4 h of co-culture, TILs began to aggregate around the organoids, and this chemotactic phenomenon reached its peak in 7 h. After 24 h, obvious structural disintegration and apoptosis of the organoids occurred. Through quantitative analysis of fluorescence intensity, we further revealed the killing kinetics of TILs. The changes in the mean fluorescence intensity (MFI) from 0 h to 48 h clearly demonstrated the killing effect of TILs (Fig. 3D). At 48 h, the Mean Fluorescence Intensity (MFI) was significantly higher in the treatment group (M = 13.47, SD = 2.65) than in the control group (M = 1.62, SD = 1.42). A Welch’s t test confirmed this difference was statistically significant, t(3.07) = 6.83, p = 0.006, with a large effect size (Cohen’s d = 5.57). The 95% confidence interval for the mean difference was [6.39, 17.30].
To quantify the cytotoxic impact of TILs, we performed flow cytometry on the sample from patient LCP81. As depicted in Fig. 3E, the percentage of viable organoid cells (CFSE⁺DAPI⁻) was quantified under three conditions: organoids cultured alone, co-cultured with PBMCs, and co-cultured with TILs. The representative gating strategy for this analysis is shown in Supplementary Fig. 2. The percentage of viable cells was dramatically lower in the ‘Organoids + TILs’ co-culture group (M = 5.37%, SD = 3.33%) compared to the ‘Organoids alone’ group (M = 43.10%, SD = 8.65%). This difference was confirmed to be statistically significant by a Welch’s two-sample t-test (t(2.56) = 6.96, p = 0.010). The 95% confidence interval for the mean difference was [18.71, 56.75], and the effect size was large (Cohen’s d = 5.69) (Fig. 3F). In contrast, co-culture with PBMCs (n = 2, mean = 49.2%) exerted negligible cytotoxicity and showed results similar to the ‘Organoids alone’ group. These results underline the specific and highly effective anti-tumor capabilities of autologous TILs.
To further investigate the specificity of this cytotoxic effect, we performed parallel co-culture experiments using organoids derived from autologous normal lung tissue, and also assessed the response against tumor organoids derived from other patients. The TIL response was highly specific to tumor cells (Supplementary Fig. 3A). As measured by an LDH release assay, the killing efficiency against tumor organoids was significantly greater than against autologous normal lung organoids, with the difference being statistically significant and representing a very large effect (t(2) = 21.47, p = 0.002; Cohen’s d = 12.40). This functional disparity was corroborated by cytokine secretion profiles. Co-culture with tumor organoids induced significantly higher secretion of key effector molecules compared to co-cultures with normal organoids (Supplementary Fig. 3B). Specifically, this was observed for GZMB (p = 0.044, Cohen’s d = 2.67), IFN-γ (p = 0.003, Cohen’s d = 10.34), and IL-17A (p = 0.031, Cohen’s d = 3.19), confirming a robust, tumor-specific cytokine response.
Furthermore, CFSE-based imaging experiments showed that while TILs from one patient (LCP8) effectively lysed their autologous tumor organoids, they exhibited no significant killing activity against tumor organoids from other patients (LCP5 and LCP7) (Supplementary Fig. 4). Collectively, these data indicate that the cytotoxic activity of the expanded TIL population is highly specific for autologous tumor cells.
Single-cell transcriptomics reveals a coordinated T cell response during TIL-organoid interaction
In order to evaluate the cellular dynamics and underlying molecular mechanisms during the TIL-organoid interaction, we conducted single-cell RNA sequencing on patient samples LCP89 - LCP95(LCP89, LCP90, LCP92, LCP94, LCP95), including tumor organoids (T_ORG), organoids derived from normal lung tissue (N_ORG), TILs, and tumor organoid - TIL co-culture samples. TSNE clustering analysis unveiled 28 distinct cell populations (Fig. 4A). Based on the expression patterns of 20 key genes, all cells were categorized into 5 major types: B cells, epithelial cells, fibroblasts, mast cells, and T cells (Fig. 4B, C). The clustering of samples from different patients, tissue origins, and pathological types is depicted in Fig. 4D–F.

A t-distributed Stochastic Neighbor Embedding (t-SNE) plot of all single cells analyzed, showing 28 distinct cell clusters. B A heatmap displaying the expression of canonical marker genes used to identify major cell lineages across the clusters. C The t-SNE plot from panel (A), with cells colored and annotated by their identified major cell type. D The t-SNE plot showing the integrated distribution of cells from different patient samples following batch effect correction. E The t-SNE plot with cells colored by experimental condition: before co-culture (Before-co), after co-culture (After-co), normal organoids (N_ORG), and tumor organoids (T_ORG). F The t-SNE plot with cells colored by the pathological subtype of the tumor of origin. G Violin plots comparing the expression of representative effector genes in T cells before versus after co-culture. H Bar chart showing the results of KEGG pathway enrichment analysis for genes differentially expressed in T cells after co-culture.
First, we analyzed the overall changes in the total T cell population. Gene expression analysis of samples before versus after co-culture showed that the expression of GZMB (Granzyme B) was significantly upregulated, along with genes such as CCL4, CTSL, CTSD, and TNFRSF1B (Fig. 4G), while there was no significant difference in the expression of GZMH and other genes after co-culture (Supplementary Fig. 5). These genes are involved in various immune-related activities, suggesting that co-culturing TILs with tumor organoids could activate multiple immune-related pathways, thereby enhancing the anti-tumor activity of immune cells24,25. Functional enrichment analysis of the differentially expressed genes(DEGs) demonstrated significant activation of multiple immune-related pathways during the co-culture process (Fig. 4H), including the TNF signaling pathway, the apoptosis pathway, and the antigen processing and presentation pathway26,27. Among them, the antigen processing and presentation pathway was the most prominent, as antigen processing and presentation are the cornerstones for activating the adaptive immune response, thus reflecting the strengthening of the key mechanism by which TILs recognize tumor cells28. The enrichment of chemokine signaling pathways and cytokine-cytokine receptor interactions indicated the strengthened recruitment and activation of TILs29.
To dissect these global changes at a higher resolution, we then analyzed the CD4+ and CD8 + T cell subsets separately. This deeper analysis revealed that the anti-tumor program was a coordinated effort. Consistent with the overall T cell findings, key effector genes such as CTSL, CCL4, and TNFRSF1B were upregulated in both subsets (Supplementary Fig. 6A, C). Pathway analysis also confirmed that core immune pathways, including TNF and IL-17 signaling, were significantly enriched in both CD4+ and CD8+ T cells (Supplementary Fig. 6B, D); this is consistent with previous studies indicating the crucial role of CD8 + T cells in TNF-mediated cytotoxic activity30. Interestingly, we identified a distinct feature in CD8+ T cells: a significant enrichment of metabolic-related pathways, likely reflecting the high energy demand required to fuel their sustained cytotoxic activity31.
Collectively, these transcriptomic data suggest the formation of a coordinated immune killing network—encompassing antigen recognition, direct cytotoxicity, metabolic support, and inflammatory signaling—that provides a molecular basis for the effective killing of tumor cells by TILs.
Analysis of the dynamic changes in T cell subpopulations
Through single-cell clustering analysis of TIL-T cells in the co-culture system, 25 distinct T cell subpopulations were successfully identified (Fig. 5A). Based on the expression of well-known marker genes, these subpopulations were classified as effector memory CD4+ T cells (CD4+Tem), regulatory T cells (Treg), Th1, Th2, CD8+ proliferating T cells (CD8+Tpf_HAVCR2, CD8+Tpf_GZMH), CD4+ proliferating T cells (CD4+Tpf_MKI67, CD4+Tpf_BIRC5), pre-exhausted T cells (CD4+Tpex, CD8+Tpex), exhausted CD8+ T cells (CD8+Tex), and γδT cells (Tgd) (Fig. 5B, C).

A A t-SNE plot of all T cells, revealing 25 distinct T-cell sub-clusters. B The t-SNE plot from panel (A), with clusters colored and annotated into functional T-cell subpopulations. C A dot plot showing the expression level and percentage of cells expressing key marker genes across the identified T-cell subpopulations. D Box plots comparing the proportional abundance of four representative T-cell subpopulations before versus after co-culture, using data from five biologically independent patient samples (n = 5). Statistical significance was determined by a one-tailed paired t-test. Subpopulation abbreviations: CD4⁺Tem, CD4⁺ effector memory T cells; Th2, T helper 2 cells; CD4⁺Tpf_MKI67, proliferating CD4⁺ T cells expressing MKI67; CD8⁺Tpf_HAVCR2, proliferating CD8⁺ T cells expressing HAVCR2.
Further analysis of T cell subpopulations revealed dynamic changes during the co-culture process (Fig. 5D). The proportion of CD4+Tem (p = 0.047, Cohen’s d = 0.98) and Th2 (p = 0.005, Cohen’s d = 2.06) cells increased significantly. On the other hand, the proportions of the CD4+Tpf_MKI67 (p = 0.026, Cohen’s d = −1.22) and CD8+Tpf_HAVCR2 (p = 0.025, Cohen’s d = −1.25) subpopulations were significantly decreased. The decrease of CD8+Tpf_HAVCR2 cells might reflect their transition to an exhausted state (Tex) or their undergoing activation-induced cell death32,33. Meanwhile, the decrease of CD4+Tpf_MKI67 cells might imply that the initial T cell expansion stage had transitioned to a more specialized effector or memory stage34. On the other hand, the proportion of CD4+Tem increased from 30 to 40% before co-culture to 40–50% after co-culture. Conversely, the proportions of CD4+Tpf_MKI67 and CD8+Tpf_HAVCR2 decreased significantly, dropping from 10−15% to 5–10% and from 5–7.5% to 2.5–5%, respectively. MKI67 and HAVCR are associated with the activation and proliferation of T cells and are often regarded as markers of tumor-reactive T cells35,36. It has been found that proliferating T cells contain cytotoxic subpopulations, for example, there are cytotoxic subpopulations expressing GZMB or GZMK in Ki67 + CD4 + T cells37. The dynamic change patterns observed in our study reveal the changes in T cell subpopulations during the TIL-organoid interaction, and the co-culture conditions promote the transition of T cells from a proliferating state to a more mature effector and memory state. Consistent with this transition towards a more differentiated state, we observed that TILs upregulated several key exhaustion-associated genes after co-culture with tumor organoids. Specifically, in CD8⁺ T cells, we noted a significant upregulation of the co-inhibitory receptors PDCD1 (PD-1), LAG3, HAVCR2 (TIM-3), and TIGIT, as well as the key exhaustion-associated transcription factor TOX. A similar trend was observed in CD4⁺ T cells, which also showed significantly increased expression of CTLA4, PDCD1, LAG3, HAVCR2, etc. (Supplementary Fig. 7). These molecular findings suggest that upon activation, the TILs progress towards a state that is accompanied by features of T cell exhaustion, an intrinsic regulatory process common in tumor immunity. Cell interaction analysis based on CellChat suggested that both CD4+Tem and CD4+Tpf_MKI67 had strong communication with CD8+ T cells (Supplementary Fig. 8)38.
Discussion
In this study, we established and utilized a patient-specific NSCLC organoid-TIL co-culture platform to provide a multi-faceted characterization of the anti-tumor immune response, with our overall workflow and key findings summarized in Fig. 6. Our findings demonstrate that in vitro expanded TILs mediate potent and specific killing of autologous tumor organoids, a function that is associated with a dynamic shift in T cell subpopulations from a proliferative to an effector memory phenotype. At the molecular level, this cytotoxic response was linked to the activation of key signaling pathways, including the TNF pathways and an enrichment of tumor-associated TCR clonotypes.

This schematic summarizes the study’s workflow. Autologous TILs and tumor organoids are derived from NSCLC patients for co-culture. The resulting model is evaluated by in-vitro functional analyses (left; IHC, killing validation, flow cytometry) and scRNA-seq analyses (right; cell clustering, DEGs, pathway enrichment). These integrated analyses identify key genes, T cell population dynamics, and signaling pathways (center and bottom) that characterize the TIL-mediated anti-tumor response.
A key finding of our work is that the TIL expansion protocol acts as a selective process, leading to a focused T-cell receptor (TCR) repertoire enriched with tumor-associated clones. Indeed, the foundational principle of TIL therapy is that ex vivo expansion selects for and massively amplifies the rare, pre-existing tumor-reactive T cells within a patient’s tumor39,40. Our data, showing a contraction in TCR diversity alongside an increased dominance of shared clones and a higher “Lung Cancer TCR Score”, are consistent with this principle. This focusing of the TCR repertoire in the final cell population is a key feature of the TIL manufacturing process, and a less diverse, more clonal repertoire is often associated with positive clinical outcomes in patients41,42. In renal cell carcinoma, clonal T-cell expansion is similarly considered a signature of response to tumor antigens43. The clinical significance of this principle was recently established by the landmark M14TIL trial, which demonstrated the superiority of TIL therapy in ICI-resistant melanoma44, leading to the 2024 FDA approval of the first TIL therapy, lifileucel45. Promisingly, the application of adoptive TIL therapy in NSCLC is also showing promise. A clinical trial by Creelan et al., for instance, confirmed its safety and demonstrated objective remissions in ~23% of patients with metastatic disease46. our study provides multi-layered evidence that the clonally focused populations we generated are indeed tumor-specific: our functional assays demonstrate their potent and specific killing of autologous tumor organoids, while our single-cell analysis reveals they possess the molecular signature of tumor-reactive T cells. This is particularly relevant for the future of TIL therapy. For patients who may lack a sufficient number or quality of pre-existing tumor-reactive clones to benefit from non-selective approaches, strategies to prospectively identify and “actively focus” the expansion on the most potent, neoantigen-specific T cells will be critical47,48. Our platform provides a potential framework for developing such advanced TIL therapies.
The execution of this specific killing function of TILs against lung tumor organoids derived from the same patient is driven by an intricate molecular program. Through in-depth analysis of gene expression changes in TILs before and after co-culture with NSCLC organoids and signaling pathway enrichment analysis on DEGs, we revealed upregulated expression of multiple immune regulatory genes and activation of TIL killing pathways. These efforts have revealed the enhanced expression of multiple immune regulation genes and the activation of TIL killing pathways. The GZMB gene, which encodes Granzyme B, a key effector molecule of cytotoxic lymphocytes (CTLs), induced apoptosis of target cells through proteolysis-mediated mechanisms to combat pathogens49. The upregulation of GZMB after TIL-organoid contact reflected the activation of CTLs, and such activated CTLs in turn released Granzyme B into the immune synapse, triggering apoptosis of target cells50. Other immune-related genes such as CTSL, BIRC3, and CCL4 were also upregulated in our study. The upregulation of CTSL enhances the ability of antigen processing and presentation; the upregulation of BIRC3 improves the survival ability of T cells, and the upregulation of CCL4 enhances the secretion of chemokines51,52,53. Together, the modulation of these gene expression patterns indicates that TILs are effectively activated after exposure to lung cancer organoids, not only enhancing their own effector functions and survival abilities but also potentially expanding the anti-tumor immune response by recruiting additional immune cells. In addition, pathway enrichment analysis showed that DEGs were mainly associated with antigen processing and presentation and IL-17, TNF signaling pathways. TNF, as a multifunctional cytokine, can activate signaling pathways such as NFκB and MAPK, demonstrating the ability to directly kill tumor cells and regulate inflammatory responses26,54,55. This corroborates the significant killing effect of TILs on lung cancer organoids. A notable finding is the concurrent upregulation of the IL-17 signaling pathway. In our experimental system, we observed the activation of the IL-17 pathway concurrently with a definitive anti-tumor response. Therefore, while acknowledging the context-dependent nature of IL-17 signaling, we hypothesize that in our specific in vitro model, its activation is a component of a productive and multi-faceted anti-tumor program56,57. However, direct functional validation is required to confirm this hypothesis.
Using single-cell sequencing technology, we comprehensively characterized the dynamic changes in T cell subpopulations during co-culture. The results demonstrated a significant increase in CD4+ effector memory T cells (Tem) and a decrease in proliferative T cells (CD4+Tpf_MKI67 and CD8+Tpf_HAVCR2). This suggests that the initial T cell expansion stage had transitioned to a more specialized effector or memory stage. The enrichment of Tem is particularly notable, as memory T cells can rapidly produce inflammatory factors and participate in anti-tumor immune responses58, and both CD4+ and CD8+ Tem are reported to be significantly enriched in metastatic lymph nodes of lung cancer patients59. Within this remodeled T cell population, our analysis revealed distinct and complementary roles for the major subsets. Consistent with their classical function as cytotoxic effectors, CD8⁺ T cells upregulated metabolic-related signaling pathways, which may provide the necessary energy for their sustained anti-tumor function31. Concurrently, differentially expressed genes (DEGs) in CD4⁺ T cells were enriched in pathways related to cytokine receptors, cell adhesion, and chemokine signaling, suggesting a role in intercellular communication and cell migration60. However, recent findings challenge a purely supportive role for CD4⁺ T cells in this context. A key study on TIL therapy for melanoma found that neoantigen-specific T cell clones in the infusion product were often dominated by CD4⁺ T cells. Crucially, these CD4⁺ T cells were not merely supportive; they were capable of directly killing tumor cells in an MHC-II-dependent manner and secreting high levels of effector cytokines such as IFN-γ and TNF-α61. Therefore, the strong enrichment of cytokine and chemokine signaling pathways in the CD4⁺ T cells in our study may not only reflect a supportive function but could also be indicative of a potent, direct anti-tumor effector program. Future investigations are warranted to explore the mechanisms underlying the alternation of T cell subpopulations and their roles in tumor progression and regression.
We acknowledge several limitations of our study. The findings are based on a modest sample size, and our in vitro system does not recapitulate the full complexity of the in vivo tumor microenvironment. Furthermore, the functional roles of specific pathways suggested by our data require direct validation through future blocking experiments, and the high effector-to-target ratios used reflect a therapeutic rather than a physiological setting.
Last but not the least, it is our hypothesis that successful anti-cancer treatment strategy will have to appropriately stimulate both humoral and cellular immunity. We identified anti-tumor effector B cells, as evidenced by multiple publications from our group62. We reported that in vivo sensitized and in vitro activated B cells mediated tumor regression in cancer immunotherapy63; effector B cells could kill tumor cells directly without CDC and ADCC in an antigen-specific way64. Further investigation of additional TIL immune cells, such as B cells, NK cells, macrophages as well as T cells and their interactions with tumor-derived organoids may help develop more effective TIL therapies in cancer treatment.
In conclusion, our work establishes a patient-specific organoid-TIL co-culture platform capable of dissecting anti-tumor immune responses at high resolution. By integrating analyses of TCR repertoire dynamics, single-cell transcriptomics, and direct cytotoxicity, we have delineated the clonal and molecular signatures that characterize a productive TIL-mediated response in NSCLC. This multi-layered approach provides a valuable framework for the preclinical assessment of TILs and offers mechanistic insights that may guide the future development of more effective, personalized adoptive cell therapies.
Methods
Samples
The patient lung cancer samples were obtained from the Department of Thoracic Surgery at Peking University Shenzhen Hospital. Written informed consent was obtained from all participants prior to sample collection. These include tumor tissues and adjacent normal tissues obtained in surgery, along with patient blood samples. The study protocol for this study and all procedures involving human participants were approved by the Ethics Committee of Peking University Shenzhen Hospital (NO.2020-057). All ethical regulations relevant to human research participants were followed. Organoids were successfully constructed from 12 samples. The main reagents used in this study included DPBS buffer (Sigma, USA), Fetal Bovine Serum (FBS) (Thermo Fisher, USA), Penicillin-Streptomycin (P/S) (Gibco, USA), TrypLE™ Express Enzyme (Gibco, USA), Matrigel (Corning, USA), Advanced DMEM/F-12 (Gibco, USA), HEPES (Thermo Fisher, USA), RPMI 1640 (Thermo Fisher, USA), IL-2 (ELITE-MEDIA, China) and GlutaMAX™ Supplement (Gibco, USA).
Tissue processing and digestion
To obtain high-quality tumor cell suspensions, tissue blocks were first evaluated and non-target tissues were removed. Subsequently, the tissues were repeatedly washed with DPBS containing 1% penicillin/streptomycin (P/S). Prior to digestion, all tissue specimens were carefully inspected to manually remove any necrotic, hardened, or non-tumor components (e.g., adipose or muscle tissue). This step was critical for enriching the proportion of tumor cells in the initial culture. The digestion solution was prepared according to the manufacturer’s instructions for the Miltenyi tumor tissue dissociation kit. The tissues were minced (1–2 mm³) and added to the solution, then subjected to shaking digestion at 37 °C and 1000 rpm. Following the addition of FBS to terminate the reaction, the mixture was filtered through a 70 μm strainer and thoroughly rinsed with DPBS. When necessary, red blood cell lysis buffer was applied for 5 min. Finally, viable cells were counted using acridine orange/propidium iodide (AO/PI) dye, and cell pellets were collected by centrifugation at 300–400 × g for 5–10 min.
In vitro expansion and maintenance of TILs
The in vitro expansion of TILs utilized a two-phase strategy, intended to support both the initial activation and the long-term maintenance of the T cells. The initial activation and expansion phase began with fresh tumor fragments (~1 mm³) cultured in 24-well plates. These were maintained in a medium containing a high concentration of IL-2 (6000 IU/mL; ELITE-MEDIA, China), supplemented with 10% FBS and 1% Penicillin-Streptomycin in an RPMI-1640 base. The goal of this phase, which typically lasted 1-3 weeks, was to use high-dose IL-2 to promote the activation and rapid proliferation of TILs migrating from the tissue. Once a sufficient number of TILs were observed around the fragments, the tissue remnants were removed for the continued culture of the purified TIL population.
After approximately four weeks, the protocol transitioned to a maintenance and functional stabilization phase. To better approximate a physiological cytokine milieu and to mitigate potential T cell exhaustion from prolonged high-dose IL-2 stimulation, the medium was switched to a formulation containing a lower concentration of IL-2 (100 IU/mL) supplemented with the homeostatic cytokines IL-7 (100 IU/mL) and IL-15 (100 IU/mL). These cytokines are known to be important for the long-term survival and maintenance of memory T cell phenotypes. Once the TIL population expanded to ~10⁷ cells, they were cryopreserved for subsequent functional assays.
Construction and passaging of organoids
For organoid construction, cell pellets were suspended in Matrigel (Corning) at a concentration of at least 70% to provide sufficient matrix rigidity for three-dimensional growth. To prevent premature solidification of the temperature-sensitive Matrigel, all pipette tips were pre-chilled. For plating in a 6-well plate, ~200 µL of the cell-Matrigel suspension was dispensed as 7–10 small, individual droplets per well to facilitate nutrient and oxygen exchange. After incubating at 37 °C for 30 min to solidify the droplets, 2 mL of pre-warmed organoid culture medium was added. The specialized culture medium was formulated based on published literature and contained key factors such as R-spondin-1, Noggin, FGF7, and FGF10 to provide a supportive growth environment. Organoid growth was observed and recorded daily, with medium changes every 2–3 days. When organoids filled the gel drops or exceeded 300 μm in diameter, TrypLE Express dissociation reagent was used to disperse the Matrigel and cells by incubating at 37 °C for 30–60 min until complete dissociation. FBS was added to stop the digestion, cells were collected by centrifugation and resuspended in new Matrigel for passaging at a 1:2 to 1:6 ratio. Optionally, single clones were selected and cultured separately in 48- or 96-well plates.
Identification of organoids
Lung cancer organoid identification comprised three main steps: H&E staining, immunohistochemistry (IHC) staining, and sequencing-based identification. Initially, organoids were digested using Matrigel-specific recovery solution at 4 °C for 30 min, followed by centrifugation at 200 × g for 5 min. The organoids were then mixed with 0.5% agarose solution, solidified, and embedded in paraffin before being sectioned into 5–10 μm thick slices. The H&E staining protocol included xylene deparaffinization (5–10 min), gradient ethanol hydration, hematoxylin staining (3–5 min), differentiation with 1% hydrochloric acid-ethanol (3–5 s), eosin staining (3–5 s), dehydration, and mounting. For IHC staining, following deparaffinization and hydration, antigen retrieval was performed, followed by blocking with 3% hydrogen peroxide for 10 min and goat serum for 10 min. Primary antibodies were incubated overnight at 4 °C, followed by secondary antibody incubation for 10 min at room temperature. The following primary antibodies were used: Napsin A (PTGCN, Cat# 16558-1-AP), Cytokeratin 5/6 (PTGCN, Cat# 68395-1-Ig), p63 (Abcam, Cat# ab124762), and TTF-1 (Abcam, Cat# ab76013). DAB was used for color development (3–10 min), and hematoxylin for counterstaining (1–2 min). IHC-specific markers included CK5/6 and p63 for squamous cell carcinoma, and TTF-1 and NAPSIN for adenocarcinoma. This multi-step validation approach (combining H&E, IHC, and sequencing) was employed to rigorously confirm the tumor origin of the established organoid lines, a critical step given the potential for overgrowth by normal cells from the primary tissue.
Organoid-TIL co-culture and data analysis
The co-culture experiment of lung cancer organoids and TILs consisted of three main steps: organoid collection, co-culture plate preparation, and co-culture. Two methods were used for organoid collection: enzymatic digestion using TrypLE™ Express at 37 °C for 30–60 min, or a special digestion method using Corning Cell Recovery Solution at 4 °C for 30–60 min. For co-culture plate preparation, 50–70% concentration Matrigel was used to coat the well bottoms and solidified at 37 °C. During co-culture, organoids were first labeled with CFSE or Caspase 3/7 dye, then mixed with TILs at effector-to-target ratios of 5:1 or 10:1 and cultured in 500 μL of specific medium65,66. Cytotoxic effects were observed and recorded under a fluorescence microscope every 2–3 h.
Following the co-culture period, the results were analyzed using several complementary methods. For flow cytometry, cells were digested with TrypLE™ Express, stained with DAPI for viability, and analyzed using a flow cytometer, with data analysis performed using FlowJo software67. To quantify cytotoxicity biochemically, lactate dehydrogenase (LDH) release was measured from the culture supernatant using the LDH-Glo™ Cytotoxicity Assay kit (Promega, Cat. No. G1780) according to the manufacturer’s protocol. The percentage of specific cytotoxicity was calculated with the following formula, which accounts for background from both target and effector cells: % Cytotoxicity = 100 × [(Experimental Release − Effector Spontaneous Release − Target Spontaneous Release)/(Target Maximum Release − Target Spontaneous Release)]. In addition, the concentrations of secreted cytokines in the supernatant were quantified by ELISA. The levels of Granzyme B (GZMB; MultiSciences, Cat. No. EK1217HS), Interferon-gamma (IFN-γ; MultiSciences, Cat. No. EK180HS), and Interleukin-17A (IL-17A; MultiSciences, Cat. No. EK158) were measured using their respective commercial kits following the manufacturers’ instructions. For transcriptomic analysis, single-cell sequencing data were processed using the Seurat (v4.0.6) for quality control, normalization, dimensionality reduction, clustering, and differential expression analysis.
TCR sequencing and clonality analysis
T-cell receptor (TCR) sequencing was performed on genomic DNA isolated from TIL samples at Day 0 and Day 14 of the expansion culture. High-throughput sequencing of the TCR loci was conducted, and the resulting data were processed to identify and quantify the frequency of unique TCR clonotypes.
Several metrics were used to characterize the TCR repertoire. Repertoire diversity was assessed by the total number of unique TCR clonotypes. The Shannon index, a classic diversity metric, was also calculated to provide a composite measure of clonal richness and evenness. To evaluate functional relevance, a “Lung Cancer TCR Score” was calculated; this score refers to the enrichment of clonotypes by aligning the repertoire against a reference dataset of lung cancer tissue-enriched TCR sequences(https://www.lungtcr.com). Finally, “shared clones” were defined as clonotypes present at both Day 0 and Day 14, and their cumulative frequency was calculated to assess changes in clonal dominance during the expansion process.
Single-cell RNA-seq with DNBelab C4 system
The DNBelab C4 Single-Cell Library Prep Set from MGI in Shenzhen was utilized for the preparation of single-cell RNA sequencing libraries as previously described68. Initially, single-cell suspensions were encapsulated in droplets with barcoded beads, followed by cell lysis and mRNA capture. Subsequent steps included emulsion breaking, bead recovery, and reverse transcription. The libraries were quantified using the Qubit ssDNA Assay Kit (Thermo FisherScientific, USA) and sequenced on the DNBSEQ-T7 platform at the China National GeneBank (CNGB).
Single-cell data analysis
Single-cell RNA sequencing data analysis was performed using the Seurat (v4.0.6) package in R environment. After quality control and filtering, data were normalized using the “LogNormalize” method. Highly variable feature genes (HVGs) were identified using the “FindVariableFeatures” function. Principal component analysis (PCA) was performed on the matrix of HVGs to reduce noise and reveal the main axes of variation. To eliminate batch effects, the “RunHarmony” function was used for data integration. The first 20 principal components were selected to construct the SNN network, and the Louvain algorithm was used for cell clustering. Finally, tSNE was used for dimensionality reduction visualization, with parameters set to “dims = 1:20”.
Cell type annotation was based on known marker genes: FOXP3, KLRD1, KLRF1, CD4, CD3D, IL2RA, CTLA4, CD8A (T cells); CD79A, JCHAIN, MZBA, MS4A1 (B cells); EPCAM, KRT18, KRT19 (epithelial cells); COL1A1, COL1A2, DCN (fibroblasts); GATA2, MS4A2 (mast cells). For T cell subgroup identification, the following marker genes were used: CD8A, CD8B (CD8+ T cells); CD4 (CD4+ T cells); FOXP3, IL2RA (regulatory T cells); HAVCR2, LAG3 (exhausted T cells); MKI67, BIRC5, TOP2A (proliferating T cells); TRDC, TRGC1 (Tgd cells); GPR183, S100A4 (memory T cells); KLRD1, KLRF1 (Cytotoxic cells).
Functional enrichment analysis of DEGs
DEGs before and after co-culture were selected and analyzed using KOBAS software (http://bioinfo.org/kobas/genelist) based on KEGG database. Gene symbols were input to assess gene functions and signaling pathway involvement.
Statistics and reproducibility
In this study, ‘technical replicates’ represent multiple measurements of the same sample within a single experiment (e.g., replicate wells), whereas ‘biological replicates’ represent samples derived from different patients. All key experiments were performed with at least three technical or biological replicates, as specified in the relevant figure legends. Sample sizes were chosen based on experimental feasibility and standards established in previous, similar studies.
All statistical analyses were performed using R software (version 4.1.0) and GraphPad Prism (version 10.12). For all statistical tests, significance was denoted as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ‘ns’ indicates a non-significant difference. An alpha level of 0.05 was used for all statistical tests.
To compare the percentage of CFSE⁺DAPI⁻ cells between the ‘organoids alone’ and ‘organoids + TILs’ experimental conditions, a two-tailed unpaired Welch’s t test was used. For the comparison of fluorescence intensity between the treatment and control groups at 48 h, statistical significance was determined using a Welch’s two-sample t test, which does not assume equal variances between groups. For the LDH cytotoxicity and ELISA data, a two-tailed paired Student’s t test was used to assess the difference between autologous tumor and normal organoid groups. For T cell subpopulation analysis before versus after co-culture in five patient samples, one-tailed paired Student’s t tests were performed.
For single-cell RNA sequencing analysis, quality control parameters were set to “nFeature_RNA > 500 & nFeature_RNA < 8000 & percent.mt < 10”. Differential expressed genes (DEGs) were identified using the “FindAllMarkers” function with parameters set to “min.pct = 0.25, logfc.threshold = 0.25”. The selection criteria for DEGs were FDR < 0.01 and |log2|> 0.585. For T cell subgroup analysis, the “SubsetData” function was used followed by re-dimensionality reduction and clustering. Functional enrichment analysis was performed using KOBAS software, with FDR < 0.05 defined as significantly enriched. The Benjamini–Hochberg method was used for multiple testing correction.
Data availability
The sequencing data generated in this study have been deposited in the EBI BioStudies database under accession number E-MTAB-15784 and the China National Center for Bioinformation (CNCB) database under accession numbers OMIX008571 and HRA013880. The source data underlying the main figures are provided as Supplementary Data 1 with this paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Li, Y., Yan, B. & He, S. Advances and challenges in the treatment of lung cancer. Biomed. Pharmacother. 169, 115891 (2023).
Mlika, M. et al. Prognostic impact of tumor-infiltrating lymphocytes in non-small cell lung carcinomas. Asian Cardiovasc Thorac. Ann. 30, 177–184 (2022).
Lin, B. et al. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed. Pharmacother. 132, 110873 (2020).
Rakaee, M. et al. Association of Machine Learning-Based Assessment of Tumor-Infiltrating Lymphocytes on Standard Histologic Images With Outcomes of Immunotherapy in Patients With NSCLC. JAMA Oncol. 9, 51–60 (2023).
Park, S. et al. Artificial Intelligence-Powered Spatial Analysis of Tumor-Infiltrating Lymphocytes as Complementary Biomarker for Immune Checkpoint Inhibition in Non-Small-Cell Lung Cancer. J. Clin. Oncol. 40, 1916–1928 (2022).
Paijens, S. T., Vledder, A., de Bruyn, M. & Nijman, H. W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 18, 842–859 (2021).
Suwalska, A., Zientek, L., Polanska, J. & Marczyk, M. Quantifying Spatial Heterogeneity of Tumor-Infiltrating Lymphocytes to Predict Survival of Individual Cancer Patients. J Pers Med 12 (2022). https://doi.org/10.3390/jpm12071113
Xu, H., Jiao, D., Liu, A. & Wu, K. Tumor organoids: applications in cancer modeling and potentials in precision medicine. J. Hematol. Oncol. 15, 58 (2022).
Zhang, Q. & Zhang, M. Recent advances in lung cancer organoid (tumoroid) research (Review). Exp. Ther. Med. 28, 383 (2024).
Li, Y., Gao, X., Ni, C., Zhao, B. & Cheng, X. The application of patient-derived organoid in the research of lung cancer. Cell Oncol. 46, 503–519 (2023).
Tang, X. Y. et al. Human organoids in basic research and clinical applications. Sig. Transduct. Target Ther. 7, 168 (2022).
Kim, M. et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 10, 3991 (2019).
Dijkstra, K. K. et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 174, 1586–1598 e1512 (2018).
Podaza, E. E. A. Novel co-culture strategies of tumor organoids with autologous T-cells reveal clinically relevant combinations of immune-checkpoint and targeted therapies. bioRxiv https://doi.org/10.1101/2023.07.05.546622 (2023).
Gonzalez-Silva, L., Quevedo, L. & Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 6, 13–19 (2020).
Gawad, C., Koh, W. & Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet 17, 175–188 (2016).
Passaniti, A., Kleinman, H. K. & Martin, G. R. Matrigel: history/background, uses, and future applications. J. Cell Commun. Sign. 16, 621–626 (2022).
Fatima, N., Cohen, C., Lawson, D. & Siddiqui, M. T. TTF-1 and Napsin A double stain: a useful marker for diagnosing lung adenocarcinoma on fine-needle aspiration cell blocks. Cancer Cytopathol. 119, 127–133 (2011).
Kaufmann, O., Fietze, E., Mengs, J. & Dietel, M. Value of p63 and cytokeratin 5/6 as immunohistochemical markers for the differential diagnosis of poorly differentiated and undifferentiated carcinomas. Am. J. Clin. Pathol. 116, 823–830 (2001).
Jia, Q., Chu, H., Jin, Z., Long, H. & Zhu, B. High-throughput single-small es, Cyrillicell sequencing in cancer research. Signal Transduct. Target Ther. 7, 145 (2022).
Quah, B. J. & Parish, C. R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. https://doi.org/10.3791/2259 (2010).
Dyrkheeva, N. S. et al. 3’-5’ exonuclease activity of human apurinic/apyrimidinic endonuclease 1 towards DNAs containing dNMP and their modified analogs at the 3 end of single strand DNA break. Biochem. Mosc. 71, 200–210 (2006).
Iwama, H. et al. Cathepsin B and D deficiency in the mouse pancreas induces impaired autophagy and chronic pancreatitis. Sci. Rep. 11, 6596 (2021).
Muller, D. Targeting Co-Stimulatory Receptors of the TNF Superfamily for Cancer Immunotherapy. BioDrugs 37, 21–33 (2023).
van Loo, G. & Bertrand, M. J. M. Death by TNF: a road to inflammation. Nat. Rev. Immunol. 23, 289–303 (2023).
Yuan, J. & Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 25, 379–395 (2024).
Lee, M. Y., Jeon, J. W., Sievers, C. & Allen, C. T. Antigen processing and presentation in cancer immunotherapy. J Immunother Cancer 8 (2020). https://doi.org/10.1136/jitc-2020-001111
Ozga, A. J., Chow, M. T. & Luster, A. D. Chemokines and the immune response to cancer. Immunity 54, 859–874 (2021).
Buzzatti, G., Dellepiane, C. & Del Mastro, L. New emerging targets in cancer immunotherapy: the role of GITR. ESMO Open 4, e000738 (2020).
Ma, S., Ming, Y., Wu, J. & Cui, G. Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol. Immunol. 21, 419–435 (2024).
Dixon, K. O., Lahore, G. F. & Kuchroo, V. K. Beyond T cell exhaustion: TIM-3 regulation of myeloid cells. Sci. Immunol. 9, eadf2223 (2024).
Sun, L., Su, Y., Jiao, A., Wang, X. & Zhang, B. T cells in health and disease. Sig. Transduct. Target Ther. 8, 235 (2023).
Adu-Berchie, K., Obuseh, F. O. & Mooney, D. J. T Cell Development and Function. Rejuvenation Res. 26, 126–138 (2023).
Sauer, N. et al. TIM-3 as a promising target for cancer immunotherapy in a wide range of tumors. Cancer Immunol. Immunother. 72, 3405–3425 (2023).
Wu, S. Y. et al. Correlation of MKI67 with prognosis, immune infiltration, and T cell exhaustion in hepatocellular carcinoma. BMC Gastroenterol. 21, 416 (2021).
Oh, D. Y. et al. Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 181, 1612–1625 e1613 (2020).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Rosenberg, S. A., Restifo, N. P., Yang, J. C., Morgan, R. A. & Dudley, M. E. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 8, 299–308 (2008).
Betof Warner, A., Corrie, P. G. & Hamid, O. Tumor-Infiltrating Lymphocyte Therapy in Melanoma: Facts to the Future. Clin. Cancer Res. 29, 1835–1854 (2023).
Cardinale, A., De Luca, C. D., Locatelli, F. & Velardi, E. Thymic Function and T-Cell Receptor Repertoire Diversity: Implications for Patient Response to Checkpoint Blockade Immunotherapy. Front. Immunol. 12, 752042 (2021).
Hopkins, A. C. et al. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 3, e122092 (2018).
Xu, Y. et al. Integrated TCR repertoire analysis and single-cell transcriptomic profiling of tumor-infiltrating T cells in renal cell carcinoma identifies shared and tumor-restricted expanded clones with unique phenotypes. Front. Oncol. 12, 952252 (2022).
Rohaan, M. W. et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 387, 2113–2125 (2022).
Kwong, M. L. M. & Yang, J. C. Lifileucel: FDA-approved T-cell therapy for melanoma. Oncologist 29, 648–650 (2024).
Creelan, B. C. et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat. Med. 27, 1410–1418 (2021).
Tsimberidou, A. M. et al. T-cell receptor-based therapy: an innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 14, 102 (2021).
van den Berg, J. H. et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother Cancer 8, https://doi.org/10.1136/jitc-2020-000848 (2020).
Aubert, A., Lane, M., Jung, K. & Granville, D. J. Granzyme B as a therapeutic target: an update in 2022. Expert Opin. Ther. Targets 26, 979–993 (2022).
Arias, M. et al. The Untold Story of Granzymes in Oncoimmunology: Novel Opportunities with Old Acquaintances. Trends Cancer 3, 407–422 (2017).
Dana, D. & Pathak, S. K. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 25, https://doi.org/10.3390/molecules25030698 (2020).
Mukaida, N., Sasaki, S. I. & Baba, T. CCL4 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 1231, 23–32 (2020).
Suo, F., Wu, Y., Zhou, Q., Li, L. & Wei, X. BIRC3-HSP90B1 Interaction Inhibits Non-Small Cell Lung Cancer Progression through the Extracellular Signal-Regulated Kinase Pathway. ACS Omega 9, 19148–19157 (2024).
Qian, Y. et al. TRIM47 is a novel endothelial activation factor that aggravates lipopolysaccharide-induced acute lung injury in mice via K63-linked ubiquitination of TRAF2. Sig. Transduct. Target Ther. 7, 148 (2022).
Yu, H., Lin, L., Zhang, Z., Zhang, H. & Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: mechanism and clinical study. Sig. Transduct. Target Ther. 5, 209 (2020).
Amatya, N., Garg, A. V. & Gaffen, S. L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 38, 310–322 (2017).
Swaidani, S., Liu, C., Zhao, J., Bulek, K. & Li, X. TRAF Regulation of IL-17 Cytokine Signaling. Front. Immunol. 10, 1293 (2019).
Daniels, M. A., Luera, D. & Teixeiro, E. NFkappaB signaling in T cell memory. Front. Immunol. 14, 1129191 (2023).
Kwiecien, I. et al. Effector Memory T Cells and CD45RO+ Regulatory T Cells in Metastatic vs. Non-Metastatic Lymph Nodes in Lung Cancer Patients. Front. Immunol. 13, 864497 (2022).
Kunzli, M. & Masopust, D. CD4(+) T cell memory. Nat. Immunol. 24, 903–914 (2023).
Hall, M. S. et al. Neoantigen-specific CD4(+) tumor-infiltrating lymphocytes are potent effectors identified within adoptive cell therapy products for metastatic melanoma patients. J. Immunother. Cancer 11, https://doi.org/10.1136/jitc-2023-007288 (2023).
Qin, Y., Lu, F., Lyu, K., Chang, A. E. & Li, Q. Emerging concepts regarding pro- and anti tumor properties of B cells in tumor immunity. Front. Immunol. 13, 881427 (2022).
Li, Q., Teitz-Tennenbaum, S., Donald, E. J., Li, M. & Chang, A. E. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J. Immunol. 183, 3195–3203 (2009).
Li, Q. et al. Adoptive transfer of tumor reactive B cells confers host T-cell immunity and tumor regression. Clin. Cancer Res. 17, 4987–4995 (2011).
Cattaneo, C. M. et al. Tumor organoid-T-cell coculture systems. Nat. Protoc. 15, 15–39 (2020).
Lyons, A. B. & Parish, C. R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods 171, 131–137 (1994).
Darzynkiewicz, Z. et al. Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis). Cytometry 27, 1–20 (1997).
Chen, C. et al. Single-cell and spatial transcriptomics reveal POSTN(+) cancer-associated fibroblasts correlated with immune suppression and tumour progression in non-small cell lung cancer. Clin. Transl. Med. 13, e1515 (2023).
Acknowledgements
We thank Guo Wei and Wenkui Dai for technical assistance. This research was supported by the Shenzhen Medical Research Fund under Grant No. A2403061, Shenzhen Science and Technology Program under Grant No. JCYJ20240813120107010, the Sanming Project of Medicine in Shenzhen under Grant No. SZSM202211035, the Shenzhen Key Medical Discipline under Grant No. SZXK078, and the National Key R&D Program of China under Grant No. 2022YFA1206000.
Author information
Authors and Affiliations
Contributions
Ziwen Qin: Investigation, Methodology, Validation, Visualization, Data curation, Writing-original draft. Hongchang Zhang: Investigation, Methodology, Validation, Visualization, Data curation, Writing-original draft. Yanling Li: Investigation. Jianyi Yang: Investigation. Haozhen Liu: Investigation. Zhuojue Guan: Investigation. Qinghua Hou: Investigation. Haocheng Du: Investigation. Xiaoqiang Li: Investigation. Xian Lin: Writing-review & editing. Qumiao Xu: Investigation, Resources. Qiao Li: Writing-review & editing. Junhui Chen: Conceptualization, Funding acquisition, Supervision. Jixian Liu: Conceptualization, Funding acquisition, Resources, Supervision. Chao Chen: Conceptualization, Funding acquisition, Formal analysis, Supervision, Writing-review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Pavithra Chavali and Johannes Stortz.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qin, Z., Zhang, H., Li, Y. et al. Transcriptome profiling of tumor-infiltrating lymphocyte-mediated cytotoxicity against patient-derived lung cancer organoids. Commun Biol 8, 1835 (2025). https://doi.org/10.1038/s42003-025-09188-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09188-0
