
Abstract
Influenza-associated bacterial super-infections in the lung lead to increased morbidity and mortality. Previous studies have investigated how preceding viral infection causes dysregulation of the innate and adaptive immune systems, leading to increased susceptibility of developing secondary bacterial pneumonia. However, these previous studies cannot account for the spatial context of immune cells in lung. In our study, we employ a spatial transcriptomics platform (10X Genomics Visium) to systematically characterize the coordination of immune cells during super-infection. We compare deconvoluted spatial transcriptomics data between super-infection and single influenza and methicillin-resistant Staphylococcus aureus infections. Consequently, we find that the recruitment of neutrophils and interstitial macrophages from lung parenchyma to the airways is inhibited in super-infection, likely impairing pathogen clearance. Additionally, by analyzing cell colocalization and signaling, we find that the interaction between CD4+ T cells, B cells, and dendritic cells is disrupted by secondary bacterial super-infection. These findings are confirmed by immunofluorescence staining. Our study constructs a spatial sequencing atlas of lung super-infection, highlighting how secondary bacterial challenge significantly impacts recruitment and signaling of immune cells. These findings provide insight into the aberrant inflammation in the super-infected lung and may aid development of therapeutics that target key immune cell recruitment pathways.
Similar content being viewed by others


Introduction
Lower respiratory tract infections are a significant cause of morbidity and mortality globally1,2. Common causes of viral pneumonias include influenza A & B viruses, respiratory syncytial virus, adenoviruses, and parainfluenza virus. Viral infections damage the airway epithelium and create an inflammatory environment in the lung, increasing susceptibility to acquiring secondary bacterial pneumonia3,4,5,6. Secondary bacterial pneumonias are primarily caused by Streptococcus pneumoniae, Haemophilus influenzae, and Staphylococcus aureus with methicillin resistant S. aureus (MRSA) emerging as a leading cause2,7. Secondary bacterial pneumonia with MRSA has resulted in increased morbidity and mortality during both seasonal influenza seasons and during the pandemics of 1918 and 20098,9. Super-infection is characterized by excessive lung inflammation, rather than immune deficiency. Previous studies have characterized shifts in immune cell populations in the lung during super-infection5,10,11,12,13; however, these studies do not systemically characterize transcriptomic changes of immune cell populations within the spatial context of lung tissue, which may contribute to aberrant host defense.
The spatial distribution of immune cells in the lung has a significant impact on the immune response. The structure of the lung is complex and composed of a diverse framework of epithelial, stromal, and immune cells, making it difficult to study the immune cells in the lung within their tissue context. Influenza virus initially replicates in epithelial cells lining the airways with most pathologic changes occurring near the airways with severe infection resulting in interstitial inflammation, diffuse alveolar damage, and necrotizing bronchitis1. While bacterial infections begin in the airways, bacterial pneumonia is primarily characterized by inflammation of the lung parenchyma and alveolar spaces2. This emphasizes the heterogeneity of the spatial organization of immune cells in virus or bacteria-infected lung tissue. Infection in the lung recruits immune cells, such as neutrophils, macrophages, dendritic cells (DCs), T cells, and B cells, causing inflammation in both airways and lung parenchyma. However, it is unclear how influenza and S. aureus superinfection change the spatial organization and coordination of these immune cells, which would consequently impact pathogen clearance, tissue pathology, and immune cell responses. The goal of this study was to systematically address two major questions with spatial information: (1) How does preceding influenza infection disrupt subsequent anti-bacterial responses? And (2) how does secondary bacterial infection impact antiviral immunity?
Currently, it is a challenge to observe immune cell populations and their gene expression in a spatial context within tissue. Histological approaches are commonly employed to examine pathological changes of the lung structure following infection, but they cannot characterize the identity and gene expression of cells. Methods to profile different subsets of immune cells primarily include flow cytometry and transcriptomic approaches, such as single-cell RNA sequencing (scRNA-seq), but these methods do not preserve spatial information. Examining the position of immune cells relative to airways or neighboring cells would provide profound insight into how super-infection alters cell phenotype, location, and signaling in the lung. Spatial transcriptomics (ST) is a technology which can sequence the whole transcriptome and preserve the spatial information of cells simultaneously14. In our study, we used 10x Genomics Visium, a ST platform allowing spatial sequencing and hematoxylin and eosin (H&E) staining on the same tissue slide14. With this technology, we aimed to investigate how influenza and bacteria super-infection change the spatial coordination of immune cells in the lung compared to single viral or bacterial infection. We then confirmed findings using traditional immunofluorescence (IF) staining. This study revealed how super-infection alters the location and crosstalk of key immune cells in the lung, such as neutrophils, macrophages, DCs, T cells, and B cells, compared to single influenza or S. aureus infection and highlights potential pathways for novel therapeutic agents.
Results
Spatial transcriptomics profiling of infected mouse lungs
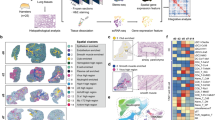
To examine changes in the spatial location and gene expression of immune cells during post-influenza bacterial super-infection, C57BL/6 mice were challenged with either PBS or influenza A/Puerto Rico/8/34 H1N1 virus for 6 days, followed by challenge with either PBS or USA300 MRSA for an additional 24 h (day 7) (Fig. 1A). This model of non-lethal single or super-infection in mice has been previously published in several studies15,16,17,18. As expected, a higher bacterial burden was observed in mice with superinfection compared to those with single infection (Supplementary Fig. 1). Each experimental mouse group had a total of eight replicates, and lung tissues of two replicates underwent formalin-fixed paraffin-embedded (FFPE) processing for ST sequencing and H&E staining. The lung tissues of other replicates were sectioned for IF and immunohistochemistry staining (Fig. 1B).

A Experimental design and treatment groups. The Naïve group was treated with PBS without any pathogens on days 0 and 6. The Staph (S. aureus) only group was only infected with S. aureus on day 6. The Flu only (influenza virus) group was only infected with the influenza virus on day 0. The Flu/Staph group was infected with the influenza virus on day 0 and S. aureus on day 6. The samples are harvested on day 7. Each group had eight mice. The mouse lungs were formalin-fixed and paraffin-embedded and sliced for spatial transcriptomics sequencing or immunofluorescence staining. B ST and data analyses. A set of tissue slides was stained on protein level. Another set was used for ST sequencing. Tissue slides were manually annotated with histology images. C Region-specific analysis focusing on airway and inflammatory regions, spatial communication between spots based on expression of ligands and receptors, and spatial colocalization analysis reveal the spatial coordination of immune cells in the lung. Created in BioRender. Liu, T. (2025) https://BioRender.com/eefoi3e.
Using the ST data from all 8 lung tissue samples, gene expression and spatial location for each spot were decoded. The spots with unique molecular identifier (UMI) counts higher than 100, and the number of genes with non-zero UMI counts higher than 100 were retained for downstream analysis. The number of spots in each sample ranged from 3190 to 4069 (Supplementary Table 1). The proportion (density) of each cell type in each spot was estimated with robust cell type decomposition (RCTD)19, based on gene expression profile. Regions of airway (bronchus) and parenchymal inflammation were annotated in a semi-manual manner with the information of both the H&E histology image and proportion of cell types (estimated with gene expression) (Fig. 1B). Downstream analysis focused on airway and inflammatory regions separately. Spatial communication and cell colocalization were employed to dissect the spatial coordination of immune cells (Fig. 1C).
Deconvolution of spatial transcriptomics data identified spatial distribution of cells (including airway and inflammatory regions) in infected mouse lungs
After combining the spots of ST data from 8 samples, apparent batch effects were observed between treatment groups, and minor batch effects were observed between the two replicates from the same condition (Supplementary Fig. 2A). To evaluate the quality of data, we corrected the batch effect. The spots were then classified into 8 clusters (Fig. 2A). These clusters were annotated as specific anatomical structures according to their gene expression (Supplementary Fig. 2B). Notably, the annotation of these clusters agreed with the histological features of the corresponding H&E image (Supplementary Fig. 3, Fig. 2A).

A Unsupervised clustering of spot-level spatial gene expression identified different anatomical structures of the infected lung. B Spatial deconvolution with RCTD identified the proportion or density of detailed cell types. C Marker genes employed to identify different cell types and estimation of cell type proportions. D The deconvolution-estimated proportion of airway epithelial cells (left, sum of goblet, club, and ciliated cells) and immune cells (right, sum of all types of immune cells) in different manually annotated regions in the lung. E The final annotation of different regions combining both manual annotation and cell type proportions (See Methods).
In addition to the rough annotation of anatomical structures, we also did cell-type-level annotation by estimating the proportion (or density) of each cell type in each spot in ST data (Fig. 2B). The density was estimated with spatial deconvolution with a reference scRNA-seq mouse lung dataset with influenza virus infection (Supplementary Fig. 4A)20. The estimated proportion of interstitial macrophages (IMs), airway epithelial cells (goblet cell, club cell, and ciliated cell), and other cell types aligned well with the observation on histology images (Fig. 2B, Supplementary Fig. 4B–D). The markers used to identify these cell types were shown in Fig. 2C. A higher proportion of immune cells and airway epithelial cells were observed in manual-annotated inflammatory regions and airway regions (Fig. 2D, Supplementary Data 1).
The information of cell type proportion (gene expression information) and manual annotation (histological morphology) was combined to annotate the airway and inflammatory regions (Fig. 2E, Supplementary Fig. 4E). Downstream analyses focused on regions near the airway and parenchymal inflammatory regions because they are the initial target of pathogens and the primary region of immune cell infiltration. Distinct patterns of inflammation in these two regions were observed between single bacterial and single viral infection (Supplementary Fig. 5). During influenza infection, we observed increased pathology and presence of influenza virus near the airways; however, infection with S. aureus resulted in a more diffuse pattern with some bacteria invading the airways (Supplementary Fig. 6). Following infection, immune cell populations were elevated in regions both adjacent to and distant from the large airways, but the alveolar epithelial cells (AT1, AT2) were reduced in infected mice (Supplementary Fig. 7). This suggests that airway associated infection and inflammation ablated parenchymal alveolar cells. Overall, these data demonstrate that the spatial distribution of cells was significantly changed by infection, necessitating the analysis of airway and parenchymal inflammatory regions separately. We next compared the spatial distribution and coordination of immune cells between single-infection and super-infection samples.
Neutrophil recruitment to the lung in response to S. aureus is attenuated in super-infection
Neutrophils are activated in the early stage of viral or bacterial infection and are an important component of innate immunity for host defense. The airway epithelium is the primary site of viral infection (Supplementary Fig. 6). Immune cells migrate from the pulmonary vasculature to the central airways in both bacterial and viral infection. Thus, we focused on airway regions and inflammatory regions in vascular-rich lung parenchyma, which are major sites of immune cell infiltration.
The proportion of neutrophils in regions near airways was reduced in super-infection compared with single bacterial infection, suggesting decreased neutrophil recruitment to airways during super-infection (Fig. 3A, Supplementary Data 2, Supplementary Fig. 8A). Similarly, the proportion of neutrophils in the inflammatory regions of the lung parenchyma also decreased during super-infection compared with single bacterial infection (Fig. 3B, Supplementary Data 3, Supplementary Fig. 8B). This is supported by quantification of neutrophils (Ly6G+) by IF staining (Fig. 4). A significant reduction in the density and area of neutrophils (Ly6G+ cells) was observed in regions near large airways and the parenchyma (Fig. 4F, G) in super-infected lungs versus single infection. This was readily apparent in regions where both the influenza virus and S. aureus were present (co-infection). Impaired neutrophil recruitment to important sites of infection (airway, parenchyma) may contribute to dysregulation of immunity and increased bacterial burden in superinfection5.

A The proportion (ranges from 0 to 1) of neutrophils in the regions within 50 µm from the airway epithelium. P values generated by the two-sample T test. B The proportion (ranges from 0 to 1) of neutrophils in inflammatory regions in the lung parenchyma. The line inside the box was the median value. The top and bottom lines of the box were the 25th and 75th quantiles. The lines extend 1.5 interquartile ranges from the 25th and 75th quantiles. C The strength of the CXCL5-CXCR2 signal sent from the airway epithelium in S (S. aureus only) and FS (influenza virus/S. aureus). D The strength of the CXCL1-CXCR2 signal received by inflammatory regions in the lung parenchyma. E The strength of the NF signal received by inflammatory regions in the lung parenchyma. The signal strength in C, D, E is estimated with COMMOT, which considers the spatial location (See “Methods”).

Representative 200× pictures show Ly6G neutrophils near bronchi of A mock-infected mice, B influenza A-infected bronchi, C influenza-infected bronchi with low bacterial infection, D influenza A-infected bronchi and parenchymal tissue with areas of profuse bacterial infection, and E parenchymal tissue infected with S. aureus. Bronchi are depicted with a white asterisk. Scale bars represent 100 µm. Morphometric analysis shows F neutrophils per mm2 and G area covered by Ly6G stain in peri-bronchial areas of non-infected and infected mice. Mice were mock infected (filled circles) or infected with influenza A (empty circles), S. aureus (filled diamonds), or co-infected with influenza A and S. aureus: peribronchial areas with dominant influenza infection (filled squares) or with viral/bacterial co-infection (empty squares). n = 1–3 200× pictures per mice from 4 mice per group. Statistical significance was calculated by a two-tailed t-test. *p < 0.05; **p < 0.005; ****p ≤ 0.0001. The error bar indicates the standard deviation of the data from different mice.
Further, we estimated the spatial cell communication in the airway epithelium and parenchymal inflammatory regions with COMMOT21. We identified that three signals correlated with neutrophils (CXCL5-CXCR2, CXCL1-CXCR2, and TNF-TNFR, Supplementary Fig. 9) were also reduced in super-infection. Signaling via the CXCL5-CXCR2 axis, a crucial pathway for neutrophil recruitment originating from the airway epithelium, was significantly reduced in the airway epithelium in samples with super-infection compared to those subjected to bacterial infection alone (Fig. 3C, Supplementary Fig. 10). Additionally, the signal received in parenchymal inflammatory regions through CXCL1-CXCR2 and the TNF-TNFR axis was also reduced in the super-infection group compared with single bacterial infection (Fig. 3D, E, Supplementary Fig. 10, Supplementary Data 4). The decreased signals related to CXCR2 in the airway and parenchymal regions indicated a chemotactic deficiency for recruitment of neutrophils. The reduction of TNF signals received by parenchymal inflammatory regions suggests a weakened defense against S. aureus. Our data show that the recruitment of neutrophils to the sites of infection is inhibited in super-infection infection, which could impair the host anti-bacterial immune responses.
Macrophage recruitment in response to influenza infection is aberrant in super-infection
IMs, found in the lung parenchyma and bronchial interstitium, are a crucial immune cell population for controlling viral spread and inflammation in the lung22. The proportion of IMs in regions proximal to airways was reduced in super-infection compared to single influenza virus infection (Fig. 5A, Supplementary Data 5, Supplementary Fig. 11A). Similarly, the proportion of IMs in inflammatory regions of the lung parenchyma was also lower in super-infection compared to single viral infection (Fig. 5B, Supplementary Data 6, Supplementary Fig. 11B). These data indicate that secondary bacterial challenge inhibits recruitment of IMs to infected regions in the lung.

A The proportion (ranges from 0 to 1) of macrophages in the regions within 50 μm from the airway epithelium. P values generated by the two-sample t-test. B The proportion of (ranges from 0 to 1) macrophages in inflammatory regions in the lung parenchyma. The line inside the box was the median value. The top and bottom lines of the box were the 25th and 75th quantiles. The lines extend 1.5 interquartile range from 25th and 75th quantiles. C The strength of the CCL5-CCR5 signal sent from the airway epithelium in sample F (influenza virus only) and FS (influenza virus/S. aureus). D The strength of the CCL2-CCR2 signal received by inflammatory regions in the lung parenchyma. E The strength of the VCAM signal received by inflammatory regions in the lung parenchyma. The signal strength in C, D, E is estimated with COMMOT, which considers the spatial location.
Critical cell-to-cell signals for the recruitment of monocytes/IMs were also reduced in super-infection compared to single influenza infection. The CCL5-CCR5 signaling pathway was reduced in the airway epithelium during super-infection (Fig. 5C, Supplementary Fig. 12). The inflammatory regions of the lung parenchyma also received less signal through VCAM and CCL2-CCR2, which direct macrophages to infected regions (Fig. 5D, E, Supplementary Data 7, Supplementary Fig. 12). VCAM is important for the adhesion of monocytes to endothelial cells, which facilitates the recruitment of monocytes from blood to tissue and their differentiation into IMs. Notably, these three signals are also correlated with IMs in the ST data (Supplementary Fig. 9). The reduction of these 3 signals in regions near airway or parenchymal inflammatory regions likely impairs chemotaxis and extravasation of monocytes and IMs, reducing recruitment of IMs in the super-infected lung.
Resident alveolar macrophages (AMs) in the lung proliferate in the early stages of infection and may play a role in our model. We observed a lower proportion of AMs in influenza infection and super-infection, compared with lung tissues without infection or those with only bacterial infection (Supplementary Fig. 13). This indicated that AMs were depleted by preceding viral infection, which is consistent with previous findings in whole lung samples11.
Spatial coordination between CD4+ T cell and B cells or DCs is disrupted in super-infection
After observing deficiencies in innate immune cell spatial recruitment during super-infection, we next aimed to characterize changes in adaptive immune cells during super-infection compared to single viral infection. Similar as IMs, the proportion of CD8+ T cells was decreased in super-infection compared with single influenza infection (Supplementary Fig. 14).
The maturation of adaptive immune cells largely relies on signals from other immune cells, so we focused on the spatial coordination between lymphocytes and other immune cells (CD4+ T cells, B cells, DCs, and IMs). We calculated the spatial correlation between all immune cells in the inflammatory regions of each sample (Supplementary Fig. 15). High spatial correlation indicates that both types of cells tend to colocalize in one spot. We then focused on samples with influenza infection, where activation of adaptive immunity was expected. The colocalization between CD4+ T cells and some antigen-presenting cells (B cells, DCs, and IMs) observed during influenza infection was reduced in super-infection compared with single influenza infection.
Specifically, the spatial correlation between CD4+ T cells and B cells was reduced in super-infection samples compared with single virus infection samples (Fig. 6A, C, Supplementary Data 8, Supplementary Fig. 16B). CD4+ T cells interact with B cells to promote the formation of B cell memory and antibody production, and this interaction also impacts the maturation and differentiation of CD4+ T cells. Similarly, the spatial correlation between CD4+ T cells and DCs (Fig. 6B, Supplementary Fig. 16A, C, D) and that between CD4+ T cells and IMs (Supplementary Fig. 17) were reduced in super-infection compared to single viral infection, but the colocalization between CD4+ T cells and B cells was more significantly reduced (Fig. 6A, C). The disruption of CD4+ T cell and B cell interaction was further evidenced by a reduction of ICOS and CD40 signaling (Fig. 6D, Supplementary Data 9, Supplementary Fig. 16E), which are critical for CD4+ T cell polarization and B cell antibody production, in super-infection compared to single viral infection23.

A Spatial correlation between CD4+ T cells and B cells in inflammatory regions in samples infected with the influenza virus or infected with both the influenza virus and S. aureus. The score was derived from marker genes for CD4+ T cells and B cells (See Methods). P values generated by linear regression. B Spatial correlation between the score for CD4+ T cells and dendritic cells in inflammatory regions in samples infected with the influenza virus or infected with both influenza virus and S. aureus. C The proportion of CD4+ T cells and B cells in spots in inflammatory regions. D The strength of the signal ICOS-ICOSL in the inflammatory regions. The size of the dot indicates the proportion of spots with non-zero signal strength. The color of the dot indicates the average signal strength in spots in one sample. E The expression of Th17-related genes (Rorc, Il17a, Ccr6, Il22, Il17f) and Th1-related genes (Ccl5, Ms4a4b, Cxcr3, Ifng, Tbx21, Gzmk) in regions near airways for each sample.
The interaction between CD4+ T cells and other immune cells with antigen-presenting functions, including B cells, DCs, and IMs, is critical for polarization of CD4+ T cells. Thus, we also profiled the spatial distribution of polarized CD4+ T cells, Th1 and Th17 cells, which are important in defense against viral and bacterial infections. Th1 cells are important for cellular immunity to viral infection. The expression of transcriptomic markers for Th1 cells in airway regions decreased in super-infection compared to single viral infection (Fig. 6E, Supplementary Data 10, 11). The markers for Th17 cells, which are significant in bacterial infection, also decreased during super-infection compared to single bacterial infection (Fig. 6E). These data show that the colocalization of CD4+ T cells with other immune cells (B cells, DCs, and IMs) is disrupted during super-infection, which may negatively impact the interaction between T and B cells.
Super-infection inhibits influenza virus-induced formation of follicle-like immune hubs in the mouse lung
Interactions between CD4+ T cells, DCs, and B cells are common in lymphoid follicles, which are sites of CD4+ T cell and B cell activation and memory formation, and these lymphoid follicles can form in the lung during infection24,25,26. Thus, we identified the follicle-like immune hubs (precursor to inducible bronchus-associated lymphoid tissue, iBALT) in the inflammatory regions of infected mouse lung with the expression of CXCL13. CXCL13 is an important chemokine expressed by follicular DCs, a key immune cell population for the formation of lymphoid follicles. We observed that these follicle-like immune hubs, characterized by high expression of CXCL13 and enrichment of CD4+ T cells, B cells, and DCs, were found only in super-infection and single viral infected samples (Fig. 7A, B, Supplementary Data 12). The abundance of these follicle-like immune hubs was significantly reduced in super-infected samples compared to single viral infected samples. The signal of CXCL13-CXCR5, which is important for the formation of lymphoid follicles, was also reduced in super-infection (Fig. 7C, D, Supplementary Data 13). Specifically, the CXCL13-CXCR5 signal was sent from immune hubs to nearby regions in influenza virus-infected samples. However, the directionality of this signal in super-infected samples was not clear due to a weak signal (Fig. 7C). Further, the signal for other immune hub-associated ligand and receptor pairs was similar in influenza virus versus super-infected lungs (Fig. 7E, Supplementary Data 14). These cytokine or chemokine pairs are spatially correlated with T cells, B cells, and lymphatic endothelial cells (Supplementary Fig. 18). It is possible that inhibited formation of these immune hubs may decrease the formation of iBALT in the lung, which are important sites for antigen-specific T and B cell formation27. Decreased formation of immune hubs enriched with T cells (CD3+) and B cells (CD19+) was also observed in IF-stained slides (Fig. 8A). There was a significant reduction in the abundance of T cells and B cells in super-infection compared with single influenza infection (Fig. 8B, C, Supplementary Data 15).

A Expression of CXCL13 in the inflammatory regions in samples only infected with the influenza virus or infected with both the influenza virus and S. aureus. B The regions of immune hubs characterized by the expression of CXCL13 and the proportion of CD4+ T cells, B cells, and dendritic cells. C The directional CXCL13-CXCR5 signal is shown as arrows. The length of the arrows indicates the strength of the signal. No arrow was observed in super-infection samples (FS_1, FS_2) since the signal was very weak. D The strength of the CXCL13-CXCR5 signal in the inflammatory regions in all 8 samples. The size of the dot indicates the proportion of spots with non-zero signal strength. The color of the dot indicates the mean signal strength of all spots in the inflammatory regions of each sample. E Signals related to cytokines or chemokines and their receptors were reduced in super-infection samples.

A The immunofluorescence staining of B cells (CD19+), T cells (CD3+), and neutrophils (Ly6G+). Representative 200x pictures were selected. B, C Morphometric evaluation for the area of B cells and T cells in different infection conditions. P values generated by the two-sample T test. The error bar indicates the standard deviation of the data from different mice.
Discussion
Given the continued prevalence of seasonal influenza and the emergence of COVID-19, which are frequently coupled with secondary bacterial infection, there is increased interest in examining how viral infection impacts secondary bacterial pneumonia. Further, how bacterial super-infections alter immunity against preceding viral pathogens is less understood. Previous knowledge regarding super-infections in the lung has primarily relied on the use of single cell suspensions or homogenized lung tissue15,16,17,18. To our knowledge, this is the first bioinformatic study to systematically examine the immunological interactions that occur during superinfection within the spatial architecture of the lung.
Previous studies have shown that immune responses to bacterial infections in the lung are impaired by preceding viral infections10,17,28,29,30,31,32. Several mechanisms for susceptibility have been proposed, including dysregulation of innate and adaptive immune mediators that promote anti-bacterial immunity. Recruitment of neutrophils to the lung is impaired during superinfection compared to single viral or bacterial infection5,11,13. Alternatively, neutrophil numbers in bronchoalveolar lavage were shown to be greater in super-infected mice versus single infection with either influenza virus or S. aureus17. However, we observed decreased recruitment of neutrophils to the airway or inflammatory regions during super-infection compared to single bacterial infection in a region-specific manner (both ST and IF staining). This is likely due to impaired cell-to-cell communication (CXCL5-CXCR2, CXCL1-CXCR2) in the lungs during superinfection compared to single bacterial infection. Interestingly, these signals were directly associated with neutrophils in our data, suggesting a key role in the regulation of these cells. Additionally, expression of CXCL5 is concentrated around the airways, whereas the chemokine receptor is found in parenchymal regions with inflammation, suggesting migration of neutrophils towards the airways during bacterial infection. This is consistent with previous findings that observed decreased neutrophil recruitment in the lungs during post-influenza super-infection with S. pneumoniae due to type I IFNs induced by influenza infection, attenuating neutrophil chemokine pathways13. Additionally, rescuing these pathways by adding exogenous KC (CXCL1) and MIP2 (CXCL2) during super-infection restored neutrophil function and reduced lung bacterial burden13. Therefore, targeting the chemokine-chemokine receptor pathways found in our analysis may also restore proper neutrophil localization.
In addition to the aberrant recruitment of neutrophils in superinfection, we also profiled macrophages. The proportion of AMs was decreased in influenza infection and super-infection compared to uninfected controls and single bacterial infection. This finding is consistent with prior work and may increase susceptibility to secondary bacterial infection11. Additionally, we observed an increased proportion of IMs near airways and immune regions during single influenza infection compared with super-infection and all other groups. The CCL5-CCR5, CCL2-CCR2, and VCAM-VLA4 pathways, which are key regulators of macrophage migration in the lung, were impaired during super-infection compared to single influenza infection33. While CCL5 can be expressed by a variety of cell types, the main expressers during influenza virus infection appear to be airway epithelial cells and other cells proximal to the airways, such as T cells and macrophages34. However, expression of VCAM and CCR2 is upregulated in parenchymal areas with inflammation during influenza virus infection. This suggests that the impaired recruitment of IMs in the lung during superinfection is likely due to impaired cell-to-cell communication networks. Interestingly, these key mediators are most associated with IMs in our data. IMs are key immune cells for control of viral infection, and deficiency could result in increased viral burden in superinfection22. Overall, we demonstrated that recruitment of key innate immune cells involved in bacterial or viral clearance was impaired in super-infection, suggesting a mechanism for impaired innate immunity during influenza-associated secondary bacterial pneumonia5.
We have also profiled adaptive immune cells. Following influenza infection, lymphocyte aggregation occurs primarily near the bronchi in the lung, eventually forming iBALT27. While iBALT is typically not observed until 14 days post-influenza infection, we observed aggregation of lymphocytes (follicle-like immune hubs) in the super-infected and single influenza virus-infected samples at day 7 post-infection. During the early stages of iBALT formation, CXCL13-producing follicular DCs attract CXCR5+ lymphocytes27. Therefore, the concentrated CXCL13 expression in the immune hubs of the lung samples is consistent with early formation of iBALT. During the formation of iBALT or other lymphocyte aggregations, B cells co-localize with CD4+ T cells and follicular DCs, which can be seen in the influenza virus-infected samples. Interestingly, co-localization of CD4+ T cells with B cells and ICOS signaling decreased during super-infection compared to single virus infection, potentially impacting T and B cell differentiation and responses to subsequent infection. Additionally, the overlap of CXCL13 and CXCR5 decreased in super-infection compared to influenza virus infection, further suggesting that the integrity of these immune hubs within the tissue is impaired due to secondary bacterial pneumonia. Compromised immune hub integrity could hinder the polarization of CD4+ T cells to Th1 and Th17 phenotypes. Previous findings from Kudva et al. demonstrated that previous influenza infection impairs the Th17 pathway (IL-17A, IL-22, IL-23, and ROR\(\alpha\)) activation during super-infection, which is a crucial pathway for promoting immunity to S. aureus17. Prior work employed analysis of lung homogenates. We also observed a decrease in Th17 signal in the super-infection samples compared to the single bacterial infection samples in this ST study, potentially increasing susceptibility to severe bacterial pneumonia. Similarly, we observed that secondary bacterial challenge decreased Th1 expression compared with single viral infection samples, likely impacting anti-viral responses during super-infection. Additionally, we observed a decrease in the proportion of CD8+ T cells in super-infection compared to single virus infection, which could impact protection for preceding viral infection12,35. Overall, we found that secondary bacterial challenge impacts the spatial location of adaptive immune cells in the lung, which may affect T and B cell memory formation and differentiation.
We have recognized limitations of this study. The 10x Genomics Visium ST platform can only measure gene expression in spots with a 55 µm diameter, so each spot will capture multiple cells. This could limit the accuracy of analyses. Although some high-resolution technologies have come out, such as 10x Genomics Visium HD and Xenium, these technologies suffer from the problem of low efficiency in capturing molecules. As a result, we used the 10x Genomics Visium platform for this study. To reduce the effect of low resolution, we used scRNA-seq reference data to deconvolute the cell type proportion of each spot20. With the future advancement of ST technologies, we will be able to further elucidate the molecular mechanisms driving the disruption of important pathways in the lung during superinfection.
Overall, our study presents a spatial sequencing atlas of the mouse lung with single viral or bacterial infection or super-infection. We demonstrated that super-infection significantly alters the recruitment and cell-to-cell signaling of both the innate and adaptive immune cell compartments. This immune dysregulation correlates with altered bacterial burden and increased severity of superinfection. Additionally, the disruption of immune hub integrity during super-infection may impact adaptive immune cell activation and differentiation, affecting future responses to infection in the lung. This ST study further characterizes the dysregulation of immune cells in superinfection, compared with bacterial or viral single infection, providing insight into the development of novel therapeutic agents. Based on the findings from our study, we propose that developing therapeutics to target crucial immune cell recruitment pathways may improve patient morbidity and mortality outcomes from superinfection.
Methods
Mouse model of super-infection
Mice used in this study were 10–12-week-old male C57BL/6 mice purchased from Jackson Laboratories (Bar Harbor, ME). On day 0, mice were inoculated by oropharyngeal aspiration with 50 µL of PBS vehicle or 1000 PFUs of influenza A/Puerto Rico/8/34 H1N1 virus. On day 6, mice were inoculated by oropharyngeal aspiration with 50 µL of PBS vehicle or 5 × 107 CFUs of USA300 MRSA suspended in PBS and harvested a day later. In total 32 mice were used with 8 mice per infection group. ST was performed on 2 mice per condition selected from 4 mice based upon histology clarity and depth. Immunostaining was performed on 4 mice per condition. No animals or data points were excluded from analyses. Formal randomization was not performed, confounders were not formally controlled, and samples were not blinded for ST. For immunostaining, images were obtained and scored in a sample blinded manner. All mice were maintained under specific-pathogen free conditions at UPMC Children’s Hospital of Pittsburgh, and all animal studies were conducted with approval from the University of Pittsburgh Institutional Animal Care and Use committee (protocol #23073501). Humane endpoints include weight loss greater than 25% of initial weight. No mice met this condition in the study. Mice were euthanized via pentobarbital injection followed by exsanguination. We have complied with all relevant ethical regulations for animal use.
Sample collection and processing
Following euthanasia, the left lung lobe was inflated and fixed in 10% neutral buffered formalin solution for 48 h before transfer to 70% ethanol for tissue processing. Tissues were sent to the Histology Core at UPMC Children’s Hospital of Pittsburgh, where they were paraffin-embedded and sectioned on 10x Genomics Visium slides and H&E stained following 10x Genomics Visium Spatial FFPE tissue processing protocol (Pleasanton, CA). H&E-stained slides were imaged at 40× magnification on a Leica Aperio CS2 digital slide scanner. The images were analyzed in QuPath36, an open-source software used to analyze whole image slides. Qupath has built-in algorithms to detect cells and tissues, which can be classified using a machine learning based algorithm to identify objects using training datasets. For this study, control (uninfected) and influenza virus-infected lung sections were used to train Qupath 0.5.0 to identify healthy parenchyma, inflammation/immune cell infiltration, airways, and blood vessels. Lung sections were manually traced in the Qupath software, and the Qupath cell detection algorithm was applied to identify all lung nucleated cells. The trained machine learning algorithm was then applied to all lung sections included in this study to identify which lung nucleated cells were airways, blood vessels, healthy parenchyma, and inflammation/immune cell infiltration. A heatmap indicating areas of inflammation was overlayed onto the lung images. In addition, lung pathology was scored in a sample-blinded fashion using a 0–4 scale for inflammation associated with airways, vasculature, or parenchyma.
IHC staining of mouse lung lobes
Unstained 5 µm sections of left lobes from mice infected as above were de-paraffinized and rehydrated in subsequent washes of xylene, 100%, 95%, 70%, and 50% ethanol and diH2O. Antigen retrieval was performed using sodium citrate buffer (10 mM sodium citrate, 0.05% Tween, pH = 6.0) at 60 °C. Slides were blocked for 1 h at room temperature (RT) in 1x Tris-buffered saline (TBS) with 1% bovine serum albumin (BSA) and 10% donkey serum. Slides were then incubated overnight at 4 °C with primary antibody in 1x TBS with 1% BSA at the following dilutions: 1:300 of goat polyclonal anti-influenza A virus (ab20841, Abcam, Waltham, MA) or 1:200 rabbit polyclonal anti-S. aureus antibody (ab20920, Abcam, Waltham, MA). Slides were washed three times for 5 min with 1x TBS. Slides were incubated for 1 h at RT with secondary antibody in 1x TBS with 1% BSA at the following dilutions: 1:1000 donkey anti-goat IgG H&L (ab6886, Abcam, Waltham, MA) or 1:500 donkey anti-rabbit IgG H&L (ab6803, Abcam, Waltham, MA). Slides were developed with Fast Red substrate kit (ab64254, Abcam, Waltham, MA) for 15 min (influenza) or 20 min (S. aureus). Slides were counterstained with H&E counterstain kit (ab245880, Abcam, Waltham, MA) and were mounted with aqueous mounting media (ab64230, Abcam, Waltham, MA) and allowed to dry overnight before imaging.
Immunofluorescence staining
Five micrometers formalin-fixed paraffin sections were incubated at 60 °C overnight for deparaffinization. The tissue sections were quickly transferred to xylene and gradually hydrated by sequential transfer to absolute alcohol, 96% alcohol, 70% alcohol, and finally water. Subsequently, the sections were immersed in Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA) and boiled for 2 h. Nonspecific binding was blocked with 5% normal donkey serum in TBS containing 0.5% Triton X-100 for 40 min at RT in a humidified chamber. Primary antibodies at appropriate concentrations were then added to these sections and incubated at 4 °C overnight. This was followed by washing with PBS and incubation with a secondary antibody at RT for 2 h. Finally, the slides were rinsed for 1 h in PBS and mounted with Vectashield antifade mounting medium with DAPI (OM-1200, Vector Laboratories, Burlingame, CA). Images were acquired with a Zeiss Axioplan 2 microscope connected to a Hamamatsu camera.
Raw sequencing data processing
Raw sequencing data were processed with Space Ranger (10x Genomics, Pleasanton, CA). Sequencing reads were mapped to the mouse genome reference. The output from Space Ranger contained a gene \(\times\) spot matrix and the x and y location for each spot. The raw counts of genes of all spots from 8 samples were pooled together and normalized. The sum of counts for all genes was normalized in each spot.
Clustering of ST data and correction of batch effect
The top 2000 highly variable genes were selected with the function “FindVariableFeatures” in Seurat package (Version 4.4.0)37,38. Principal component analysis was performed on these highly variable genes. The FindNeighbors function in Seurat package was employed to construct the nearest neighbor graph. Then the spots in each sample were classified into several clusters with “FindClusters” in the Seurat package. The clusters were classified by their characteristics of gene expression in an unsupervised way. The summary of gene expression for each cell was visualized in two dimensions with function “RunUMAP”. To evaluate the ability of ST to classify different anatomical structures, different samples were integrated and homogeneous clusters were identified. The package harmony (version 0.1.1)39, rliger (version 2.1.0)40, PRECAST (version 1.6.6)41 and “MNN” function of the package batchelor (version 1.16.0)42 were employed to correct the batch effect. The parameters for batch corrections models were tuned with the performance to avoid over-correction. The parameters are listed (harmony: Lambda = 1, theta = 0.7, rliger: k = 25, lambda = 4, PRECAST: K = 11, MNN: k = 20, d = 30). After integrating all the samples, the marker genes for each cluster were identified with the “FindMarker” function in Seurat with Wilcoxon test. The clusters were annotated according to the marker genes for specific anatomical regions.
Cell type deconvolution for each spot
The proportion of each cell type in each spot was estimated with RCTD19. The reference scRNA data were employed to derive the gene expression features for each cell type, which were employed to infer the cell type composition in each spot. The reference of scRNA data was a public dataset of mouse lung with influenza virus infection20. The cell type in scRNA data was reannotated according to the marker gene for each cluster. RCTD deconvolution was performed with R package spacexr (version 2.2.1) with default parameters.
Manual annotation for airway and inflammatory regions
First, airway regions were manually annotated; then, gene expression information was included by considering the cell type proportion estimated from RCTD deconvolution. The spots with at least one type of airway epithelial cell in the top 2 dominant cells of these spots were classified as airway regions. Manual annotation was compared with the annotation based on gene expression side by side. The slides were relabeled in loupe browser with the consideration of both gene expression and histology images. The airway regions classified by gene expression have overlap with the airway regions on histology image in re-check. Some small airways were missed in the first manual annotation. These regions were added to airway regions on re-check. The inflammatory regions were also annotated follow the same workflow. The spots with the sum of proportion of all immune cells larger than 35% were classified as inflammatory regions.
Airway and parenchyma inflammatory region classification
The distance from each spot to the nearest airway region was calculated. The spots within 50 µm from airway regions were classified as peri-airway regions. The proportion of IM and neutrophils of spots in peri-airway regions were compared between different groups. The inflammatory regions in lung parenchyma were defined as inflammatory regions which are more than 50 µm away from the airway regions. The proportion of neutrophils and IMs in these regions was compared between different groups.
Spatial cell communication and signaling analysis
Spatial communication between two spots was estimated with COMMOT (Version 0.0.3)21. All ligand receptor pairs in the CellChat database which are expressed in more than 5% of the spots were included in the analyses43. This method provides the signal strength for each ligand receptor pair and between every two spots. COMMOT considered both the spatial relationship between spots and the expression of genes corresponding to these ligand receptor pairs. The strength of specific ligand receptor pairs of spots in peri-airway regions or those of spots in parenchymal inflammatory regions was compared between different groups.
Spatial colocalization analysis
Pearson’s correlation between the proportions of two types of immune cells across spots in inflammatory regions in each sample was calculated. The proportions of each cell type in inflammatory regions within each sample were independent variables. High correlation between two types of cells indicated that they exist in one spot. However, the proportion of two types of cells in those spots was a dependent variable. So, the normally distributed gene module scores for marker genes of CD4+ T cell (Ccr7, Tcf7, Cd4, Il7r, Cd3d), B cell (Ms4a1, Cd19, Ighm, Bank1, Pax5), and classical DC (Itgae, Xcr1, Clec9a, Itgax, Cd74, H2-Eb1, H2-Ab1, H2-Aa) were employed to model the colocalization between cells. The gene module scores of two types of cells were normally distributed and independent, which made it reasonable to use a linear regression model. Similarly, the high correlation between gene module scores of the two types of cells indicated that these two types of cells colocalize in one spot. All analyses were performed with R (Version 4.3.0).
Statistics and reproducibility
We used a two-sample t-test to evaluate the difference between cell type proportions in between each experimental condition (two mice per condition). Spatial deconvolution with RCTD estimated the proportion of each cell type in each spot. t-test was performed on the spot level proportions of each sample with ST data. For IF data, the area and density of specific markers were quantified in each lung section. 1–3 pictures were taken for each mouse, and 4 mice were included from each condition. Two-sample t-test was performed on area or density of specific markers. The data met the following assumptions: they were continuous, independent, normally distributed, and the variances were equal. The study was repeated in groups of 16 mice (4 per condition). R software was used throughout.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All spatial transcriptomics data of mouse lung in this study is available in zenodo (https://zenodo.org/records/17486453)44. The source data for figures is provided in the Supplementary Data file.
Code availability
All code, documentation, and tutorial are available at https://github.com/TianhaoLiu829/Superinfection_ST.git.
References
Pritt, B. S. & Aubry, M. C. Histopathology of viral infections of the lung. Semin. Diagn. Pathol. 34, 510–517 (2017).
Torres, A. et al. Pneumonia. Nat. Rev. Dis. Prim. 7, 25 (2021).
Atkin-Smith, G. K., Duan, M., Chen, W. & Poon, I. K. H. The induction and consequences of influenza A virus-induced cell death. Cell Death Dis. 9, 1002 (2018).
Cipolla, E. M., Huckestein, B. R. & Alcorn, J. F. Influenza sequelae: from immune modulation to persistent alveolitis. Clin. Sci. 134, 1697–1714 (2020).
Robinson, K. M., Kolls, J. K. & Alcorn, J. F. The immunology of influenza virus-associated bacterial pneumonia. Curr. Opin. Immunol. 34, 59–67 (2015).
Rynda-Apple, A., Robinson, K. M. & Alcorn, J. F. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect. Immun. 83, 3764–3770 (2015).
Morris, D. E., Cleary, D. W. & Clarke, S. C. Secondary bacterial infections associated with influenza pandemics. Front. Microbiol. 8, 1041 (2017).
Pickens, C. O. et al. Bacterial superinfection pneumonia in patients mechanically ventilated for COVID-19 pneumonia. Am. J. Respir. Crit. Care Med. 204, 921–932 (2021).
Morens, D. M., Taubenberger, J. K. & Fauci, A. S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–970 (2008).
Small, C. L. et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 184, 2048–2056 (2010).
Ghoneim, H. E., Thomas, P. G. & McCullers, J. A. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 191, 1250–1259 (2013).
Blevins, L. K. et al. Coinfection with Streptococcus pneumoniae negatively modulates the size and composition of the ongoing influenza-specific CD8⁺ T cell response. J. Immunol. 193, 5076–5087 (2014).
Shahangian, A. et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Investig. 119, 1910–1920 (2009).
Rao, A., Barkley, D., França, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Antos, D. et al. Cell-intrinsic regulation of phagocyte function by interferon lambda during pulmonary viral, bacterial super-infection. PLoS Pathog. 20, e1012498 (2024).
Duray, A. M. et al. Nrf2 regulates inflammation by modulating dendritic cell-T cell crosstalk during viral-bacterial superinfection. J. Immunol. 213, 1834–1843 (2024).
Kudva, A. et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 186, 1666–1674 (2011).
Robinson, K. M. et al. Survival during influenza-associated bacterial superinfection improves following viral- and bacterial-specific monoclonal antibody treatment. JCI Insight 4, https://doi.org/10.1172/jci.insight.125554 (2019).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Kasmani, M. Y. et al. A spatial sequencing atlas of age-induced changes in the lung during influenza infection. Nat. Commun. 14, 6597 (2023).
Cang, Z. et al. Screening cell-cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023).
Li, H., Wang, A., Zhang, Y. & Wei, F. Diverse roles of lung macrophages in the immune response to influenza A virus. Front. Microbiol. 14, 1260543 (2023).
Mahajan, S. et al. The role of ICOS in the development of CD4 T cell help and the reactivation of memory T cells. Eur. J. Immunol. 37, 1796–1808 (2007).
Moyron-Quiroz, J. E. et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat. Med. 10, 927–934 (2004).
Fleige, H. et al. Induction of BALT in the absence of IL-17. Nat. Immunol. 13, 1 (2011).
Rangel-Moreno, J. et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat. Immunol. 12, 639–646 (2011).
Silva-Sanchez, A. & Randall, T. D. Role of iBALT in respiratory immunity. Curr. Top. Microbiol. Immunol. 426, 21–43 (2020).
Nakamura, S., Davis, K. M. & Weiser, J. N. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Investig. 121, 3657–3665 (2011).
Robinson, K. M. et al. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1β production in mice. J. Immunol. 191, 5153–5159 (2013).
Li, W., Moltedo, B. & Moran, T. M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J. Virol. 86, 12304–12312 (2012).
Sun, K. & Metzger, D. W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 14, 558–564 (2008).
Damjanovic, D., Lai, R., Jeyanathan, M., Hogaboam, C. M. & Xing, Z. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. Am. J. Pathol. 183, 868–880 (2013).
Dawson, T. C., Beck, M. A., Kuziel, W. A., Henderson, F. & Maeda, N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am. J. Pathol. 156, 1951–1959 (2000).
Marques, R. E., Guabiraba, R., Russo, R. C. & Teixeira, M. M. Targeting CCL5 in inflammation. Expert Opin. Ther. targets 17, 1439–1460 (2013).
Miller, L. M. et al. B cell deficiency induces cytotoxic memory CD8+ T cells during influenza-associated bacterial pneumonia. J. Clin. Investig. 135, https://doi.org/10.1172/JCI188342 (2025).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Welch, J. D. et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e1817 (2019).
Liu, W. et al. Probabilistic embedding, clustering, and alignment for integrating spatial transcriptomics data with PRECAST. Nat. Commun. 14, 296 (2023).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Liu, T., Leigh, M. M., Chongyue, Z., Chen, W., & Alcorn, J. F. Mouse_lung_superinfection_ST (2025).
Acknowledgements
This study was supported by NIH R01 HL107380 and R01 HL167449 (JFA), Children’s Hospital of Pittsburgh Research Advisory Committee Fellowship (LMM), NIH T32 AI060525 (BPD), and NIH P01 AI106684 (WC). We would like to thank Dr. William A. MacDonald and the University of Pittsburgh Health Sciences Sequencing Core for assistance in conducting the ST work. We would also like to thank the Histology Core at UPMC Children’s Hospital of Pittsburgh for their work producing lung sections for the study. This study was supported in part by the University of Pittsburgh Center for Research Computing, RRID:SCR_022735, through the resources provided. Specifically, this work used the HTC cluster, which is supported by the NIH award number S10OD028483.
Author information
Authors and Affiliations
Contributions
Conceptualization, W.C. and J.F.A.; methodology, L.M.M., B.P.D., C.Z., Y.P., A.R.G., M.L.G.H., J.R.M., and T.L.; data analysis, L.M.M., J.R.M., and T.L.; writing-original draft, T.L. and L.M.M.; writing—review and editing, W.C. and J.F.A.; supervision, W.C. and J.F.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Laura Denney and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Si Ming Man, Dario Ummarino and Mengtan Xing. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, T., Miller, L.M., Dresden, B.P. et al. Influenza virus and Staphylococcus aureus super-infection disrupts spatially coordinated cellular immunity in the mouse lung. Commun Biol 8, 1837 (2025). https://doi.org/10.1038/s42003-025-09268-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09268-1
