
Abstract
Disruption of blood-brain barrier (BBB) is the critical step of Streptococcus suis (S. suis) meningitis. Here we show that Ephrin type-A receptor 2 (EphA2) plays an important role in BBB against S. suis meningitis. S. suis downregulates EphA2 and EphA2 deficiency aggravates S. suis-induced BBB disruption in vitro and in vivo. We identify that EphA2-mediated autophagy alleviates S. suis-induced BBB disruption. To destroy the protective effect of EphA2 in BBB, S. suis recruits E3 ligase DCAF15 to interact with EphA2, which induces EphA2 ubiquitination and degradation. Mechanically, DCAF15 promotes K48- and K63-linked ubiquitination of EphA2 at K646, K649 and K754. In addition, S. suis serine/threonine protein kinase (STK) is indispensable for EphA2 degradation. STK phosphorylates histone deacetylase SIRT1 at S48 to trigger the deacetylation of DCAF15, enhancing DCAF15-EphA2 interaction and promoting ubiquitinated degradation of EphA2, thereby disrupting BBB. Moreover, acetylation of DCAF15 at K552 and K581 sites is involved in mediating EphA2 ubiquitination and BBB stability. Our study suggests a protective role of EphA2 in S. suis-induced BBB disruption, which provides a valuable insight on the development of EphA2 as a potential therapeutic target to treat bacterial meningitis.
Similar content being viewed by others

Introduction
Bacterial meningitis is the neurologic infection, such as pneumococcal meningitis and streptococcal meningitis. Invasion of bacteria circulation in the blood into the brain leading to the release of inflammatory factors and toxic compounds is the critical step for the development of meningitis, which requires bacteria penetrating the blood-brain barrier (BBB)1. As a critical defensive structure, BBB is primarily composed of brain microvascular endothelial cells (BMECs), glial cells and pericytes, of which BMECs are responsible to remain BBB structure and functional integrity by tight junctions (TJs) including occludin, claudin, junctional adhesion molecules (JAMs), and zonula occludens (ZO)-11. Once interacting with BMECs, bacteria penetrate BBB by different ways, such as by invading brain endothelial cells (transcellular route), by disrupting intercellular junctions or inducing cell damage (paracellular route), or by infected phagocytes (Trojan-horse mechanism)2.
Streptococcus suis (S. suis) is an emerging zoonotic pathogen causing meningitis3. In recent years, several studies have been performed to understand the mechanism of S. suis meningitis by the identification of key bacterial virulence factors. For example, S. suis small RNA rss04 was found to induce meningitis by regulating capsule synthesis and transcriptional virulence factors, thus promoting S. suis invasion4. In addition to small RNA, protein regulators have been also verified to contribute S. suis meningitis, such as serine/threonine protein kinase (STK) inducing ubiquitinated degradation of TJs protein claudin-5 in human BMECs (hBMECs), enolase interaction with ribosomal protein SA (RPSA) on the surface of porcine BMECs inducing cell apoptosis and fimbria-like component SssP1 interaction with vimentin of hBMECs promoting bacterial adhesion, BBB penetration and robust inflammatory response as well as suilysin inducing hBMECs apoptosis5,6,7,8. Furthermore, upregulation of inflammatory cytokines including IL-1β, IL-6, IL-8 and TNF-α also contributes to S. suis meningitis9. Our previous study has reported that NLRP3 facilitates BBB disruption in S. suis-infected hBMECs9. These findings revealed the process of destroying BBB integrity, but the precise mechanism by which S. suis disrupts BBB in vivo and whether other host factors are involved in S. suis-induced BBB disruption are poorly understood.
Ephrin type-A receptor 2 (EphA2) as a member of the Eph receptor tyrosine kinase family mediates diverse cell signals such as tissue development, cytoskeleton remodeling, endothelial cell barrier integrity, cell adhesion and migration. EphA2 is highly expressed in cancer cell types and involved in cancer and neurological disorders10. In addition, EphA2 could be a receptor of some pathogens including Candida albicans, Chlamydia trachomatis and hepatitis C virus to invade the host11,12,13. Recent studies reveal that EphA2 plays an important role in pathogens across BBB. For example, EphA2 activation via CD44 promotes migration of Cryptococcus neoformans across the BBB14. Besides, EphA2 is critical for Plasmodium-induced BBB breakdown15. These findings demonstrate that EphA2 could be developed as a therapeutic target for anti-tumor/cancer or anti-infective therapies.
In this study, we reported that EphA2 serves as an important defensive factor in the host against S. suis-induced BBB disruption. EphA2-mediated autophagy protects BBB against S. suis infection. S. suis STK is involved in ubiquitinated degradation of EphA2 via E3 ubiquitin-protein ligase DCAF15. Strikingly, S. suis STK induces DCAF15 deacetylation via the direct phosphorylation on SIRT1 to augment EphA2-DCAF5 interaction, thus promoting ubiquitinated degradation of EphA2, thereby triggering BBB disruption.
Results
S. suis downregulates expression of EphA2 and tight junctions (TJs) protein ZO-1 to disrupt blood-brain barrier
Brain endothelial cells are the first defense line against bacteria across BBB. EphA2 expression on the endothelial cells has been reported to regulate cells junction formation16. Since meningitic bacteria including S. suis and Streptococcus pneumoniae (S. pneumoniae), have been known to adhere to brain endothelial cells and disrupt their integrity17, we determined the effect of S. suis on the expression of EphA2 and TJs protein ZO-1 in brain endothelial cells. S. suis SC19 and S. pneumoniae D39 downregulated expression of EphA2 and ZO-1 both in human and mice brain endothelial cells (hCMEC/D3 and bEnd.3) as well as in mice brain (Fig. 1A-F and Supplementary Fig. 1A-D). Consistently, we also observed that SC19 and D39 downregulated EphA2 expression through immunofluorescence microscopy (Fig. 1G). However, SC19 and D39 did not affect mRNA expression of EphA2 (Fig. 1H), indicating that SC19 targets degradation of the EphA2 protein.

A–F Immunoblot analysis of ZO-1 and EphA2 in hCMEC/D3 cells (A, C), brain (B, D) and bEnd.3 cells (E, F). Cells were infected with SC19 (A, E) and D39 (MOI = 10) (C, F) for 0, 2, 4, 6 h. For in vivo infection, mice (total N = 36, each group n = 3) were intraperitoneally infected with SC19 (B) and D39 (2.5 × 108 CFU) (D) for 48 h. G Representative images of immunofluorescence staining of EphA2. Scale bar, 50 μm. H mRNA expression of EphA2 in SC19 and D39-infetced hCMEC/D3 cells. GAPDH served as an internal control. I Immunoblot analysis of EphA2 in SC19-infected hCMEC/D3 cells with treatment of MG132 or chloroquine (CQ). J Co-IP analysis of ubiquitination of EphA2 in SC19-infected hCMEC/D3 cells. Data are representative of three independent experiments with triplicate samples in vitro or three mice per group in vivo. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
To further investigate the mechanism by which SC19 induces degradation of EphA2, MG132 and chloroquine (CQ) were used to inhibit ubiquitin-proteasome and lysosome pathway, respectively. As shown in Fig. 1I and Supplementary Fig. 1E, MG132 reversed SC19-induced downregulation of EphA2 but CQ did not affect such downregulation. Furthermore, SC19 enhanced EphA2 ubiquitination at 4 h and 6 h post infection (Fig. 1J), indicating that SC19 targets degradation of EphA2 via ubiquitin-proteasome pathway.
EphA2 deficiency augments S. suis-induced disruption of blood-brain barrier in vitro and in vivo
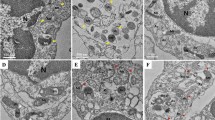
EphA2 is involved in neurological disorders via regulating multiple cell signals. In addition, EphA2 has been reported to play dual role in response to different pathogens. To explore the function of EphA2 in S. suis breaking through BBB, we generated EphA2−/−hCMEC/D3 cells using CRISPR-Cas9 knockout technology (Supplementary Fig. 2A, B) and knocked down and overexpressed EphA2 in hCMEC/D3 cells (Supplementary Fig. 2C, D). We found that EphA2 knockout elevated SC19 and D39-induced reduction of ZO-1 protein expression while overexpression of EphA2 in EphA2−/−hCMEC/D3 cells rescued such reduction (Fig. 2A-D and Supplementary Fig. 2E-H). Similarly, EphA2 knockdown using specific small interfering RNAs (siRNAs) also promoted SC19 and D39-induced reduction of ZO-1 protein expression in hCMEC/D3 cells while EphA2 overexpression reversed such reduction (Supplementary Fig. 2I-L). In addition, BBB transwell infection model showed that EphA2−/−cells exhibited lower trans-endothelial electrical resistance (TEER) values compared to EphA2+/+ cells (Fig. 2E, F), indicating EphA2 protects endothelial barrier integrity. Next, we explored the role of EphA2 in SC19 and D39-induced BBB disruption in vivo using EphA2−/− and EphA2+/+ mice. We found that EphA2−/− mice exhibited lower expression level of ZO-1 in brain and more bacterial load in brain and blood compare to EphA2+/+ mice (Fig. 2G-J). Additionally, compared to EphA2+/+ mice, EphA2−/− mice showed higher permeability of blood brain barrier with increased Evans Blue (EB) diffusion in the brain (Fig. 2K). Furthermore, EphA2−/− mice exhibited stronger meningeal destruction along with inflammatory cell infiltration (green arrows), necrotic cell debris (blue arrows), and reactive gliosis (yellow arrows) (Fig. 2L). These results demonstrate that EphA2 plays a protective role in maintaining BBB integrity during S. suis and S. pneumoniae meningitis.

A, B Immunoblot analysis of ZO-1 and EphA2 in EphA2+/+ or EphA2−/−hCMEC/D3 cells infected with SC19 (A) or D39 (B) for 0, 2, 4 h. C, D Immunoblot analysis of ZO-1 and EphA2 in EphA2−/−hCMEC/D3 cells transfected with Flag-tagged EphA2 and infected with SC19 (C) or D39 (D) for 0, 2, 4 h. E, F The TEER value in the absence and presence of EphA2 in transwell SC19 (E) or D39 (F) infection model for indicated time. For animal experiments, EphA2+/+ (total N = 90) and EphA2−/− mice (total N = 75) were intraperitoneally inoculated with SC19 and D39 for 48 h. After infection, brains were collected for indicated assays. G, H Immunoblot analysis of ZO-1 and EphA2 (total N = 36, each group n = 3). I, J bacterial load in the blood and brain. K blood-brain barrier permeability via Evans Blue staining (total N = 75, each group n = 5). L Representative images of histopathological change of brains via H&E staining (total N = 54, each group n = 3). Data are representative of three independent experiments with triplicate samples in vitro or three or five mice per group in vivo. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
EphA2 is required for LKB1/AMPK-mediated autophagy to protect S. suis-induced disruption of blood-brain barrier
We identified the protective role of EphA2 in maintain the BBB integrity against S. suis infection, but molecular mechanism underlying this protective effect remains unclear. It has been reported that EphA2 regulates autophagy to maintain cell homeostasis in lens epithelial cells and cancer cells18,19,20. We therefore hypothesized the importance of EphA2-mediated autophagy against SC19-induced disruption of BBB since autophagy also plays a critical role in the host defense against bacterial infection. Next, we investigated EphA2-mediated autophagy and autophagic flux in SC19-infected hCMEC/D3 cells. EphA2−/− hCMEC/D3 cells exhibited reduced LC3II expression but increased P62 accumulation compared to EphA2+/+ hCMEC/D3 cells (Fig. 3A). Conversely, overexpression of EphA2 in EphA2⁻/⁻ hCMEC/D3 cells reversed these alterations in the expression level of LC3II and P62 (Supplementary Fig. 3A). The presence of autophagosomes was further confirmed by Transmission electron microscopy (TEM) (Fig. 3B, red arrows). EphA2-mediated autophagic flux was investigated in hCMEC/D3 cells infected with adenovirus expressing mRFP-GFP-LC3. EphA2−/−cells exhibited more green puncta and yellow puncta compared to EphA2+/+cells, indicating the impairment of autophagic flux (Fig. 3C). The essential role of EphA2 in the autophagic flux was confirmed using bafilomycin A1 (BafA1) to inhibit autophagosome-lysosome fusion. BafA1 treatment significantly elevated LC3II level in EphA2+/+cells but failed to do so in EphA2−/− cells (Fig. 3D). To further investigate the role of autophagy in BBB, 3-methyladenine (3MA) and rapamycin (Rapa) were used to inhibit and activate autophagy, respectively. 3MA treatment augmented reduction of ZO-1 expression and enhanced accumulation of P62 in EphA2+/+ cells and EphA2−/− cells (Fig. 3E and Supplementary Fig. 3B). However, Rapa reversed SC19-induced reduction of ZO-1 and promoted reduction of P62 in EphA2+/+ cells but promoted accumulation of P62 in EphA2−/− cells (Fig. 3E, Supplementary Fig. 3C, D), suggesting that EphA2 contributes to the basal maintenance of BBB integrity but it is not the only signal molecular regulating autophagy. BBB transwell infection model showed that 3MA treatment induced lower TEER values while Rapa treatment induced higher TEER values compared to DMSO treatment (Fig. 3F). These results suggest that EphA2-mediated autophagy plays a protective role in maintaining BBB permeability during SC19 infection.

A Immunoblot analysis of LC3 and P62 in EphA2+/+ or EphA2−/−hCMEC/D3 cells infected with SC19 for indicated time. B Representative TEM images of EphA2+/+ or EphA2−/−hCMEC/D3 cells infected with SC19. Red arrows indicate autophagosomes and autolysosomes. Scale bars: 2 μm, 500 nm. C Analysis of autophagic flux using mRFP-GFP-LC3 adenovirus-infected hCMEC/D3 cells, showing impaired flux in EphA2−/− cells evidenced by increased green and yellow puncta. D Immunoblot analysis of LC3-II and P62 about the autophagic flux in SC19-infected EphA2+/+ and EphA2−/−cells treated with or without bafilomycin A1 (BafA1). E Immunoblot analysis of ZO-1 and P62 in SC19-infected hCMEC/D3 cells for indicated time with treatment of DMSO, 3MA or Rapamycin (Rapa). F The TEER value in transwell infection model with treatment of DMSO, 3MA or Rapamycin (Rapa). G Immunoblot analysis of pERK1/2, ERK1/2, pAMPK, AMPK, pAKT, AKT and EphA2 in EphA2+/+ or EphA2−/−hCMEC/D3 cells infected with SC19 for indicated time. H Immunoblot analysis of pLKB1 and LKB1 in EphA2+/+ or EphA2−/−hCMEC/D3 cells infected with SC19 for indicated time. I, J Co-IP analysis of interaction between the endogenous EphA2 and LKB1 in hCMEC/D3 cells infected with SC19 for indicated time. K Phosphorylation of LKB1 induced by EphA2 in vitro. L, M Immunoblot analysis of ZO-1, pLKB1 and LKB1 in SC19-infected hCMEC/D3 cells for indicated time with treatment of DMSO, Crocetin (L) and Pim1 (M). N, O The TEER value in transwell infection model with treatment of Crocetin (N) and Pim1 (O). P The flowchart showing that mice (total N = 25, each group n = 5) were pretreated with Crocetin (20 mg/kg) via tail veil for 12 h prior to SC19 infection. After 48 h infection, samples were collected for the determination of bacterial load and ZO-1 expression in brain. This figure was created using Figdraw. Q bacterial load in blood and brain (Total N = 27, each group n = 3). R ZO-1 expression in brain. Data are representative of three independent experiments with triplicate samples in vitro or three or five mice per group in vivo. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
The ERK/AKT/AMPK signaling pathway is important for autophagy activation and EphA2 has been reported to regulate ERK/ATK/AMPK pathways18,21,22. We investigated ERK/AMPK/AKT pathways in SC19-infected EphA2+/+ and EphA2−/−hCMEC/D3 cells. EphA2−/−hCMEC/D3 cells showed less phosphorylation level of AMPK compared to EphA2+/+hCMEC/D3 cells but EphA2 knockout did not affect phosphorylation level of ERK and AKT (Fig. 3G and Supplementary Fig. 3E). In addition, EphA2 overexpression upregulated phosphorylation level of AMPK but did not affect phosphorylation level of ERK and ATK (Supplementary Fig. 3F), indicating that EphA2 mediates autophagy through AMPK signaling pathway. AMPK could be directly phosphorylated or activated by LKB1, which is a highly conserved serine/threonine kinase involved in many cellular processes, including cell metabolism, division, migration and proliferation23. We proposed that EphA2 directly activates LKB1 to regulate AMPK during SC19 infection. We found that EphA2−/−hCMEC/D3 cells showed less LKB1 phosphorylation compared to EphA2+/+hCMEC/D3 cells while EphA2 overexpression in EphA2−/−hCMEC/D3 cells rescued such reduction (Fig. 3H, Supplementary Fig. 3G, H). To further investigate whether EphA2 directly regulates LKB1 activity, we verified the interaction of EphA2-LKB1 via co-immunoprecipitation (Co-IP) assay in SC19-infected hCMEC/D3 cells (Fig. 3I, J). Their exogenous interaction was also identified by transfection with Flag-EphA2 and HA-LKB1 in HEK293T cells (Supplementary Fig. 3I, J). Structural modeling analysis further revealed the detailed interactions between EphA2 and LKB1 (Supplementary Fig. 3K). Importantly, we also observed that EphA2 directly phosphorylated LKB1 through in vitro phosphorylation assay (Fig. 3K).
To further investigate the role of LKB1 in EphA2-mediated autophagy, we used crocetin and Pim/AKK1-IN-1 (Pim1) to activate and inhibit LKB1 activity, respectively. We identified the activated and inhibitory role of crocetin and Pim1 in autophagy and both of them did not affect EphA2 expression (Supplementary Fig. 3L), demonstrating their specific target on LKB1-AMPK-autophagy axis. Crocetin reversed SC19-induced reduction of ZO-1 expression, LKB1 phosphorylation and TEER value but Pim1 aggravated such reduction (Fig. 3L-O and Supplementary Fig. 3M-O). In addition, Fig. 3P illustrates a schematic diagram using crocetin to protect SC19-induced BBB disruption. crocetin decreased bacterial load and upregulated ZO-1 expression in mice brain (Fig. 3Q, R). Taken together, these results suggest that EphA2 regulates AMPK activity through interaction with LKB1 to mediate autophagy, thus maintaining the stability of BBB.
S. suis recruits DCAF15 to interact with EphA2
As shown in Figs. 1, 2, SC19-induced ubiquitinated degradation of EphA2 leads to disruption of BBB. Although the functional role of EphA2 in maintaining BBB integrity via autophagy has been elucidated, the mechanism by which SC19 induces EphA2 degradation remains unknown. To further investigate the mechanism of ubiquitinated degradation of EphA2, we screened potential E3 ubiquitin ligases using hCMEC/D3 cells expressing Flag-EphA2 or Empty Vector (EV) (Fig. 4A). Protein bands corresponding to Flag-EphA2-interacting proteins immunoprecipitated by Flag were analyzed by Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) (Fig. 4B, left panel). Based on peptide spectrum match (PSM) scores, DCAF15 was identified as the top-ranking E3 ligase candidate among the highest-scoring interactors (Fig. 4B, right panel). DCAF15 has been found to ubiquitinate and target for degradation of key substrates, including IKZF1, IKZF3, ZFP91, RBM23, RBM39 and ZEB124,25,26,27. Next, we further investigated the interaction of DCAF15 and EphA2. SC19 enhanced endogenous interaction between EphA2 and DCAF15 (Fig. 4C). Similarly, exogenous interaction between EphA2 and DCAF15 was evident by co-transfection with Flag-DCAF15, HA-EphA2 or Flag-EphA2, HA-DCAF15 in HEK293T cells (Fig. 4D, E). In vitro glutathione-S-transferase (GST) affinity isolation assay also showed the direct interaction between EphA2 and DCAF15 (Fig. 4F). Confocal microscopy showed co-localization of DCAF15 and EphA2 at 4 h post infection (Fig. 4G). Interestingly, we observed DCAF15 distributed in the nucleus at 2 h post infection but in the cytoplasm at 4 h post infection (Fig. 4G). Nuclear and cytoplasmic fractionation assay showed that SC19 reduced DCAF15 expression in the nucleus but increased its expression in the cytoplasm (Fig. 4H), suggesting SC19 might induce DCAF15 redistribution from nuclear to cytoplasm to promote its interaction with EphA2. Subsequently, to further investigate the key domains of DCAF15 interaction with EphA2, we constructed different DCAF15 truncated mutants including DCAF15 (1-258), DCAF15 (259-385), DCAF15 (386-598), DCAF15 (1-385), and DCAF15 (259-598) and transfected these Myc-tagged DCAF15 mutants in HEK293T cells (Fig. 4I). The interaction between DCAF15 (1-258)/DCAF15 (1-385) and EphA2 was observed (Fig. 4J). Structural modeling analysis further revealed the detailed interaction between DCAF15 (1-385) and EphA2 (Fig. 4K). Several key interaction sites were observed, including hydrogen bond between ALA-149, GLN-241 on DCAF15 and GLU-626 and VAL-618 on EphA2 (Fig. 4K). Taken together, these results suggest that S. suis recruits DCAF15 to interact with EphA2.

A Schematic illustration of the quantitative proteomic to screen proteins binding to EphA2. This figure was created using Figdraw. B Identification of EphA2-interacting proteins by Co-IP and LC–MS/MS analysis in SC19-infected hCMEC/D3 cells expressing Flag-EphA2 (left). DCAF15 was identified as a top-ranking E3 ligase candidate based on peptide spectrum match (PSM) scores (right). C Co-IP analysis of the interaction between DCAF15 and EphA2 in SC19-infected hCMEC/D3 cells for indicated time. D, E Co-IP analysis of HEK293T cells co-transfected with HA-EphA2 and Flag-DCAF15 or HA-DCAF15 and Flag-EphA2. F DCAF15-EphA2 interaction via GST affinity-isolation assay. G Representative immunofluorescence images of colocalization of GFP-DCAF15 and EphA2 in SC19-infected hCMEC/D3 cells for indicated time. H DCAF15 expression in the nucleus and cytoplasm of SC19-infected hCMEC/D3 cells transfected with Flag-DCAF15. I Sketch map of full-length DCAF15 along with its truncated mutants. J Co-IP analysis of HEK293T cells co-transfected with Flag-EphA2, Myc-DCAF15 or its mutants, including Myc-DCAF15 (1-258), Myc-DCAF15 (259-385), Myc-DCAF15 (386-598), Myc-DCAF15 (1-385), Myc-DCAF15 (259-598). K Structural model of EphA2-DCAF15 interaction. Blue represents the electropositive region and red represents electronegative region. 3D display of interaction key amino acids. Yellow dotted lines represent hydrogen bonds. Data are representative of three independent experiments with triplicate samples per group. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
S. suis recruits DCAF15 to promote the K48 and K63-linked ubiquitination of EphA2 to degrade EphA2
Since E3 ubiquitin ligases are known to be involved in proteins degradation, we next investigated whether DCAF15-EphA2 interaction functionally regulates EphA2 stability by transfection with Myc-EphA2 and Flag-DCAF15 in HEK293T or hCMEC/D3 cells. DCAF15 inhibited EphA2 protein expression in a dose dependent manner (Fig. 5A and Supplementary Fig. 4A). DCAF15 overexpression aggravated SC19-induced reduction of ZO-1 and EphA2 but DCAF15 knockdown alleviated reduction of ZO-1 and EphA2 (Fig. 5B, C and Supplementary Fig. 4B-D). Furthermore, under the treatment of cycloheximide (CHX) which is a protein synthesis inhibitor and shortens life span of protein, EphA2 levels were gradually reduced but DCAF15 overexpression enhanced such reduction (Fig. 5D and Supplementary Fig. 4E). These results demonstrate that DCAF15 is indispensable for EphA2 degradation. Next, we proposed that DCAF15 as an E3 ubiquitin ligase mediates EphA2 stability via ubiquitin-proteasome-mediated degradation. To explore whether DCAF15 ubiquitinates EphA2, Co-IP assay was performed in HEK293T cells transfected with Myc-EphA2, HA-Ub or Flag-DCAF15. We observed that DCAF15 promoted EphA2 ubiquitination (Fig. 5E). Subsequently, we detected ubiquitin linkage types conjugated onto EphA2 driven by DCAF15. DCAF15 catalyzed K48- and K63-linked polyubiquitination of EphA2 (Fig. 5F). Furthermore, we identified ubiquitination sites on EphA2 through mass spectrometry analysis (Supplementary Fig. 4F). As shown in Fig. 5G, H, ubiquitination of EphA2 mutants including K646R, K649R, K754R and triple-mutant (EphA2-3KR) in which lysine was replaced with arginine were reduced in DCAF15-expressed cells compared with those of WT EphA2. These data demonstrated that DCAF15 induced the K48- and K63-linked ubiquitination of EphA2 at K646, K649 and K754. Taken together, these results suggest DCAF15 degrades EphA2, though inducing its ubiquitination to trigger S. suis-induced disruption of BBB.

A EphA2 expression in HEK293T cells transfected with Myc-EphA2 and different concentrations of Flag-DCAF15. B Immunoblot analysis of ZO-1 and EphA2 in SC19-infected hCMEC/D3 cells transfected with empty vector or Flag-DCAF15. C Immunoblot analysis of ZO-1 and EphA2 in DCAF15 knockdown hCMEC/D3 cells using SiRNA under SC19 infection. D DCAF15 shortened the half-life of EphA2. HEK293T cells were co-transfected with Myc-EphA2, Flag-EV or Flag-DCAF15 for 24 h and then treated with cycloheximide (CHX, 100 μg/ml) for indicated time. E Co-IP analysis of ubiquitination of EphA2 in HEK293T cells transfected with Myc-EphA2, HA-Ub in the absence or presence of Flag-DCAF15. F Co-IP analysis of polyubiquitination of EphA2 in HEK293T cells transfected with Myc-EphA2, HA-Ub or its mutants K6, K11, K29, K33, K48, K63 in the absence or presence of Flag-DCAF15. G, H Co-IP analysis of ubiquitination of EphA2 or its mutants in HEK293T cells transfected with Flag-DCAF15, HA-Ub, Myc-EphA2 or its indicated mutants. Data are representative of three independent experiments with triplicate samples per group. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
S. suis STK contributes to EphA2 degradation
Bacterial protein kinases such as stk, tyrosine kinase (cpsD) and histidine kinase (vicK) are the important virulence factors of pathogenic bacteria. It has been reported that STK regulates S. suis to penetrate the BBB5. Therefore, we investigate whether STK is involved in EphA2 degradation. Deletion of stk did not reduce expression of EphA2 while deletion of cpsD and vicK reduced its expression in hCMEC/D3 cells (Fig. 6A-D). In addition, SC19 Δstk alleviated ZO-1 downregulation (Fig. 6E and Supplementary Fig. 5A) compared to WT SC19. Similarly, SC19 Δstk did not reduce EphA2 expression in vivo (Fig. 6F). Furthermore, SC19 Δstk induced less EphA2 ubiquitination compared to WT SC19 (Fig. 6G), suggesting that STK is required for S. suis to degrade EphA2.

A–C Immunoblot analysis of EphA2 in hCMEC/D3 cells infected with SC19 Δstk (A), ΔcpsD (B) and ΔvicK (C). D Representative immunofluorescence images of EphA2 in hCMEC/D3 cells infected with WT SC19 or Δstk. E, F Immunoblot analysis of ZO-1 in hCMEC/D3 cells (E) and mice brain (F) infected with WT SC19 or Δstk (total N = 27, each group n = 3). G Co-IP analysis of ubiquitination of EphA2 in hCMEC/D3 cells infected with WT SC19 or Δstk. Data are representative of three independent experiments with triplicate samples per group. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
S. suis facilitates DCAF15 deacetylation via SIRT1 to enhance EphA2 ubiquitination for disruption of blood-brain barrier
Based on our findings that S. suis STK is required for EphA2 ubiquitinated degradation (Fig. 6), we sought to elucidate the underlying mechanism by which STK signaling promotes this process. We focused on acetylation, a widespread post-transcriptional modification that plays a key role in regulating protein stability and enzymatic activity28. We found that SC19 Δstk induced a higher acetylation level of DCAF15 in hCMEC/D3 cells compared to WT SC19 (Fig. 7A), suggesting that S. suis STK induces deacetylation of DCAF15. To further verify the specific deacetylase of DCAF15, we first identified P300 as acetyltransferase of DCAF15 and the interaction between P300 and DCAF15 (Supplementary Fig. 6A-C). Then, Flag-DCAF15 and HA-P300 were co-transfected into HEK293T cells and then these cells were treated with trichostatin A (TSA), a broad-spectrum inhibitor of histone deacetylase (HDAC) family deacetylases and nicotinamide (NAM), an inhibitor of inhibitor of sirtuin (SIRT) family deacetylases. NAM enhanced acetylation of DCAF15 while TSA did not affect its acetylation, indicating SIRT family members may participate in the deacetylation of DCAF15 (Fig. 7B). In order to investigate whether acetylation of DCAF15 affects ubiquitination of EphA2. Next, Myc-EphA2, HA-Ub or Flag-DCAF15 were co-transfected into HEK293T cells. We observed that DCAF15 enhanced EphA2 ubiquitination while NAM treatment attenuated its ubiquitination (Fig. 7C), demonstrating DCAF15 deacetylation enhances EphA2 ubiquitination. To further explore the mechanism by which DCAF15 is deacetylated. We screened SIRT family (SIRT1-SIRT5) deacetylases in HEK293T cells co-transfected with Flag-DCAF15 or HA-SIRTs under NAM treatment. We found that SIRT1 and SIRT2 reduced acetylation of DCAF15 (Fig. 7D). We also observed exogenous SIRT1-DCAF15 interaction in HEK293T cells and direct deacetylation of DCAF15 by SIRT1 in vitro (Fig. 7E and Supplementary Fig. 6D). However, we did not find SIRT2-DCAF15 interaction (Supplementary Fig. 6E). Structural modeling analysis further revealed the detailed interactions between DCAF15 and SIRT1 (Supplementary Fig. 6F). SIRT1 expression was upregulated with SC19 infection time and its role in the BBB was identified using siRNA, with the validated knockdown efficiency (Supplementary Fig. 6G, H). SIRT1 knockdown significantly enhanced acetylation of DCAF15 and reduced ubiquitination of EphA2 but alleviated the disruption of ZO-1 (Supplementary Fig. 6I, Fig. 7F, G), indicating that SIRT1 is essential for BBB damage during S. suis infection. Subsequently, we found three potential acetylation sites at K552, K560, and K581 of DCAF15 through MS analysis (Supplementary Fig. 6J). These three sites were evolutionary conserved in mammals (Fig. 7H). To verify these three sites, Flag-tagged DCAF15 mutants in which each lysine residue or two lysine residues were replaced with arginine (to attenuate acetylation) or glutamine (to enhance acetylation), respectively, were transfected into HEK293T cells via site-directed mutagenesis. Compared with WT DCAF15, DCAF15 K552R and K581R showed reduced acetylation and the double-mutant DCAF15-K552R/K581R (DCAF15-2KR) even showed much less acetylation, but the double-mutant DCAF15-K552Q/K581Q (DCAF15-2KQ) showed higher level of acetylation (Fig. 7I, J). Next, we further explore the role of acetylation sites at K552 and K581 of DCAF15 in EphA2-mediated disruption of BBB. Compared to WT DCAF15, DCAF15-2KQ reduced its interaction with EphA2 in HEK293T cells transfected with Myc-EphA2, Flag-DCAF15 or its mutant (Fig. 7K). However, upon SC19 infection, DCAF15-2KR enhanced EphA2 ubiquitination in hCMEC/D3 cells (Fig. 7L and Supplementary Fig. 6K). In addition, DCAF15-2KR promoted reduction of ZO-1 expression and TEER values while DCAF15-2KQ rescued reduction of ZO-1 expression and TEER values (Fig. 7M, N). Taken together, these data demonstrate that deacetylation of DCAF15 via SIRT1 promotes DCAF15-EphA2 interaction to enhance EphA2 ubiquitination, thus inducing EphA2 degradation, thereby resulting in disruption of BBB.

A Co-IP analysis of acetylation of DCAF15 in hCMEC/D3 cells infected with WT SC19, Δstk or CΔstk. B Co-IP analysis of acetylation of exogenous DCAF15 in HEK293T cells co-transfected with Flag-DCAF15, HA-P300 in the treatment of deacetylase inhibitors TSA or NAM. C Co-IP analysis of ubiquitination of exogenous EphA2 in HEK293T cells co-transfected with Myc-EphA2, HA-Ub, Flag-DCAF15 in the absence or presence of NAM. D Co-IP analysis of acetylation of exogenous DCAF15 in HEK293T cells co-transfected with Flag-DCAF15 or HA-SIRT1, HA-SIRT2, HA-SIRT3, HA-SIRT4, HA-SIRT5 in the presence of NAM. E Deacetylation of DCAF15 in vitro from HEK293T cells co-transfected with HA-P300 and Flag-DCAF15 followed by immunoprecipitated with Flag-agarose beads and then incubated with His-SIRT1. F Co-IP analysis of ubiquitination of EphA2 in HEK293T cells co-transfected with Flag-DCAF15, Myc-EphA2, HA-Ub under the condition of SIRT1 knockdown. G Western blot analysis of ZO-1 expression in SC19-infected hCMEC/D3 cells with SIRT1 knockdown. H Sequence alignment of the conserved K552 and K581 containing region in DCAF15 orthologs of different species. I, J Co-IP analysis of acetylation of DCAF15 or its mutants in HEK293T cells transfected with Flag-DCAF15 or its mutants including K552R, K560R, K581R (I) and DCAF15-2KR or DCAF15-2KQ (J) in the presence of NAM. K Co-IP analysis of the interaction between EphA2 and DCAF15 or its mutant (DCAF15-2KQ) in HEK293T cells transfected with Myc-EphA2, Flag-DCAF15 or its mutant. L Co-IP analysis of ubiquitination of EphA2 in SC19-infected hCMEC/D3 cells transfected with Flag-EV, Flag-DCAF15 or its mutants including DCAF15-2KR or DCAF15-2KQ. M ZO-1 expression in SC19-infected or non-infected hCMEC/D3 cells transfected with Flag-DCAF15 or its indicated mutants. N The TEER value in transwell infection model transfected with Flag-EV, Flag-DCAF15 or its indicated mutants. Data are representative of three independent experiments with triplicate samples per group. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
S. suis STK phosphorylates SIRT1 to degrade EphA2 for disruption of blood-brain barrier
Since SIRT1-mediated deacetylation of DCAF15 leads to EphA2 ubiquitination and BBB damage, we next sought to investigate whether S. suis STK regulates SIRT1 activity. Deletion of stk reduced SIRT1-DCAF15 interaction in hCMEC/D3 cells compared to SC19 (Fig. 8A). This interaction was also confirmed in HEK293T cells co-transfected with Flag-DCAF15, HA-SIRT1 or Myc-STK- (Fig. 8B). STK-SIRT1 interaction was evident in HEK293T cells co-transfected with Myc-STK and HA-SIRT1 through co-immunoprecipitation and pull-down assay (Fig. 8C-E). Interestingly, phosphorylation in vitro assay revealed that SIRT1 was phosphorylated by STK (Fig. 8F). These results demonstrate that STK plays an important role in triggering the activity of SIRT1. Subsequently, we found the potential phosphorylation site at S48 on SIRT1 through MS analysis (Fig. 8G). This site is evolutionarily conserved in mammals (Fig. 8H). To verify the function of the specific site, HA-tagged SIRT1 mutants, in which a serine residue was replaced with alanine, were generated by site-directed mutagenesis and then transfected into HEK293T cells. STK-induced phosphorylation of SIRT1 was diminished in HA-SIRT1 mutant-transfected HEK293T cells (Fig. 8I). Importantly, SIRT1 mutant significantly impaired the ubiquitination of EphA2 and alleviated the disruption of ZO-1 (Fig. 8K). These results demonstrate that the phosphorylation of SIRT1 induced by STK is critical for the disruption of BBB during S. suis infection.

A Co-IP analysis of SIRT1-DCAF15 interaction in hCMEC/D3 cells infected with WT SC19, Δstk or CΔstk. B Co-IP analysis of SIRT1-DCAF15 interaction in HEK293T cells co-transfected with Flag-DCAF15, HA-SIRT1 and Myc-STK. C, D Co-IP analysis of STK-SIRT1 interaction in HEK293T cells co-transfected with Myc-STK and HA-SIRT1. E STK-SIRT1 interaction via GST affinity-isolation assay. F Phosphorylation of SIRT1 induced by STK in vitro. G Identification of phosphorylated sites of SIRT1 using mass spectrometry. H Sequence alignment of the conserved Ser48 containing region in SIRT1 orthologs of different species. I Phosphorylation level of SIRT1 in HEK293T cells co-transfected with Myc-STK, HA-SIRT1 or HA-SIRT1-S48A. J Ubiquitination of EphA2 in HEK293T cells co-transfected with HA-Ub, Myc-EphA2, Flag-DCAF15, His-STK, HA-SIRT1-WT or HA-SIRT1-S48A. K Western blot analysis of ZO-1 expression in SC19-infected hCMEC/D3 cells transfected with HA-SIRT1-WT or HA-SIRT1-S48A. Data are representative of three independent experiments with triplicate samples per group. All data are presented as mean ± SD. P ≤ 0.05 was considered as statistical significance.
Collectively, these data establish the STK-SIRT1-DCAF15 signaling axis where S. suis STK phosphorylates and activates SIRT1, promoting DCAF15 deacetylation to enhance EphA2 ubiquitination, leading to BBB disruption (Fig. 9).

Upon S. suis infection, EphA2 expression is downregulated. S. suis recruits DCAF15 to interact with EphA2 and induce K48- and K63-linked ubiquitination of EphA2 at K646, K649 and K754. This modification reduces EphA2 stability, which associates with LKB1/AMPK-mediated autophagy to attenuate S. suis-induced disruption of blood-brain barrier. In addition, S. suis STK phosphorylates SIRT1 to deacetylate DCAF15, which augments DCAF15-EphA2 interaction and EphA2 degradation, thereby breaking down blood-brain barrier. This figure was created using Figdraw.
Discussion
Although many studies have reported that EphA2 is mainly involved in the development of cancer and tumor, several studies have shown that EphA2 plays an important role in mediating integrity of BBB. In neurological disorders, high expression of EphA2 exacerbates the damage of BBB in a mouse or rat model of focal stroke or diffuse axonal injury, while inhibition or knockout of EphA2 protects BBB integrity with alleviating the loss of tight junction proteins in human BMECs29,30. For pathogenic infection, EphA2 has been proposed as a pattern recognition receptor to recognize pathogenic infection including viruses, fungi, parasites and chlamydia. In the BBB infection model, EphA2 promotes Cryptococcus neoformans fungal cells and Plasmodium falciparum parasites across BBB with the loss of junction proteins on mouse and human BMECs. Recent studies have shown that EphA2 is identified on oral epithelial cells, neutrophils and macrophages to induce anti-fungal activity and inflammatory response11,31,32. In contrast to fungal infection, EphA2 could be an entry receptor for viral infection and facilitates viral replication, which suppresses antiviral response through the inhibition of inflammatory response33,34,35. These studies suggest that EphA2 exerts different biological functions in response to different pathogens infection.
In this study, EphA2−/− mice and EphA2−/− hCMEC/D3 cells were used to investigate the exact role of EphA2 in S. suis across BBB. EphA2−/− mice brain and EphA2−/−hCMEC/D3 cells showed higher BBB permeability and severer BBB damage through the detection of tight junction protein, Evans Blue staining and TEER value during S. suis and S. pneumoniae infection, demonstrating the protective role of EphA2 in maintaining BBB integrity during bacterial meningitis.
These varied function roles of EphA2 in the host could be considered as pathogens’ lifestyle, its specific expression and signaling. For example, infection with Cryptococcus neoformans or Plasmodium falciparum, both of which utilize EphA2 as an entry receptor for cellular invasion, leads to the upregulation of EphA2 expression, and its role in facilitating pathogen internalization directly compromises BBB integrity11,31,32. In contrast, S. suis, an extracellular bacterium that does not depend on receptor-mediated internalization, promotes EphA2 protein degradation. This downregulation impairs the protective function of EphA2 in maintaining endothelial cell junctions, ultimately leading to BBB breakdown. Thus, the functional outcome of EphA2 is not only intrinsic to the receptor itself but also determined by the pathogens’ invasion strategy. This distinction highlights the importance of specific pathogen-host interaction mechanisms in defining the role of EphA2 in CNS infection.
Based on dual roles of EphA2 in the host, EphA2 has been developed as a potential therapeutic target. For example, as the viral entry receptor, targeting EphA2 is a promising strategy in anti-viral infection34. Furthermore, EphA2 phosphorylates NLRP3 and inhibits its activation in the airway epithelial cells35. Due to EphA2’s function in inflammation, small-molecule ALW-II-41-27 targeting EphA2 inhibits Pneumocystis carinii β-glucans-induced inflammatory response in macrophages32. However, EphA2-mediated inflammatory response in bacterial meningitis needs to be further studied. Our present study reveals the protective role of EphA2 in maintaining BBB integrity via stabilizing endothelial cell junctions and EphA2 degradation leads to BBB disruption during bacterial meningitis, suggesting that enhancing EphA2 signaling could be a promising therapeutic strategy via the development of EphA2 agonists to stabilize its expression.
Autophagy has been reported to be involved in multiple diseases and mediates the BBB dysfunction by different mechanisms. For example, autophagy can alleviate BBB impairment by regulating claudin 5 redistribution36. ULK-mediated autophagy alleviates S. pneumonia-induced microglial pyroptosis37. During ischemia/reperfusion (I/R) injury, promoting autophagy is an effective therapy for maintaining BBB integrity38. EphA2-mediated autophagy has been reported in the corneal epithelium and cancer model18,19,20. Our present study reveals that EphA2 is required for autophagy and EphA2-mediated autophagy plays an important role in BBB integrity during S. suis infection. We further found that EphA2 regulated LKB1-AMPK-mediated autophagy, which has been reported that LKB1-AMPK is one of the major autophagy pathways and plays an important role in regulating neurological function39. In contrast, autophagy activation also contributes to endothelial cell death and disrupts BBB during Group C streptococci infection40. These studies demonstrate the dual role of autophagy in the BBB and EphA2-meidated autophagy could be developed as a specific target to alleviate bacterial meningitis. However, whether EphA2-mediated autophagy leads to redistribution of tight junction of endothelial cells needs to be further studied.
According to the protective role of EphA2 against S. suis-induced BBB disruption, our data showed that S. suis downregulated EphA2 protein expression but did not affect its mRNA expression. Following the mechanistic investigation by which S. suis degrades EphA2, our study focused on the mechanism on S. suis-induced ubiquitinated degradation of EphA2. Deubiquitination of EphA2 via deubiquitinase USP3 plays an important role in the progression of various tumors, while ubiquitination of EphA2 via E3 ligase RNF5 limits its anti-tumor function, indicating that EphA2 plays dual functions in tumorigenesis41,42. Our study screened E3 ligase DCAF15 recruited by S. suis to interact with EphA2, which resulted in EphA2 ubiquitination. So far, DCAF15 catalyzes different substrates, including RBM39, RBM23 and ZEB1 to enhance their ubiquitination and proteasomal degradation, which exerts anti-cancer or anti-tumor activity24,25,26,43. This present study found EphA2 as a substrate of DCAF15 during S. suis infection and DCAF15 catalyzed K48-/K63-linked ubiquitination of EphA2 at K646, K649 and K754, which led to EphA2 degradation. DCAF15 knockdown rescued S. suis-induced BBB disruption and EphA2 degradation. While K48-linked polyubiquitination is well-established as a canonical signal for proteasomal degradation, the K63-linked ubiquitination of EphA2 suggests the regulatory complexity. Unlike K48-linked chains, K63-linked ubiquitination mainly exerts non-proteolytic functions, including modulation of protein-protein interactions, regulation of subcellular localization, and participation in inflammatory signaling pathway activation44,45. The concurrent formation of K48 and K63-linked polyubiquitin chains on EphA2 may not only target the protein proteasomal for degradation but also actively disrupt its functional scaffolding at the endothelial junction, ultimately amplifying BBB breakdown. These findings suggest a previously unreported effect of DCAF15 on the modulation of EphA2 ubiquitination and degradation induced by S. suis infection. However, the exact regulatory role of K63-linked ubiquitination of EphA2 in BBB needs to be further studied.
Considering the importance of S. suis virulence factor across BBB and Ser/Thr protein kinase (STK) has been reported to be involved in the disruption of BBB tight junction protein claudin-55, we investigated the effect of protein kinases, including STK, tyrosine kinase (CpsD) and histidine kinase (VicK) on EphA2 degradation. We found that S. suis STK deacetylated DCAF15 to enhance EphA2 ubiquitinated degradation. Acetylation plays an essential role in protein-protein interactions and protein degradation. Cohesin deacetylation by deacetylases HDAC8 promotes DCAF15-mediated ubiquitinated degradation of cohesin regulatory factor PDS5A, leading to defective DNA replication and apoptosis46. Intriguingly, we found that K552 and K581 residues of DCAF15 played important roles in the process of acetylation, promoting BBB disruption. Our findings suggest an important role of DCAF15 in direct deacetylation-mediated protein degradation and BBB stability. Notably, it is well-known that STK phosphorylates substrates in S. suis to regulate bacterial virulence. Recent study has identified that STK in S. pneumoniae phosphorylates beclin-1 to induced autophagic degradation of occluding in alveolar epithelial barrier47. Our study showed that STK phosphorylated SIRT1 to induce DCAF15 deacetylation, facilitating BBB disruption. These findings suggest the central role of STK in linking bacteria to communicating with host target proteins.
In conclusion, we reveal a critical role of EphA2 in the BBB against bacterial meningitis and elucidate the interplay between EphA2, DCAF15 and SIRT1 in the regulation of S. suis-induced BBB disruption. The post-translational modifications of EphA2 and DCAF15, including ubiquitination and acetylation, play crucial roles in modulating BBB stability. These findings increase our knowledge on the underlying mechanism that meningitic bacteria break though BBB.
Materials and methods
Antibodies and reagents
The following antibodies were used in this study: anti-EphA2 (6997), anti-LC3B (3868 T), anti-AKT (9272S), anti-Phospho-AKT (Ser473) (4060S), anti-Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (4370 T), anti-p44/42MAPK (ERK1/2) (4695 T), anti-AMPKα (5831), Anti-Phospho-AMPKα (Thr172) (2535) and Phospho-LKB1 (Ser334) (3055) antibodies were from Cell Signaling Technology; anti-DYKDDDDK tag (20543-1-AP), anti-P62/SQSTM1 (18420-1-AP), anti-STK11/LKB1 (10746-1-AP), anti-ZO-1 (21773-1-AP), anti-SIRT1 (60303-1-Ig), anti-HA tag polyclonal antibody (51064-2-AP), anti-Myc (16286-1-AP), anti-DYKDDDDK (66008-4-Ig) and histone H3 (17168-1-AP) antibodies were from protein tech; Anti-Phospho-(Ser/Thr) (ab117253) and anti-P300 (ab275378) antibodies were from Abcam; anti-Ubiquitin (sc-8017) antibody was from santa cruz; anti-Acetyl lysine antibody (PTM-105RM) was from PTM BIO; DCAF15 antibody was from Sigma; 3-methyladenine (3MA, HY-19312), Rapamycin (Rapa, HY-10219), MG-132 (HY-13259), Chloroquine (CQ, HY-17589A), Pim1/AKK1-IN-1 (Pim1, HY-10371), Crocetin monomethyl ester (HY-N6904), cycloheximide (HY-12320) and DMSO (HY-Y0320) were from MCE; Anti-Myc affinity gel (P2285), Protein A + G agarose (P2055), GST-tag purification resin (P2251) and NP-40(P0013F) were from Beyotime; Anti-DYKDDDDK G1 affinity resin (L00432) was from Gen Script, Lipomaster 293 transfection reagent (JC3001) and ChamQ Blue Universal SYBR qPCR Master Mix (Q312) were from Vazyme; Lipofectamine™ 3000 (L3000008) was from Thermo Scientific; SIRT1 Fusion Protein (Ag17677) and STK11 Fusion Protein (Ag1048) were from protein tech.
Plasmid constructs and transfection
The plasmids, including Flag-tagged DCAF15-K562R, DCAF15-K560R, DCAF15-K581R, DCAF15-2KR, DCAF15-2KQ acquired from BGI (Beijing, China). HA-tagged LKB1, Ub, Ub-K6, Ub-K11, Ub-K29, Ub-K33, Ub-K48, Ub-K63, P300, SIRT1, SIRT2, SIRT3, SITR4, SIRT5, KAT2A, KAT2B, KAT5 were acquired from KA&M BIO (Shanghai, China). Myc-tagged DCAF15 (1-258), DCAF15 (295-385), DCAF15 (386-598), DCAF15 (1-385), DCAF15 (259-598), EphA2-K99R, EphA2-K162R, EphA2-K468R, EphA2-K639R, EphA2-K646R, EphA2-K649R, EphA2-K684R, EphA2-K686R, EphA2-K754R, EphA2-K778R, EphA2-K882R, EphA2-954R, EphA2-3R were acquired from BGI (Beijing, China). EphA2 and DCAF15 ORFs amplified from hCMEC/D3 cells were inserted into vectors pLV3, pcNDA3.1 or pCMV expressing Flag, Myc or HA designed as Flag-EphA2, HA-EphA2, Myc-EphA2, Flag-DCAF15, HA-DCAF15 or Myc-DCAF15, respectively. These constructed mutants by standard molecular biology techniques were stored in our laboratory.
All constructs were confirmed by DNA sequencing. Plasmids were transiently transfected into HEK293T or hCMEC/D3 cell with lipofectamine 3000 reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. All primers used for plasmids construction are shown in Supplementary Data 1.
Bacterial strains
The S. suis epidemic strain SC19 (serotype 2) with high pathogenicity in humans, mice and pigs was used in the present study. Streptococcus pneumoniae D39 was kindly gifted by Kohsuke Tsuchiya (Kanazawa University, Japan). The strains including Δstk, ΔcpsD and ΔvicK were constructed under the SC19 background by deleting the gene of stk, cpsD and vicK. These bacterial strains were stored in our laboratory. All strains were cultured in Todd-Hewitt broth (THB; OXOID, CM0189B) or on Tryptic soy agar (TSA) supplemented with 10% fetal bovine serum (FBS) at 37 °C.
Cell culture and bacterial infection
All cells including HEK293T (CL-0005, Procell), human cerebral microvascular endothelial cell (hCMEC/D3, JNO-H0520, Jennio Biotech), EphA2−/− hCMEC/D3 and mice microvascular endothelial cell (bEnd.3, CL-0598, Procell) were cultured in DMEM (Gbico, USA) supplemented with 10% FCS and 1% penicillin/streptomycin (Gbico, USA) and maintained at a humidified 37 °C incubator with 5% CO2. These cells were infected at a multiplicity of infection (MOI) of 10 for indicated time. After infection, lysates were collected for assays as described below. These cells were infected at a multiplicity of infection (MOI) of 10 for indicated time. Chemicals including 3-methyladenine (3-MA) at 5 mM, bafilomycin A1 (BafA1) at 50 nM and rapamycin (Rapa) at 20 μM were added to the cells prior to infection for 1–2 h. After infection, lysates were collected for assays as described below.
Western blotting
After infection, cells were lysed in Radioimmunoprecipitation assay (RIPA) lysis buffer. Protein samples were subjected to SDS-PAGE and subsequently transferred onto a polyvinylidene difluoride (PVDF) membrane by electroblotting. Next, the membranes were blocked with 5% nonfat dry milk and then immunoblotted with indicated primary antibodies (Abs) overnight at 4 °C. Next day, the blots were incubated with horseradish peroxidase-conjugated goat anti-mouse/rabbit IgG. Finally, the distinct protein bands were detected by ECL detection reagent (Biosharp, China).
Quantitative real time polymerase chain reaction (qRT-PCR)
After infection, cells were lysed and total RNA were extracted by FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, Nanjing, China) according to the manufacturers’ instructions. Then RNA (1 μg) was reverse transcribed by HiScript IV All-in-One Ultra RT SuperMix. Next, cDNA was detected via ChamQ Blue Universal SYBR qPCR Master Mix (Vazyme). Finally, qPCR was performed using the CFX96 (Bio-Rad, USA). Relative gene expression levels were normalized against the expression levels of GAPDH. All primers used for PCR are shown in Supplementary Data 1.
The generation of EphA2 −/− hCMEC/D3 cells using CRISPR-Cas9 knockout technology
The EphA2 gene target sequence (5’-AGGCTCCGAGTAGCGCACAC-3’) was inserted into the guide RNA expression plasmid PX459. Then the recombinant construct was identified by sequencing and transfected into hCMEC/D3 cells. After 36 h transfection, cells were selected using puromycin (10 μg/ml) for 3–7 d. Finally, the monoclonal cells were obtained using the limiting dilution method. RT-qPCR and immunoblotting assays were used to identify monoclonal cell line. All primers used for PCR amplification are shown in Supplementary Data 1.
EphA2 and DCAF15 knockdown using SiRNA interference
siRNA were transfected using Lipofectamine 3000 (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Cells were transfected with 20 μM of EphA2 and DCAF15 siRNA (Sangon Biotech) for 36 h and then infected with S. suis as described above. Finally, cell lysates were collected for western blot. Knockdown efficiency was identified by qPCR and western blot analysis. All primers used for siRNA are shown in Supplementary Data 1.
Bacterial infection in vivo
WT C57BL/6 J (EphA2+/+) (total N = 180, each group n = 3 or 5) and EphA2−/−mice (total N = 75, each group n = 3 or 5) were intraperitoneally infected with 100 μL S. suis and S. pneumoniae (2.5 × 108 CFU) and PBS as blank control. For Crocetin treatment, mice were pretreated with Crocetin (20 mg/kg) via tail veil for 12 h prior to SC19 infection. After 48 h infection, mice were sacrificed by cervical vertebrae dislocation and brain tissues were collected to determine bacterial load and tight junction protein expression. In addition, brain tissues were fixed in 10% formalin and then processed in paraffin for hematoxylin and eosin staining.
Evans Blue blood-brain barrier permeability assay in vivo
WT C57BL/6 J (EphA2+/+) (total N = 45, each group n = 5) and EphA2−/−mice (total N = 30, each group n = 5) were injected with of 200 μL 1% Evans Blue (Sigma) by tail vein at 48 h post infection. After 2 h Evans Blue administration, mice were transcardially perfused with sterile PBS and then were euthanized. Subsequently, brains were removed and imaged using digital camera.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature (RT). After three wash steps, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min. Subsequently, cells were blocked with 5% Bovine Serum Albumin (BSA) in PBS for 30 min. Then, cells were stained with primary antibody containing anti-EphA2 (Cell Signaling Technology), anti-ZO-1 (protein tech) and anti-DCAF15 (Sigma) for 1 h at RT. After the wash steps, cells were incubated with second Abs containing goat anti-mouse IgG (H&L) Alexa fluor 488 and goat anti-rabbit IgG (H&L) Alexa fluor 594 (Abcam, UK) for 1 h. Anti-fade mounting medium with DAPI (Beyotime Biotechnology, Shanghai, China) was added for 5 min to visualize cell nuclei. Finally, cells were observed using confocal microscopy (Olympus, Tokyo, Japan).
Transmission electron microscopy (TEM)
After 4 h infection, EphA2+/+ and EphA2−/− hCMEC/D3 cells were digested by 0.25% trypsin-EDTA and collected. Next, cells were prefixed with a 3% glutaraldehyde for 24 h and then were postfixed in 1% osmium tetroxide, dehydrated in series acetone, infiltrated in Epon 812 and embedded. Subsequently, the semithin sections were stained with methylene blue and ultrathin sections were cut with diamond knife. Finally, sections were stained with uranyl acetate and lead citrate and examined with JEM-1400-FLASH TEM to observe autophagosome.
Endothelial cell permeability assay in vitro using transwell model
Cells were cultured on Transwell-Clear inserts with a pore diameter of 3 μM (Corning Costar, USA). Cells were incubated for 7-10 days until the stability of trans-endothelial electrical resistance (TEER) (greater than 200Ω/cm2). Next, the upper chamber was infected with S. suis for indicated time and TEER (Ω×cm2) value was detected by the Millicell-ERS electrical resistance system to evaluate its integrity.
Co-immunoprecipitation (Co-IP) and GST affinity-isolation assays
For Co-IP assay, cells were lysed in NP-40 lysis buffer containing PMSF for 4 h. Then, cell lysates were centrifuged at 12,000 × g for 10 min at 4 °C. After centrifugation, supernatants were collected and incubated with indicated Abs followed overnight at 4 °C by the addition of protein A + G agarose for 4 h. After centrifugation at 2500 × g for 5 min at 4 °C, the immunocomplexes were washed and then subjected to immunoblotting analysis. Anti-Myc, anti-HA, anti-His and anti-Flag Abs were used in this study.
For GST affinity-isolation assay, purified GST (10 μg) and GST-DCAF15 (10 μg) recombinant proteins were separately mixed with purified Flag-EphA2 (20 μg) in 500 μL NP-40 lysis buffer overnight at 4 °C followed by the addition of GST resin. After centrifugation at 1000 × g for 5 min at 4 °C, the pellets were washed with NP-40 lysis buffer and then lysed in RIPA lysis buffer (Beyotime Biotechnology, P0013B) for immunoblotting analysis.
Mass spectrometry (MS) analysis
To screen the specific protein with which EphA2 interacts, hCMEC/D3 cells were transfected with Flag-EphA2 followed by S. suis infection for 4 h. After infection, cells were harvested and lysed in NP-40 lysis buffer containing PMSF and N-ethylmaleimide (NEM) with the addition of Anti-DYKDDDDK G1 Affinity Resin (L00432, Gen Script) for 4 h at 4 °C. For the verification of ubiquitinated sites on EphA2, Myc-EphA2 and Flag-DCAF15 were transfected into 293 T cells. After 48 h transfection, cells were harvested and lysed in NP-40 lysis buffer containing PMSF and N-ethylmaleimide (NEM) with the addition of anti-Myc affinity gel (Beyotime Biotechnology, P2285) for 4 h at 4 °C. The immunoprecipitation complexes were subjected to SDS-PAGE and Coomassie Brilliant Blue R250 staining. Subsequently, the regions of the staining gels were mixed and subjected to Liquid Chromatography Mass Spectrometry (LC-MS) analysis in BGI (Beijing, China) to identify the specific interaction protein with EphA2 and ubiquitinated sites of EphA2.
Protein–protein docking analysis
The structural models for the protein interaction complexes presented in this study were generated through protein-protein docking using GRAMM. The amino acid sequences for the docking calculations were derived from the corresponding UniProtKB entries, including DCAF15 (Q66K64), EphA2 (P29317), LKB1 (Q15831), and SIRT1 (Q96EB6). The protein structures used as templates for docking were obtained from the Protein Data Bank (PDB). The resulting docking complexes were analyzed for protein-protein interactions using the PDBe PISA server (https://www.ebi.ac.uk/pdbe/pisa/) and were subsequently visualized using PyMOL (https://pymol.org/).
Statistics and reproducibility
Data are presented as mean ± SD. For in vitro experiments, data were representative of at least three independent biological replicates with triplicate samples. For in vivo experiments, data were representative of three independent biological replicates with three to five mice per group. Statistical differences were analyzed using the unpaired Student’s t-test for comparisons between two groups and one-way ANOVA with an appropriate post hoc test for comparisons among multiple groups. A P-value of ≤ 0.05 was considered as statistical significance.
Ethics statement
Female C57BL/6 J wild type (WT) mice aged 6-8 weeks were bred in-house or purchased from Byrness Weil biotech Ltd (Chongqing, China). EphA2−/− mice generated via CRISPR/Cas9 technology were kindly provided by Genome Technologies Limited (Chongqing, China). All mice were maintained under specific pathogen-free (SPF) conditions. This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Southwest University, Chongqing, China (IACUC-20231215-02). We have complied with all relevant ethical regulations for animal use.
Data availability
All data supporting the findings of this study are available within the paper, including Supplementary Information, Supplementary Table 1 and Supplementary Data. Supplementary Information contains Supplementary Figs. 1-6. Unprocessed blot images can be found in Supplementary Fig. 7, which is included in the Supplementary Information as an uncropped and unedited PDF. Supplementary Table 1 contains all primer sequence information for RT-PCR, siRNA and primers. Source data can be found in Supplementary Data 1. Biological materials (strains, plasmids, antibodies) are available from the corresponding author on reasonable request. Correspondence and requests could be addressed to rdfang@swu.edu.cn. The mass spectrometry data have been deposited to the Proteome X change Consortium via the iPro X partner repository with the dataset identifier PXD070205. URL, https://www.iprox.cn/page/PSV023.html;?url=1762158514070z3pD. Password: eIfn.
References
Al-Obaidi, M. M. J. & Desa, M. N. M. Mechanisms of blood brain barrier disruption by different types of bacteria, and bacterial-host interactions facilitate the bacterial pathogen invading the brain. Cell Mol. Neurobiol. 38, 1349–1368 (2018).
Le Guennec, L., Coureuil, M., Nassif, X. & Bourdoulous, S. Strategies used by bacterial pathogens to cross the blood-brain barrier. Cell Microbiol. 22, e13132 (2020).
Susilawathi, N. M. et al. Streptococcus suis-associated Meningitis, Bali, Indonesia, 2014-2017. Emerg. Infect. Dis. 25, 2235–2242 (2019).
Xiao, G. et al. Streptococcus suis small RNA rss04 contributes to the induction of meningitis by regulating capsule synthesis and by inducing biofilm formation in a mouse infection model. Vet. Microbiol. 199, 111–119 (2017).
Rui, L. et al. The serine/threonine protein kinase of Streptococcus suis serotype 2 affects the ability of the pathogen to penetrate the blood-brain barrier. Cell Microbiol. 20, e12862 (2018).
Liu, H. et al. Streptococcus suis serotype 2 enolase interaction with host brain microvascular endothelial cells and RPSA-induced apoptosis lead to loss of BBB integrity. Vet. Res. 52, 30 (2021).
Pan, Z. et al. SssP1, a Fimbria-like component of Streptococcus suis, binds to the vimentin of host cells and contributes to bacterial meningitis. PLoS Pathog. 18, e1010710 (2022).
Jiang, C. et al. ARF6 promotes Streptococcus suis suilysin induced apoptosis in HBMECs. Int. J. Biol. Macromol. 268, 131839 (2024).
Cao, X. et al. The critical role of NLRP3 inflammasome activation in Streptococcus suis-induced blood-brain barrier disruption. Vet. Microbiol. 295, 110161 (2024).
Kania, A. & Klein, R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 17, 240–256 (2016).
Swidergall, M. et al. EphA2 is a neutrophil receptor for candida albicans that stimulates antifungal activity during oropharyngeal infection. Cell Rep. 28, 423–433.e425 (2019).
Subbarayal, P. et al. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 11, e1004846 (2015).
Lupberger, J. et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17, 589–595 (2011).
Aaron, P. A., Jamklang, M., Uhrig, J. P. & Gelli, A. The blood-brain barrier internalises Cryptococcus neoformans via the EphA2-tyrosine kinase receptor. Cell Microbiol. 20, https://doi.org/10.1111/cmi.12811 (2018).
Darling, T. K. et al. EphA2 contributes to disruption of the blood-brain barrier in cerebral malaria. PLoS Pathog. 16, e1008261 (2020).
Zhou, N. et al. Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc. Res. 82, 113–121 (2011).
Zysk, G. et al. Pneumolysin is the main inducer of cytotoxicity to brain microvascular endothelial cells caused by Streptococcus pneumoniae. Infect. Immun. 69, 845–852 (2001).
Han, J. et al. EphA2 inhibits SRA01/04 cells apoptosis by suppressing autophagy via activating PI3K/Akt/mTOR pathway. Arch. Biochem. Biophys. 711, 109024 (2021).
Kaplan, N. et al. Ciliogenesis and autophagy are coordinately regulated by EphA2 in the cornea to maintain proper epithelial architecture. Ocul. Surf. 21, 193–205 (2021).
Mubthasima, P. P., Singh, S. A. & Kannan, A. Sesamol-mediated targeting of EPHA2 sensitises cervical cancer for cisplatin treatment by regulating mitochondrial dynamics, autophagy, and mitophagy. Mol. Biol. Rep. 51, 949 (2024).
Harly, C. et al. Human γδ T cell sensing of AMPK-dependent metabolic tumor reprogramming through TCR recognition of EphA2. Sci. Immunol. 6, https://doi.org/10.1126/sciimmunol.aba9010 (2021).
Chen, Z. et al. EPHA2 blockade reverses acquired resistance to afatinib induced by EPHA2-mediated MAPK pathway activation in gastric cancer cells and avatar mice. Int. J. Cancer 145, 2440–2449 (2019).
Trelford, C. B. & Shepherd, T. G. LKB1 biology: assessing the therapeutic relevancy of LKB1 inhibitors. Cell Commun. Signal 22, 310 (2024).
Lu, J. et al. The aryl sulfonamide indisulam inhibits gastric cancer cell migration by promoting the ubiquitination and degradation of the transcription factor ZEB1. J. Biol. Chem. 299, 103025 (2023).
Han, T. et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, https://doi.org/10.1126/science.aal3755 (2017).
Ting, T. C. et al. Aryl sulfonamides degrade RBM39 and RBM23 by recruitment to CRL4-DCAF15. Cell Rep. 29, 1499–1510.e1496 (2019).
An, J. et al. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4(CRBN) ubiquitin ligase. Nat. Commun. 8, 15398 (2017).
Narita, T., Weinert, B. T. & Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174 (2019).
Zhao, Y. et al. High expression of EphA2 led to secondary injury by destruction of BBB integrity though the ROCK pathway after diffuse axonal injury. Neurosci. Lett. 736, 135234 (2020).
Thundyil, J. et al. Evidence that the EphA2 receptor exacerbates ischemic brain injury. PLoS One 8, e53528 (2013).
Swidergall, M., Solis, N. V., Lionakis, M. S. & Filler, S. G. EphA2 is an epithelial cell pattern recognition receptor for fungal β-glucans. Nat. Microbiol. 3, 53–61 (2018).
Kottom, T. J., Carmona, E. M. & Limper, A. H. Targeting host tyrosine kinase receptor EphA2 signaling via small-molecule ALW-II-41-27 inhibits macrophage pro-inflammatory signaling responses to Pneumocystis carinii β-glucans. Antimicrob. Agents Chemother. 68, e0081123 (2024).
Zhang, H. et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 3, 1–8 (2018).
Vincenzi, M., Mercurio, F. A. & Leone, M. EPHA2 Receptor as a Possible Therapeutic Target in Viral Infections. Curr. Med. Chem. 31, 5670–5701 (2024).
Zhang, A. et al. EphA2 phosphorylates NLRP3 and inhibits inflammasomes in airway epithelial cells. EMBO Rep. 21, e49666 (2020).
Yang, Z. et al. Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5). Autophagy 17, 3048–3067 (2021).
Wang, G. et al. NOD2-RIP2 signaling alleviates microglial ROS damage and pyroptosis via ULK1-mediated autophagy during Streptococcus pneumonia infection. Neurosci. Lett. 783, 136743 (2022).
Yang, Z. et al. Circular RNA circ-FoxO3 attenuates blood-brain barrier damage by inducing autophagy during ischemia/reperfusion. Mol. Ther. 30, 1275–1287 (2022).
Wani, A. et al. Crocetin promotes clearance of amyloid-β by inducing autophagy via the STK11/LKB1-mediated AMPK pathway. Autophagy 17, 3813–3832 (2021).
Pan, F. et al. Membrane vesicle delivery of a streptococcal M protein disrupts the blood-brain barrier by inducing autophagic endothelial cell death. Proc. Natl. Acad. Sci. USA 120, e2219435120 (2023).
Li, A. et al. USP3 promotes osteosarcoma progression via deubiquitinating EPHA2 and activating the PI3K/AKT signaling pathway. Cell Death Dis. 15, 235 (2024).
Li, X., Wang, F., Huang, L., Yang, M. & Kuang, E. Downregulation of EphA2 stability by RNF5 limits its tumor-suppressive function in HER2-negative breast cancers. Cell Death Dis. 14, 662 (2023).
Dong, X. et al. Tumor suppressor DCAF15 inhibits epithelial-mesenchymal transition by targeting ZEB1 for proteasomal degradation in hepatocellular carcinoma. Aging 13, 10603–10618 (2021).
Waltho, A. et al. K48- and K63-linked ubiquitin chain interactome reveals branch- and length-specific ubiquitin interactors. Life Sci. Alliance 7, https://doi.org/10.26508/lsa.202402740 (2024).
Tracz, M. & Bialek, W. Beyond K48 and K63: non-canonical protein ubiquitination. Cell Mol. Biol. Lett. 26, 1 (2021).
Grothusen, G. P. et al. DCAF15 control of cohesin dynamics sustains acute myeloid leukemia. Nat. Commun. 15, 5604 (2024).
Cui, L. et al. Streptococcus pneumoniae extracellular vesicles aggravate alveolar epithelial barrier disruption via autophagic degradation of OCLN (occludin). Autophagy 20, 1577–1596 (2024).
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2021YFD1800800), National Natural Science Foundation of China (32473027, 32172850, 32473043), Special fund for youth team of the Southwest University (SWU-XJPY202305), National Center of Technology Innovation for pigs (NCTIP-XD/C17), the Chongqing Modern Agricultural Industry Technology System (CQMAITS202512), the Southwest University graduate students research innovation project (SWUB23079).
Author information
Authors and Affiliations
Contributions
Rendong Fang was responsible for conceptualization, study design, supervision and funding supporting; Lianci Peng participated in supervision and manuscript drafting. Xiangru Wang contributed to manuscript revision and provided funding supporting; Hang Yin conducted experiments, data collection, data analysis and manuscript drafting; Shiqi Lang and Yi Lu engaged in experimentation and data collection; Zeyu Zhou, Shuying Ren and Jianqiao Qiu conducted experimentation; Xiaoying Yu performed bioinformatics analysis; Zhiwei Li participated in study design and supervision. All authors approved and contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Kwang-Su Park and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Toshiro Moroishi, Christina Karlsson Rosenthal and Mengtan Xing. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yin, H., Lang, S., Lu, Y. et al. Dcaf15-mediated EphA2 degradation triggers disruption of the blood-brain barrier during Streptococcus suis meningitis. Commun Biol 8, 1838 (2025). https://doi.org/10.1038/s42003-025-09213-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09213-2
