
Abstract
The G protein-coupled receptor associated sorting protein 2 (GPRASP2) gene mutation is one of only three deafness genes identified to be implicated in X-linked recessive syndromic hearing loss (SHL) to date. However, the function of GPRASP2 in the auditory system has not yet been fully understood. In this study, we generated Gprasp2-deficient mice and found that they exhibited a hearing loss phenotype and depression-like behaviors. In addition, we observed a disordered arrangement of cochlear hair cells in Gprasp2-deficient mice. GPRASP2 binds to NCAM1. Gprasp2 deficiency decreased NCAM1 level and further enhanced ferritinophagy in cochlear hair cells. This study could improve our understanding of the role of GPRASP2 deficiency in auditory cells, which contributes to the pathophysiology of X-linked SHL.
Similar content being viewed by others


Introduction
Hearing loss (HL) is a prevalent sensory neuropathic disorder that significantly impacts the quality of life. Genetic factors play a pivotal role in causing HL, contributing to 50—70% of cases. Among hereditary HL cases, syndromic hearing loss (SHL) accounts for approximately 30%. Additionally, X-linked HL is a relatively rare form of hereditary HL, representing only 1–5% of cases1,2. To date, there have been only three X-linked recessive SHLs characterized at the molecular level (NDP mutations mapped to Xp113,4, COL4A5 mutations mapped to Xq21-q225, and GPRASP2 mutations located in Xq22.16. Notably, GPRASP2 was first identified as a deafness gene by our team in a Chinese family. This missense mutation of GPRASP2 results in a truncated protein, but the underlying molecular mechanism of GPRASP2 mutations in HL is still unclear. Thus, fully understanding the function of GPRASP2 in the auditory system is important for gene therapy of the X-linked recessive SHL with GPRASP2 mutations.
The G protein-coupled receptor associated sorting protein 2 (GPRASP2), encoded by GPRASP2, belongs to the GPRASP family. GPRASP2 is abundantly expressed in the brain and is intimately linked to the occurrence of nervous system diseases. Notably, the deletion of Gprasp2 has been found to induce autism-like behaviors in mice7. Recently, we have reported that the lack of Gprasp2 inhibited the proliferation of HEI-OC1 cells (an immortalized cell line from the organ of Corti of the mouse cochlea)8; this finding underscores the significance of GPRASP2 in the auditory system. GPRASP2 is found in the cytosol and nucleus, and it interacts with the cytoplasmic tails of G protein-coupled receptors (GPCRs) located in the cell membrane7. Additionally, GPRASP2 directly interacts with proteins like huntingtin, and this interaction can affect receptor trafficking, which may contribute to Huntington’s disease9. Therefore, GPRASP2 likely plays a crucial role in mediating intracellular signal transduction through its interactions with receptor proteins situated in cell membranes, thereby regulating cellular functions.
Ferroptosis is a type of regulated cell death driven by exogenous iron overload or by endogenous inhibition of the antioxidant system, resulting in uncontrolled lipid peroxidation by the Fenton reaction and ultimately causing cell death10. Among the ferroptosis-related genes, the receptors (such as TFRC, SLC39A14, and SLC40A1) localized in cell membranes maintain iron homeostasis by controlling iron uptake and export, and thus abnormal iron homeostasis could trigger ferroptosis. Additionally, ferroptosis contributes to HL by damaging cochlear hair cells11,12. Enhancing autophagy-dependent ferritinophagy also prompts ferroptosis in cochlear cells13. We thus hypothesize that there is an iron trafficking mechanism related to GPRASP2 in auditory cells.
Herein, we generated Gprasp2-deficient C57BL/6J mice by CRISPR/Cas9 gene editing technology to simulate human GPRASP2-related loss of function. We further evaluated the diseased region in the cochlea and found which cell type was more affected by Gprasp2 deficiency. Then we explored the function and molecular mechanism of Gprasp2 in the regulation of intracellular iron homeostasis using the HEI-OC1 cell line. Our findings revealed that Gprasp2-deficient mice exhibited impaired auditory function and disorganized hair cell arrangement in the cochlea. Furthermore, Gprasp2 deficiency led to increased iron influx, elevated ROS production, and ultimately induced ferroptosis in HEI-OC1 cells. These results offer valuable insights into the significance of GPRASP2 in auditory cells and pave the way for future research aimed at treating SHL associated with human GPRASP2 mutations.
Materials and methods
Chemicals and antibodies
FerroOrange (#F374) and ROS assay kit (#R253) were purchased from DOJINDO LABORATORIES (Kumamoto, Japan). Antibodies against GPRASP2 (#12159-1-AP) and MYO7A (#20720-1-AP) were from Proteintech (Wuhan, China). Antibodies against 4-HNE (#A26085), TFRC(#A22161), α-tubulin (#AC012), β-tubulin (#AC008), FTH1 (#A25458) were from ABclonal (Wuhan, China). Antibodies against GPX4 (#ET1706-45), NCOA4 (#HA722282), and NCAM1 (#HA722755) were from HUABIO (Hangzhou, China). Ferrostatin-1 (#S7243) was purchased from Selleckchem (Shanghai, China). Chloroquine (CQ, HY-17589A, 100 μM) was purchased from MCE (Shanghai, China).
Cell culture
The HEI-OC1 cell line (WT) was obtained from the cochlear hair cells of male mice, preserved by Dr. Federico Kalinec (The Regents of the University of California, USA). The Gprasp2-/y (KO) HEI-OC1 cell line was previously generated in our lab8. The HEI-OC1 cells were cultured in DMEM (Gibco, USA) supplemented with 10% FBS (Gibco, USA) in a humidified atmosphere containing 5% CO2 at 33 °C.
Generation of Gprasp2 mutant mice and genotyping analysis
Generation of Gprasp2 mutant mice: the DNA sequences of mice Gprasp2 were collected from the GenBank Database (NC_000086.7). To generate Gprasp2 mutant mice, a single guide RNA (sgRNA) was designed (http://crispr.mit.edu/). The oligo of sgRNA was synthesized, and the complementary oligos were annealed to double-stranded DNA. The AgeI-digested Cas9 vector (pSt1374-N-NLS-fl was kindly provided by Dr. Zhang Dong at Nanjing Medical University) was transcribed into mRNA using mMESSAGE mMACHINET7 Ultra Kit (#AM1345, Ambion). The mixture of Cas9 mRNAs and Gprasp2 sgRNAs was injected into a mouse embryo.
Genotyping analysis: we genotyped wild-type mice (WT), heterozygous mice, and homozygous mice by PCR. DNA was extracted from F0 generation mice tail snips for PCR analysis. The F0 generation mice were genotyped for Cas9 using the Cas9 primers (Table S1). The positive F0 generation mice were then crossed with C57BL/6J mice. The genomic DNA was extracted from the tail snips of offspring mice using the DNeasy Blood and Tissue Kit (QIAGEN, Mannheim, Germany). The PCR genotyping of Gprasp2 was detected by Gprasp2 primers (Table S1).
Animals care: all animals were raised in an animal room under SPF conditions (temperature: 24.1 °C; humidity: 55–60%; photoperiod: 12 h light/12 h dark) in the animal core facility of Nanjing Medical University. Animal management was performed strictly in accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing Medical University (Permit Number: No.1601268). All surgical procedures were performed under anesthesia to minimize animal suffering.
Auditory brainstem response (ABR)
After anesthesia by intraperitoneal injection of sodium pentobarbital (40 mg/kg), the mice underwent ABR measurements in a soundproof room. The mice were presented with click stimuli and pure tone stimuli using the Neuro-Audio/ABR system (Neurosoft, Russian) at sound pressure levels between 50 and 130 dB in 5 dB steps until a threshold intensity that evoked a reproducible ABR waveform with an identifiable Peak III was detected. The ABR thresholds were defined as the lowest stimulus intensity.
DPOAEs auditory measurements
DPOAEs auditory measurements were performed using the Neuro-Audio system (Neurosoft, Russian) and software. For DPOAEs, sound from two independently driven MF1 speakers was merged into a custom acoustic coupler for closed-field stimulation. DPOAEs were recorded before ABR measurement by placing an acoustic coupler into the ear canal. A microphone (EK 23103, Knowles) was inserted into the acoustic coupler and connected to a pre-amplifier (ER-10B+, Etymotic Research) and a processor (200 kHz sample rate) to measure sound intensity in the ear canal. Each speaker played one of two primary tones (f1 and f2) and swept in 5 dB steps from 80 to 10 dB SPL (for f2). The 2f1-f2 distortion product was measured with f2 = 2,4,6, 8,12 kHz, f2/f1 = 1.25 and stimulus intensity L1 = L2 + 10 dB SPL. DPOAE thresholds were determined as the lowest sound stimulus by which a DPOAE amplitude reached 5 dB above noise.
Noise exposure experiment
The mice were subjected to a noise exposure experiment in a foam-lined, double-walled sound-proof room by Super Fear Conditioning Analysis System (#XR-XC404, XinRuan, Shanghai, China). All young mice (6-week-old) were exposed for 20 min to octave band (8–16 kHz) at (60-100 dB) sound pressure level (SPL). Noise levels were measured with a sound-level meter (#Type2606, Bruel and Kjaer).
The rotarod test
The mice were tested on the constant speed rotarod at 6 weeks. Before the official test, the mice undergo acclimation training on the rotarod with a constant speed (10 r/min), and the rotation lasted for 60 s every day during the training. The mice passed the acclimation training, which received the official test three times at 10-min intervals. The mice were placed on the rotating rod at a constant speed at 15 r/min and 30 r/min. The mice that fell off the rod were recorded within 180 s, and the mean value was used for statistical analysis by GraphPad Prism8.
GSH detection
Different groups of HEI-OC1 cells were collected and mixed with the GSH solution. At the end of the reaction, the solution was centrifuged, and the GSH concentration was determined by the Micro Reduced GSH Assay Kit (Solarbio BC1175, Beijing, China).
Flow cytometry examination
For Intracellular Fe(II) levels quantified, different groups of HEI-OC1 cells were plated in 6-well plates and exposed to the ferroptosis inhibitor Fer-1 for 24 h. Following treatment, cells were collected using trypsin and analyzed by FerroOrange fluorescent probe (DOJINDO, Shanghai, China). The fluorescence intensity of Fe (II) content was detected on BD FACSymphony A5 Cell Analyzer (BD Biosciences, Franklin Lakes, NJ, USA) with FerroOrange (LASER excitation: 561 nm, emission: 610/20 nm). Reactive oxygen species (ROS) were detected by incubating cells with a 5 μmol/L DCFH-DA probe (MCE, NJ, USA) for 30 mins. ROS was detected on BD FACSymphony A5 Cell Analyzer (BD Biosciences, Franklin Lakes, NJ, USA) with DCFH-DA probe (LASER excitation: 488 nm, emission: 515/20 nm). The data were analyzed with FlowJo V10 software.
Western blot analysis
Protein sample preparation: HEI-OC1 cells were lysed with RIPA (#P0013B, Beyotime, China) supplemented with protease and phosphatase inhibitor cocktail at a 1:50 dilution (#P1045, Beyotime, China). After ultrasound and centrifugation, protein lysate precipitation was removed, and the supernatant was added with loading buffer (#P0015, Beyotime, China).
Western blot: SDS-PAGE gels were prepared using One-Step PAGE Gel Fast Preparation Kit (#E303-01, Vazyme, China). After protein separation by SDS-PAGE, the samples were then transferred onto PVDF membranes. The membranes were blocked and then incubated with primary antibody (1:1000 dilution) overnight at 4 °C. All bands were visualized using protein detection and analysis instruments (Protein Simple, Bio-Techne, USA).
Histology analysis
Cochleae were fixed in 4% paraformaldehyde (PFA) at 4 °C overnight and then were decalcified in 10% EDTA for 1~2 days. The tissues were embedded at the proper orientation in paraffin, sectioned at 7-μm thicknesses, and stained using H&E.
Immunostaining analysis
Cochleae were fixed in 4% PFA at 4 °C overnight and then were decalcified in 10% EDTA for 1 ~ 2 d. For whole-mount immunostaining, cochleae were dissected into basal, middle, and apical sections. Sections were penetrated with 0.1% Triton-100 and blocked with 10% goat serum. Then they were incubated with primary antibody (1:200 dilution) overnight at 4 °C. After an incubation with secondary antibody, the samples were then stained with phalloidin (A12379, Thermo Fisher, USA) and followed by a nuclear stain (DAPI). For fluorescence multiple staining, cochlear sections were stained using the tyramide signal amplification kit (#G1236, Servicebio, China). The cochleae of mice in Gprasp2-/y and control groups were stained with primary antibody (ZO-1 antibody, MYO7A antibody, LC3 antibody, β-tubulin antibody, GPRASP2 antibody, GPX4 antibody, FTH1 antibody, TFRC antibody, NCOA4 antibody, and NCAM1 antibody) and fluorescence antibody (A11008, A11012, A21245, Thermo Fisher, USA). Cochlear sections were imaged with an LSM700 confocal microscope. The quantitative assessment of hair cells was performed as described previously14.
Image acquisition and quantification
Confocal images were acquired using an LSM880 or LSM980 with Zeiss ZEN software. Acquisition parameters were selected to maximize contrast between structures of interest. The sharpest focal plane with the clearest structures was selected for analysis. HCs were manually counted using ImageJ software. Images were acquired using the same imaging settings among samples, and a customized ImageJ script was used by adjusting images to a threshold, followed by Measure Particles. The number of Myo7a+ OHCs in the cochlear was subsequently multiplied by the average particle size and normalized by dividing by the size of the smallest particle per cochlea, as recommended by the manufacturer’s technical note. Ratio of ectopic OHCs in the cochlear were collected by the number of particles below average size.
SEM analysis
Cochlea were fixed with 2.5% glutaraldehyde in SCB (sodium cacodylate buffer; 0.1 M, pH 7.2) overnight and decalcified with 10% EDTA for 2 d. Then they were cut into sections at each half turn and were placed into 2.5% glutaraldehyde in SCB again. Samples were mounted and sputter-coated with gold. All the samples in this experiment were taken at the same time point and prepared under the same conditions. The stereociliary bundles were visualized using SEM (Hitachi S-3700N, Japan).
Statistical analysis
All data were represented as mean ± standard error of the mean. Student’s t-test was used for comparisons between two independent sample groups, one-way analysis of variance (ANOVA) was used for single-factor comparisons among multiple groups, and two-way ANOVA was used for two-factor comparisons among multiple groups, P-value < 0.05 were considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Generation of homozygous Gprasp2-deficient mice
We have previously identified GPRASP2 as a causative gene of X-linked SHL in a Chinese family spanning five generations6, and have reported that a 2 bp missense mutation in human GPRASP2 (c.1717_1718GC > AA, p.A573N) resulted in the structural change of protein (Fig. S1A). According to UCSC Genome Browser (www.genome.ucsc.edu), mouse Gprasp2 is a homologous gene to human GPRASP2, which is also located on the X chromosome, and the structure of human and mouse GPRASP2 proteins is highly similar (Fig. S1B). Thus, we generated Gprasp2-deficient mice by the CRISPR/Cas9 gene-editing technique to simulate the HL phenotype in human GPRASP2 mutation.
Since the seventh exon of Gprasp2 initiates the encoding of the GPRASP2 protein, we designed a single-guide RNA (sgRNA) to target the ATG start codon present within this exon. Following this, a mixture of sgRNA and Cas9 mRNA was injected into mouse embryos (Fig. S2). DNA sequencing revealed that the offspring mice exhibited a 10-bp deletion at the target site, resulting in a frameshift mutation in Gprasp2 (Fig. S2). Considering that GPRASP2 is an X-linked recessive pathogenic gene, we selected male mice for subsequent experiments.
The existence of abnormal auditory function and depression-like behavior in Gprasp2-deficient mice
To assess whether mice lacking GPRASP2 display a hearing loss (HL) phenotype similar to humans with GPRASP2 mutations, we conducted an auditory brainstem response (ABR) test, which gauges the functional integrity of the auditory system. As shown in Fig. 1A, the adult Gprasp2-deficient mice exhibited profound HL with a sound pressure level (SPL) of >90 dB, whereas their matched wild-type (WT) mice showed normal hearing thresholds of 55-60 dB, under the click stimulation. We further evaluated the ABR threshold under tone burst stimulation and found that the adult KO mice exhibited elevated ABR threshold shifts at 16 kHz (Fig. 1B). Furthermore, Gprasp2-deficient mice consistently demonstrated significantly higher thresholds than their matched WT mice at all tested ABR frequencies (2, 4, 8, 10, 16, and 32 kHz) (Fig. 1C). To confirm whether this phenotype is caused by age, we also conducted an ABR test in one-month-old mice and obtained similar results (Fig. 1C). Additionally, in a double-walled soundproof room, we tested the mice’s acoustic startle response. Compared to the control group, the Gprasp2-deficient mice exhibited slower movement speeds and shorter movement distances in response to noise and pure tone stimulation (Fig. 1D). Moreover, we compared the auditory function of C57BL/6 J mice injected with AAV-shGprasp2 or AAV-NC for 28 days alongside WT mice. The study revealed that mice receiving AAV-shGprasp2 exhibited significantly elevated ABR thresholds and markedly reduced distortion product otoacoustic emission (DPOAE) amplitudes (Fig. S6A, B). These results suggested that Gprasp2 deficiency led to abnormal auditory function in mice.

A ABR tests of WT and Gprasp2-/y mice models. Click-evoked ABR waveforms in WT and Gprasp2-/y mice in six-month-old, n = 5; B ABR waveforms in WT and Gprasp2-/y mice at 16000 Hz. All the ABR traces were recorded at the same measurement range for latency (0–10 ms) and amplitude (0-1 μV), n = 5; C ABR thresholds in one-month-old and six-month-old WT and Gprasp2-/y mice at 2, 4, 8, 10, 16 and 32 kHz, n = 5; D Acoustic startle response on movement behavior between WT and Gprasp2-/y mice, n = 5; E Y maze tests of WT and Gprasp2-/y mouse models, n = 5; F Rotarod tests, n = 5; G Novelty suppressed-feeding tests, n = 5; H Tail suspension tests, n = 5; I Forced swim tests, n = 5. Each bar represents mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; n.s.: not significant.
We next confirmed whether Gprasp2 deficiency caused other abnormal behaviors. Y-maze test was first performed on the mice. As shown in Fig. 1E, KO mice displayed fewer entries into the open arms (OA) and more entries into the closed arms (CA). The rotarod test showed that there was no significant difference between the Gprasp2-deficient mice and that of the WT control group, indicating there was no vestibular deficit in these mice (Fig. 1F). More interestingly, we found a depression-like behavior in Gprasp2-deficient mice. The novelty suppressed feeding test results showed that the feeding latency was increased and the amount of food consumed was decreased in the Gprasp2-deficient mice compared with the control group (Fig. 1G). Furthermore, both the tail suspension test and forced swim test demonstrated an increase in immobility time for the Gprasp2-deficient mice when compared to their WT counterparts (Fig. 1H, I). These results suggested that Gprasp2-deficient mice exhibited depression-like behaviors.
Taken together, all these results indicated abnormal auditory function and depression-like behaviors in Gprasp2-deficient mice.
Gprasp2 deficiency leads to the existence of an abnormal cochlear structure
In our previous study, we conducted a clinical and mutational analysis of a family affected by X-linked syndromic hearing loss, including CT scans of the temporal bone. We reported that patients with GPRASP2 mutations exhibited narrow and atretic auditory canals6. Subsequently, we performed small animal CT scans on the inner ears of Gprasp2-deficient mice. However, the CT scan results did not reveal any significant morphological abnormalities in the overall inner ear structure of the Gprasp2-deficient mice. Therefore, we proceeded with magnetic resonance imaging (MRI) to examine the structure of the cochlea, vestibule, and semicircular canals. The MRI results indicated differences in the gray value of gadolinium-based contrast agent (GBCA) imaging within the cochlear lymphatic fluids. Specifically, Gprasp2-deficient mice exhibited a lower cochlear vestibular nerve signal compared to WT mice (Fig. S3). These findings suggested that Gprasp2 deficiency may impact the permeability of endolymphatic fluid within the cochlear microcirculation of mice.
Cochlear hair cells serve as the primary sensory receptors in the auditory system, converting sound wave-induced mechanical vibrations of cilia into electrical signals, and GPRASP2 is abundantly expressed in hair cells and spiral ganglia. Therefore, it is necessary to further observe the function of cochlear hair cells. The H&E staining of cochlear tissue showed partial loss of cochlear hair cells in the Gprasp2-/y mice (Fig. 2A). To confirm this finding, we performed H&E staining and immunofluorescent staining on cochlear tissue from C57BL/6 J mice injected with AAV-shGprasp2 or AAV-NC. As shown in Fig.S4A, the immunofluorescent staining results revealed a significant decrease in red intensity representing GPRASP2 in hair cells (HC) and spiral ganglia (SG) in AAV-shGprasp2-injected mice. However, we didn’t observe a significant loss of cochlear hair cells in shGprasp2 mice. To gain a deeper understanding of the impact of Gprasp2 deficiency on outer hair cells (OHCs), we next performed immunofluorescent staining on the cochlear whole-mount cryosections. We labeled cochlear hair cells using a hair cell-specific marker—MYO7A and cytoskeletal proteins with phalloidin. As shown in Fig. 2B, the Gprasp2-/y mice exhibited a significant loss of OHCs in the cochlear apex and base turns compared with the control group. The number of ectopic hair cells was significantly increased in the cochlear base turn of the Gprasp2-/y mice (Fig. 2C). Furthermore, we conducted a scanning electron microscopy (SEM) analysis on hair cells from both Gprasp2-deficient and WT mice. As shown in Fig. 2D, the stereociliary bundles of the OHCs in Gprasp2-deficient mice appeared distorted, and numerous stereocilia were even degenerated (Fig. 2D).

A HE staining of cochlea, scale bar:200 μm (inserts, 50μm). Representative photomicrographs taken from paraffin sections of six-month-old WT and Gprasp2-/y mice. HC, hair cells; SG, spiral ganglia region; SV, stria vascularis; B, C Fluorescence shows the basilar membrane in the WT and Gprasp2-/y mice. MYO7A (red) indicates hair cell and phalloidin (green) indicates the F-actin, scale bar:50μm, n = 6. Then, ectopic hair cells across the different regions of the cochlea were quantitatively analyzed. Each bar represents mean ± SEM. *P < 0.05; ***P < 0.001; D SEM shows OHCs at the apex, middle, and base turn in the WT and Gprasp2-/y mice, scale bar:10μm (inserts, 5μm).
In addition, the H&E staining of cochlear tissue also showed stria vascularis (SV) deterioration in the Gprasp2-deficient mice (Fig. 2A). The contact surface between the stria vascularis and the spiral ligament is rich in tight junction proteins, and ZO-1 is a common tight junction protein in the SV in the study of the inner ear. Our results showed that the intensity of ZO-1 was much lower in SV of shGprasp2 mice than that in shNC mice, although no significant difference in SV structure was found between shGprasp2 mice and shNC mice in H&E staining (Fig. S4B, C). Moreover, ZO-1 expression levels in the cochleae of 6-month-old mice were also detected by qRT-PCR and western blot. The results revealed that ZO-1 was decreased in both mRNA and protein levels in the Gprasp2-deficient mice (Fig. S4D, E). The above results suggested that the reduction of tight junctions between Gprasp2-deficient stria vascularis endothelial cells may be one of the reasons for its functional abnormalities. The SV is the site of material exchange between blood and the inner ear—it has the highest aerobic metabolic rate per unit area among the many capillary networks in the inner ear—and is a tissue network composed of endothelial cells, basement membrane, pericytes, macrophages, and other components. The regulation of endolymph by SV persists throughout life. As the reported15, abnormal cochlear ion currents caused by various factors can manifest as hearing loss, and the disruption of K+ cycling due to connexin gene mutations can lead to hereditary non-syndromic deafness.
Taken together, the above results indicated abnormal cochlear structures in Gprasp2-deficient mice.
Gprasp2 deficiency leads to oxidative damage and abnormal iron homeostasis of OHCs
We have previously reported the effect of Gprasp2 deficiency on cell apoptosis in HEI-OC1 cells8. In a similar vein, we conducted TUNEL staining on cochlear tissue from both KO and matched WT mice. Our findings revealed widespread TUNEL-positive regions in the OHCs and spiral ganglion neurons (SGNs) of the Gprasp2-deficient mice (Fig. 3A). Furthermore, we labeled 4-Hydroxynonenal (4-HNE), a biomarker for lipid peroxidation indicating oxidative stress and ferroptosis, using a specific antibody. Notably, we observed a significantly higher relative fluorescence intensity of 4-HNE in the outer hair cells (OHCs) of Gprasp2-deficient mice compared to the WT control group (Fig. 3B), suggesting that the lack of GPRASP2 might cause oxidative damage in OHCs.

A The immunofluorescent staining images of the cochlea in the WT and Gprasp2-/y mice. DAPI (blue), TUNEL (green), scale bar: 50 μm; B Immunofluorescent staining images of cochlea in the WT and Gprasp2-/y mice. 4-HNE (red), α-tubulin (green), scale bar: 100 μm. HC, hair cells; BM, Basilar membrane, n = 3. The intensity of 4-HNE per cell was quantitatively analyzed. Each bar represents mean ± SEM, *P < 0.05; C Western blot analysis of GPRASP2 and 4-HNE in the Gprasp2-/y and wild-type HEI-OC1 cells. The data are normalized to the β-tubulin control. All data are from three independent experiments. The data are presented as the mean ± SEM values, *P < 0.05, ***P < 0.001; D Flow cytometry with DCFH-DA staining of Gprasp2-/y and wild-type HEI-OC1 cells, three independent biological samples were analyzed per group; E GSH level of Gprasp2-/y and wild-type HEI-OC1 cells. Three biological independent samples were analyzed per group, the data are presented as the mean ± SEM values, ***P < 0.001; F TEM analysis of Mitochondrial morphology in the Gprasp2-/y and wild-type HEI-OC1 cells, scale bar:5μm (inserts, 500 nm). Mitochondria: MT. Yellow arrows indicate the autolysosome, black arrow indicates outer mitochondrial membranes; G Fluorescence shows Fe(II) stained with FerroOrange (red) in the Gprasp2-/y and wild-type HEI-OC1 cells. Scale bar, 100 μm, three biological independent samples were analyzed per group; H Flow cytometry with FerroOrange staining of Gprasp2-/y and wild-type HEI-OC1 cells treated with or without Fer-1 24 h, three biological independent samples were analyzed per group.
To further investigate the potential pathological mechanism of the loss of OHCs in Gprasp2-deficient mice, we next used the Gprasp2-deficient HEI-OC1 cell line (KO group), which was previously generated in our lab8, to simulate the effect of Gprasp2 deficiency in OHCs. Consistent with the in vivo results presented in Fig. 3B, our findings also revealed an elevation in the level of 4-HNE in the KO group as compared to the wild-type HEI-OC1 cell line (WT group) (Fig. 3C). We next conducted a flow cytometric analysis to assess the reactive oxygen species (ROS) in the KO group. We found that the ROS level in the KO group was increased compared with that in the WT group (Fig. 3D). Subsequently, we detected the glutathione (GSH) using a commercially available GSH biochemical kit, and found that the GSH level was decreased in the KO HEI-OC1 cells compared with that in the WT group (Fig. 3E). These findings suggested that Gprasp2 deficiency might enhance lipid peroxidation and lead to oxidative damage of OHCs.
Subsequently, a transmission electron microscopy (TEM) analysis was conducted on both the KO and WT groups. Our findings revealed the presence of autolysosomes in the KO group, along with abnormal mitochondrial morphologies (Fig. 3 F). Specifically, we noted a decrease in mitochondrial cristae and compromised outer mitochondrial membranes in the KO group (Fig. 3F), which was similar to the mitochondrial characteristics associated with ferroptosis11. Given that Fe(II) accumulation and lipid peroxidation constitute the primary key hallmarks of ferroptosis, we proceeded to measure Fe(II) levels using a fluorescent probe (FerroOrange) in both groups. We found that the fluorescence intensity of Fe(II) in the KO group was higher than that in the WT group (Fig. 3G). Similarly, the flow cytometry analysis showed that the Fe(II) level was increased in the KO group (Fig. 3H).
These findings suggested that Gprasp2 deficiency might disrupt iron homeostasis and contribute to the promotion of ferroptosis.
Gprasp2 deficiency is associated with ferritinophagy in HEI-OC1 cells
To further investigate the potential pathological mechanism of the loss of OHCs in Gprasp2-deficient mice, we re-analyze our previous RNA-seq transcriptomic data of KO and WT groups8 by using OmicStudio tools (www.omicstudio.cn). Principal component analysis (PCA) showed distinct differences in genes between the KO group and the WT group (Fig. S5A). There were 949 up-regulated genes and 1574 down-regulated genes out of 30641 detectable genes in the KO group (|log2FC | >1 and p < 0.05) (Fig. S5B, C). GO enrichment analysis revealed that the enriched clusters were related to iron ion homeostasis and iron ion transmembrane transport in biological processes and iron ion binding in molecular functions (Fig. 4A). KEGG analysis revealed that the enriched clusters were related to ferroptosis, TNF signaling pathway and IL-17 signaling pathway (Fig. 4B). We then compared changes in the expression of the ferroptosis-related genes in the two groups, and found that: the ferroptosis gene (Gpx4) was decreased, the iron influx-related genes (Tfrc and Slc39a14) were increased, the iron efflux-related gene (Slc40a1) was decreased, and the ferritinophagy genes (Ncoa4 and Fth1) were increased in the KO group compared with the WT group (Fig. 4C). To validate these findings, we conducted qRT-PCR and western blot tests. As evident from Fig. 4D, the RNA expression level of NCOA4 was increased in the KO group compared with the WT control group, while Gpx4, Tfrc, and LC3 were not significantly changed, suggesting possible activation of ferritinophagy.

A Transcriptome sequencing analysis of Gprasp2 gene-deficient HEI-OC1 cell line. DEGs enriched in biological process, cellular component or molecular function were shown in GO enrichment analysis. The height of the bar represents the enriched gene number; B DEGs enriched in the pathway were shown in the KEGG analysis. The abscissa represents the gene ratio, which is calculated as “input gene number”/ “background gene number”; C Genes enriched in the ferroptosis pathway were shown in the heat map. Data was normalized by logarithm of 10. OE: WT cells transfected with Gprasp2 plasmid; D The qRT-PCR analysis of ferroptosis-related genes in the Gprasp2-/y and wild-type HEI-OC1 cells. The data are normalized to the β-tubulin control, three biological independent samples were analyzed per group. The data are presented as the mean ± SEM values, **P < 0.01; E The Gprasp2-/y and wild-type HEI-OC1 cells were fixed and immune-stained with anti-NCOA4 (red). Scale bar, 20 μm, three biological independent samples were analyzed per group; F The Gprasp2-/y and wild-type HEI-OC1 cells were fixed and immune-stained with anti-LC3 (green) and anti-FTH1 (red). Scale bar, 20 μm, three biological independent samples were analyzed per group; G Western blot analysis of ferroptosis-related genes in the Gprasp2-/y and wild-type HEI-OC1 cells. The data are normalized to the β-tubulin control. All data are from three independent experiments. The data are presented as the mean ± SEM values, *P < 0.05; **P < 0.01; H The Gprasp2-/y and wild-type HEI-OC1 cells were treated with WM or CQ for 24 h, then they were stained with FerroOrange (red) to identify Fe(II). Scale bar, 200 μm, three biological independent samples were analyzed per group. The intensity of Fe(II) per cell was quantitatively analyzed. Each bar represents mean ± SEM, ***P < 0.001; I Western blot analysis of ferroptosis genes in the Gprasp2-/y and wild-type HEI-OC1 cells treated with WM or CQ. The data are normalized to the β-tubulin control. All data are from three independent experiments. The data are presented as the mean ± SEM values, *P < 0.05; n.s.: not significant.
Ferritinophagy is a ferroptosis pathway that depends on autophagy activation to release Fe(II) from ferritin-Fe complexs. To further verify the ferritinophagy feature in Gprasp2-deficient HEI-OC1 cells, immunofluorescent staining was performed. Our findings revealed a notably higher fluorescence intensity of NCOA4 in the KO group compared to the WT group (Fig. 4E). Subsequently, we labeled autophagosomes and ferritin using their respective markers—LC3 (for autophagosomes) and FTH1 (for ferritin). The immunostaining results indicated colocalization of LC3 with FTH1 specifically in the KO group (Fig. 4F). Western blot analysis further confirmed elevated protein levels of TFRC and NCOA4, and a decrease in GPX4 levels in the KO group relative to the control group (Fig. 4G). Then we treated the cells with wortmannin (WM, an inhibitor of autophagy that suppresses autophagosome synthesis) or chloroquine (CQ, an inhibitor of autophagy that suppresses degradation of autophagosomes in the lysosomes), followed by Fe(II) staining using FerroOrange. Notably, the Fe(II) fluorescence in the KO cells was higher than that in the WT cells, and either WM or CQ treatment led to a decrease in Fe(II) fluorescence intensity in KO group (Fig. 4H). The western blot results also showed a decrease of TFRC and NCOA4 in the KO group after autophagy inhibition with CQ (Fig. 4I). These findings suggest that Gprasp2 deficiency induces ferritinophagy in HEI-OC1 cells by enhancing autophagy and upregulating NCOA4 expression.
To ascertain whether ferroptosis was initiated and whether ferritinophagy was augmented in the OHCs when there is a lack of GPRASP2, we first performed immunofluorescent staining on cochlear tissue from C57BL/6 J mice injected with AAV-shGprasp2 or AAV-NC and labeled OHCs with MYO7A, together with ferroptosis markers (GPX4) and ferritinophagy markers (NCOA4 and TFRC). As shown in Fig. 5A, the immunofluorescent staining results revealed a significant decrease in red intensity representing GPX4 in OHCs, which were labeled with green fluorescence in shGprasp2 mice. Similarly, we found a decrease in GPX4 levels in the OHCs, the supporting cells, and the basilar membrane of the Gprasp2-deficient mice, indicating the presence of ferroptosis (Fig. 5B, C). Furthermore, our results demonstrated elevated levels of NCOA4 in the OHCs of Gprasp2-knockdown or knockout mice, indicating an enhancement of ferritinophagy (Fig. 5D, F). Taken together, the above findings suggest that Gprasp2 deficiency induces ferritinophagy in the OHCs by upregulating NCOA4 expression.

A Immunofluorescent staining image of cochlear tissue from C57BL/6 J mice injected with AAV-shGprasp2 or AAV-NC for 28 days (two-month-old). GPX4 (red), MYO7A (green), DAPI (blue), yellow arrows point to OHC, scale bar:200μm (inserts, 20μm), n = 3. B Immunofluorescent staining images of the cochlea in the WT and Gprasp2-/y mice. GPRASP2 (red) and GPX4 (green), scale bar: 100μm, n = 3. C Intensity of GPX4 per cell was quantitatively analyzed in relation to A and B. Each bar represents mean ± SEM, *P < 0.05. D Immunofluorescent staining images of cochlear tissue from C57BL/6J mice injected with AAV-shGprasp2 or AAV-NC. NCOA4 (red), MYO7A (green), DAPI (blue), yellow arrows point to OHC, scale bar:200μm (inserts, 20μm), n = 3. E Immunofluorescent staining images of the cochlea in the WT and Gprasp2-/y mice. TFRC (green) and NCOA4 (yellow), scale bar: 100 μm, n = 3. F Intensity of NCOA4 per cell was quantitatively analyzed in relation to D and E. Each bar represents mean ± SEM, **P < 0.01.
NCAM1 is required in maintaining iron homeostasis in HEI-OC1 cells
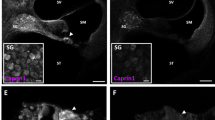
To further explore its underlying molecular mechanism, we identified 639 proteins by proteomic analysis of the GPRASP2 interactome (Fig. S5D). Using the STRING-functional protein association network database (www.string-db.org), we pinpointed 9 candidate proteins, including Neural cell adhesion molecule 1 (NCAM1) (Fig. S5E). NCAM1 is widely recognized as a protein linked to learning and memory processes16. It plays a role in the development of various psychiatric disorders, including depressive disorders17, and is intimately connected to autophagy progression18,19. Our co-immunoprecipitation (Co-IP) experiments confirmed a specific interaction between GPRASP2 and NCAM1 in HEI-OC1 cells (Fig. 6A). Western blot analysis revealed a reduction in NCAM1 protein levels in the KO cells and cochlear tissue of KO mice (Figs. 6B and S4E). To ascertain whether NCAM1 decreased in the OHCs of Gprasp2-deficient mice, we performed immunofluorescent staining. Our findings revealed a reduced intensity of NCAM1 in the OHCs of knockdown or knockout Gprasp2 mice (Fig. 6C). The above results suggested that the lack of GPRASP2 caused a reduction of NCAM1. Then we compared the auditory function of C57BL/6 J mice injected with AAV-shNcam1, AAV-shGprasp2, or AAV-NC for 28 days alongside WT mice. We found that mice receiving AAV-shNcam1 exhibited reduced DPOAE amplitudes comparable to those in the AAV-shGprasp2 group when F2 > 8 kHz (Fig. S6D). As shown in Fig. S6E, the mice receiving AAV-shNcam1 exhibited profound HL with a sound pressure level (SPL) of >90 dB, whereas AAV-shNC-injected mice or WT mice showed normal hearing thresholds, under click stimulation. This suggested potential impairment of hearing function and compromised cochlear outer hair cell activity. Moreover, immunofluorescent staining results showed that the intensity of ZO-1 was much lower in SV of shNcam1 mice than that in the shNC mice, which had a similar trend to that in shGprasp2 mice (Fig. S4C). This result suggested that Gprasp2 deficiency affected the tight junction of the inner ear through NCAM1. Our findings indicated that inhibiting the expression of NCAM1 affected auditory function.

A Co-IP analysis of endogenous GPRASP2 and endogenous NCAM1 in HEI-OC1 cells, three biological independent samples were analyzed per group. B Western blot analysis of NCAM1 in the Gprasp2-/y and wild-type HEI-OC1 cells. The data are normalized to the β-tubulin control. All data are from three independent experiments. The data are presented as the mean ± SEM values, **P < 0.01. C Immunofluorescent staining images of cochlear tissue from C57BL/6 J mice injected with AAV-shGprasp2 or AAV-NC for 28 days (two-month-old). NCAM1 (red), MYO7A (green), DAPI (blue), yellow arrows point to OHC, scale bar:200μm (inserts, 20μm), n = 3. Immunofluorescent staining images of cochlea in the WT and Gprasp2-/y mice. NCAM1 (red), a-tubulin (green), yellow arrows point to OHC, scale bar:200μm (inserts, 20 μm), n = 3. D Fluorescence shows Fe(II) stained with FerroOrange (red) in the Gprasp2-/y and wild-type HEI-OC1 cells transfected with or without Ncam1 plasmid. OE: WT cells transfected with Gprasp2 plasmid. Scale bar, 200μm. Three biological independent samples were analyzed per group. The intensity of Fe(II) per cell was quantitatively analyzed. Each bar represents mean ± SEM, ***P < 0.001; E Immunofluorescent staining images of the Gprasp2-/y and wild-type HEI-OC1 cells transfected with or without Ncam1 plasmid. NCOA4 (red). Scale bar, 20 μm. Intensity of NCOA4 per cell was quantitatively analyzed. Each bar represents mean ± SEM, *P < 0.05, ***P < 0.001; F Confocal images showing the localizations of LC3 (green) and FTH1 (red) in the Gprasp2-/y and wild-type HEI-OC1 cells transfected with or without Ncam1 plasmid. Scale bar, 20 μm. Three biological independent samples were analyzed per group. Pearson’ s R value indicates the degree of co-localization. Each bar represents mean ± SEM, *P < 0.05, n.s.: not significant; G Western blot analysis of ferritinophagy-related genes in the Gprasp2-/y HEI-OC1 cells transfected with or without Ncam1 plasmid. The data are normalized to the β-tubulin control. H Western blot analysis of cleaved caspase3 in the Gprasp2-/y and wild-type HEI-OC1 cells transfected with or without Ncam1 plasmid. The data are normalized to the β-tubulin control. I Western blot analysis of NCAM1 and cleaved caspase3 in the Gprasp2-/y, wild-type HEI-OC1 cells treated with WM or CQ. The data are normalized to the β-tubulin control; All data are from three independent experiments. The data are presented as the mean ± SEM values, *P < 0.05; **P < 0.01; n.s.: not significant.
To investigate whether GPRASP2 regulates iron homeostasis through NCAM1, we constructed a Ncam1 overexpression plasmid and transfected it into Gprasp2-deficient HEI-OC1 cells. We transfected the cells with Ncam1 plasmid or vector, followed by Fe(II) staining using FerroOrange. We found that the Fe(II) fluorescence was higher in the Gprasp2-deficient cells than that in the control group, and transfecting Ncam1 plasmid led to a decrease of Fe(II) fluorescence intensity in the KO group (Fig. 6D). Immunofluorescent staining results showed that the fluorescence intensity of NCOA4 was significantly lower in the KO group transfected with the Ncam1 plasmid compared to those transfected with the control vector (Fig. 6E). Additionally, when we labeled autophagosomes and ferritin with LC3 and FTH1, respectively, we observed significant colocalization of autophagosomes and ferritin in the KO group, and found that transfection with the Ncam1 plasmid in Gprasp2-deficient cells could not reduce this colocalization (Fig. 6F). Western blot results indicated a decrease in NCOA4 levels and TFRC levels in the Gprasp2-deficient cells transfected with the Ncam1 plasmid relative to those in the control vector-transfected group, while LC3 levels (a marker of autophagy) remained unaffected (Fig. 6G). These findings suggested that GPRASP2 interacted with NCAM1, and GPRASP2 regulated NCOA4-mediated ferritin delivery rather than autophagy through NCAM1.
Furthermore, recent research has indicated that adenovirus-induced overexpression of NCAM1 reverses CUMS-induced hippocampal neuronal apoptosis. Previously, we documented that Gprasp2 deficiency triggers apoptosis in HEI-OC1 cells8, and we noticed apoptosis in the cochlear tissue of KO mice (Fig. 3A). This led us to hypothesize whether transfecting Ncam1 plasmid could counteract cell apoptosis in HEI-OC1 cells with Gprasp2 deficiency. Western blot analysis revealed elevated cleaved caspase3 level in the KO group (Fig. 6H), echoing our previous findings. Upon treating the cells with WM or CQ, we discovered that only CQ could reverse the reduction in NCAM1 levels but WM could not, suggesting that Gprasp2 deficiency might promote the degradation process of NCAM1 via the autophagy-lysosome system (Fig. 6I). Surprisingly, we found that transfecting Ncam1 plasmid further elevated cleaved caspase3 levels in both WT and KO groups (Fig. 6H), implying that NCAM1 overexpression actually enhanced apoptosis in HEI-OC1 cells. Hence, maintaining an optimal level of NCAM1 is crucial, and GPRASP2 plays a role in maintaining this balance.
Collectively, we confirmed the HL phenotype and depression-like behaviors in Gprasp2-deficient mice. Our observations indicated a disrupted arrangement of cochlear hair cells upon Gprasp2 deletion. GPRASP2 bound to NCAM1; the absence of GPRASP2 led to enhanced ferritinophagy by downregulating NCAM1, ultimately triggering ferroptosis in cochlear hair cells.
Discussion
In this study, we confirmed that the lack of GPRASP2 led to HL in a mouse model. In the past 40 years, there have been a few X-linked disorders related to HL characterized at the molecular level4,5,20,21,22,23,24,25,26. Most of those X-linked HL cases were genetically heterogeneous deafness with syndromic phenotypes. However, the missense variation of GPRASP2 in X-linked recessive SHL families is very rare. According to computer prediction, the missense variation of GPRASP2 led to changes in its protein structure and molecular function, but this view has not been validated. In this study, we provided evidence that Gprasp2 knockout led to HL phenotype in the mouse model, suggesting that loss of function in the GPRASP2 protein is important for the onset of X-linked recessive SHL related to the missense variation of human GPRASP2.
Our data indicated that the impairment of cochlear OHCs resulted by Gprasp2 deficiency was crucial for the development of HL. The structural abnormalities of cochlear hair cells are closely associated with HL; for example, OSBPL2 deficiency leads to morphological abnormalities in cochlear HC stereocilia of the Bama mini pigs27. Because the cochlear hair cells are unable to regenerate, their loss or damage can directly affect auditory function28. In this study, we observed that GPRASP2 was enriched in the cochlear HCs and SGs. With observation of SEM detection and laser confocal microscope, we further noticed structural abnormalities or loss of cochlear hair cells in Gprasp2-deficient mice. Our findings suggest that Gprasp2 deficiency impairs OHC function.
In our study, we presented evidence demonstrating the relationship between iron homeostasis regulation and Gprasp2 in cochlear outer hair cells. Ferroptosis is characterized primarily by Fe(II) accumulation and uncontrolled lipid peroxidation. It is reported that the activity of the GPCR-cAMP signaling pathway regulates Fe(II) release from the acidified endosomes to the Fe(II) pool (LIP)29,30, suggesting a role for the GPCR family and its associated proteins in regulating iron homeostasis. Ferritinophagy, a ferroptosis pathway dependent on autophagy, involves either increasing iron uptake via TFRC upregulation or enhancing ferritin transport to the lysosome mediated by NCOA4, both of which facilitate Fe(II) release. Inhibition of autophagy flux can halt this process13,31. Our in vivo and in vitro data suggest that Gprasp2-deficiency triggers ferritinophagy. Using mass spectrometry, we discovered an interaction between GPRASP2 and NCAM1. It is reported that NCAM1 binds to ERK1/2 and promotes autophagy. It has also been reported to be associated with neuron apoptosis17. In our data, we found that Gprasp2-deficiency downregulates the NCAM1 expression, which may enhance autophagy and ferritinophagy. Furthermore, cellular ferroptosis and apoptosis are closely intertwined processes. During ferroptosis, mitochondria generate significant amounts of ROS, triggering cell apoptosis and inflammation in both normal cells and cancer cells32,33. Conversely, apoptosis can accelerate ferroptosis34. In addition, GPRASP1, another G-protein coupled receptor-associated sorting protein, has been reported to play a crucial role in mediating lysosome biogenesis and sorting35. In our data, GPRASP2 bound to NCAM1, Gprasp2-deficiency causes a decrease in NCAM1 levels, and it could be rescued by CQ, an autophagy inhibitor that prevents autophagosomes from localizing at the lysosome for protein degradation. This raises the possibility that GPRASP2 may be involved in regulating the degradation of NCAM1 in lysosomes. Additionally, recent research indicates that KLK8 cleaves the extracellular domain of NCAM1, leading to a decrease in NCAM1 levels in neurons, which promotes chronic unpredictable mild stress-induced hippocampal neuronal apoptosis17. Our study also found that pharmacological inhibition of ferroptosis or ferritinophagy increases NCAM1 levels, partially mitigating apoptosis in HEI-OC1 cells caused by Gprasp2 deficiency. Conversely, overexpressing NCAM1 leads to increased apoptosis, highlighting the importance of maintaining optimal NCAM1 levels. Moreover, our recent research demonstrates that overexpressing Gprasp2 promotes the proliferation of hair cell formation from supporting cells in a 3D-cochlear organoid model, suggesting a potential gene therapy approach for hair cell regeneration36.
In our study, we also observed depression-like behaviors in Gprasp2-deficient mice. GPRASP2 is highly expressed in brain tissue, indicating that it is also important in the CNS (central nervous system). Previous research has shown that GPRASP2 mutations are present in certain autism and schizophrenia patients, and the depletion of Gprasp2 affects mGluR function7. However, the possibility of Gprasp2 deficiency leading to depression-like behaviors has not been documented previously. It is reported that the loss of NCAM1 is associated with a pro-apoptotic mechanism in neurons, which results in depressive disorders, and overexpressing NCAM1 can rescue this phenotype16. Therefore, we hypothesize that the depression-like behaviors observed in Gprasp2-deficient mice may partly stem from the downregulation of NCAM1.
Taken together, we generated a Gprasp2-deficient mouse model and confirmed the effect of Gprasp2 knockout on auditory function. Gprasp2 deficiency led to the HL phenotype and depression-like behaviors. GPRASP2 bound to NCAM1 to regulate autophagy. The lack of GPRASP2 enhanced ferritinophagy by downregulating NCAM1 and thereby induced ferroptosis in cochlear OHCs. Through the study of GPRASP2’s function, we provide a theoretical basis for better understanding the regulatory mechanism underlying the onset of X-linked recessive SHL caused by missense variations in human GPRASP2.
Study limitation
Although GPRASP2 has been demonstrated to have multiple functions in various contexts, its underlying molecular mechanisms remain incompletely understood. We acknowledge that the present study has several limitations: some phenotypic experiments lack quantitative analysis due to insufficient samples; we did not correlate the severity of HL with the degree of hair cell damage in individual mice; the reason that causes abnormal behaviors in Gprasp2-mutant mice is not fully elucidated; and some behavioral tests (assessing repetitive behavior and social behavior) were not performed. These limitations will be addressed in future studies, which will focus on developing tools to clarify the underlying reasons for the altered behaviors.
Data availability
All source data are provided as Supplementary Data 1. Uncropped western blotting and gel images are included in the Supplementary Information. Ncam1 plasmid (ID: 252092) was deposited in Addgene. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Petersen, M. B., Wang, Q. & Willems, P. J. Sex-linked deafness. Clin. Genet. 73, 14–23 (2008).
Corvino, V. et al. X-Linked Sensorineural Hearing Loss: A Literature Review. Curr. Genomics. 19, 327–338 (2018).
Berger, W. et al. An animal model for Norrie disease (ND): gene targeting of the mouse ND gene. Hum. Mol. Genet. 5, 51–59 (1996).
Rehm, H. L. et al. Vascular defects and sensorineural deafness in a mouse model of Norrie disease. J. Neurosci. 22, 4286–4292 (2002).
Barker, D. F. et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248, 1224–1227 (1990).
Xing, G. et al. GPRASP2, a novel causative gene mutated in an X-linked recessive syndromic hearing loss. J. Med. Genet. 54, 426–430 (2017).
Edfawy, M. et al. Abnormal mGluR-mediated synaptic plasticity and autism-like behaviours in Gprasp2 mutant mice. Nat. Commun. 10, 1431 (2019).
Lu, Y. et al. Disruption of Gprasp2 down-regulates Hedgehog signaling and leads to apoptosis in auditory cells. Biochem. Biophys. Res. Commun. 574, 1–7 (2021).
Horn, S. C. et al. Huntingtin, interacts with the receptor sorting family protein GASP2. J. Neural Transm. 113, 1081–1090 (2006).
Chen, X., Kang, R., Kroemer, G. & Tang, D. Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 18, 280–296 (2021).
Hu, B. et al. Ferrostatin-1 protects auditory hair cells from cisplatin-induced ototoxicity in vitro and in vivo. Biochem. Biophys. Res. Commun. 533, 1442–1448 (2020).
Zheng, Z. et al. Liproxstatin-1 Protects Hair Cell-Like HEI-OC1 Cells and Cochlear Hair Cells against Neomycin Ototoxicity. Oxid. Med. Cell. Longev. 2020, 1782659 (2020).
Liang, L. et al. NPC1 Deficiency Contributes to Autophagy-Dependent Ferritinophagy in HEI-OC1 Auditory Cells. Front. Mol. Biosci. 9, 952608 (2022).
Jahan, I., Pan, N., Kersigo, J. & Fritzsch, B. Neurog1 can partially substitute for Atoh1 function in hair cell differentiation and maintenance during organ of Corti development. Development 142, 2810–2821 (2015).
Yang, Q. et al. OSBPL2 deficiency impaired cochlear blood-labyrinth barrier via activation of NF-κB signaling pathway. Hear Res., 467: 109432 (2025).
Liu, C. et al. HMGB1-NLRP3-P2X7R pathway participates in PM2.5-induced hippocampal neuron impairment by regulating microglia activation. Ecotoxicol. Environ. Saf. 239, 113664 (2022).
Xu, D. H. et al. Upregulation of KLK8 contributes to CUMS-induced hippocampal neuronal apoptosis by cleaving NCAM1. Cell. Death Dis. 14, 278 (2023).
Li, J. et al. NCAM regulates the proliferation, apoptosis, autophagy, EMT, and migration of human melanoma cells via the Src/Akt/mTOR/cofilin signaling pathway. J. Cell. Biochem. 121, 1192–1204 (2020).
Zhao, L. et al. ELF1 suppresses autophagy to reduce cisplatin resistance via the miR-152-3p/NCAM1/ERK axis in lung cancer cells. Cancer Sci. 114, 2650–2663 (2023).
Brunner, H. G. et al. Molecular genetics of X-linked hearing impairment. Ann. N. Y. Acad. Sci. 630, 176–190 (1991).
Berger, W. et al. Mutations in the candidate gene for Norrie disease. Hum. Mol. Genet. 1, 461–465 (1992).
Roesch, K., Curran, S. P., Tranebjaerg, L. & Koehler, C. M. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Genet. 11, 477–486 (2002).
Engl, G., Florian, S., Tranebjærg, L. & Rapaport, D. Alterations in expression levels of deafness dystonia protein 1 affect mitochondrial morphology. Hum. Mol. Genet. 21, 287–299 (2012).
Desnick, R. J., Wasserstein, M. P. & Banikazemi, M. Fabry, disease (alpha-galactosidase A deficiency): renal involvement and enzyme replacement therapy. Contrib. Nephrol. 136, 174–192 (2001).
Shiloh, Y. et al. Genetic mapping of X-linked albinism-deafness syndrome (ADFN) to Xq26.3-q27.I. Am. J. Hum. Genet. 47, 20–27 (1990).
Zong, L. et al. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder. J. Med. Genet. 52, 523–531 (2015).
Yao, J. et al. OSBPL2-disrupted pigs recapitulate dual features of human hearing loss and hypercholesterolaemia. J. Genet. Genomics. 46, 379–387 (2019).
Kwan, T., White, P. M. & Segil, N. Development and regeneration of the inner ear. Ann. N. Y. Acad. Sci. 1170, 28–33 (2009).
Camarena, V. et al. cAMP signaling regulates DNA hydroxymethylation by augmenting the intracellular labile ferrous iron pool. Elife. 6: e29750. (2017).
Huff, T. C. et al. Oscillatory cAMP signaling rapidly alters H3K4 methylation. Life Sci. Alliance 3, e201900529 (2019).
Wang, T. et al. Oxysterol 25-hydroxycholesterol activation of ferritinophagy inhibits the development of squamous intraepithelial lesion of cervix in HPV-positive patients. Cell. Death. Discov. 10, 135 (2024).
Kang, R. et al. Deoxynivalenol induced apoptosis and inflammation of IPEC-J2 cells by promoting ROS production. Environ. Pollut. 251, 689–698 (2019).
Chen, P. et al. Triptolide induces apoptosis and cytoprotective autophagy by ROS accumulation via directly targeting peroxiredoxin 2 in gastric cancer cells. Cancer Lett. 587, 216622 (2024).
Wang, Z. et al. A nuclear long non-coding RNA LINC00618 accelerates ferroptosis in a manner dependent upon apoptosis. Mol. Ther. 29, 263–274 (2021).
Marley, A. & von Zastrow, M. Dysbindin promotes the post-endocytic sorting of G protein-coupled receptors to lysosomes. PLoS One 5, e9325 (2010).
Cai, J. et al. Implication of GPRASP2 in the Proliferation and Hair Cell-Forming of Cochlear Supporting Cells. Cell Prolif. e13792. (2024).
Acknowledgements
This research was supported by the grants from the National Natural Science Foundation of China (32070587 and 82200642).
Author information
Authors and Affiliations
Contributions
Y. Lu, X. Cao. and T. Wang performed study concept and design; Y.Lu wrote the manuscript; X. Cao. and T. Wang performed review and revision of the paper; F. Sheng and H. Chen performed histochemistry and immunofluorescence staining. J. Yao and Q. Wei, provided acquisition, analysis, and interpretation of data, and statistical analysis; Z. Chen and G. Xing provided technical and material support. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Roslyn Fitch and the other anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Chris Hine and Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, Y., Sheng, F., Yao, J. et al. Abnormal iron homeostasis mediates cochlear hair cell impairment and hearing loss in Gprasp2-deficient mice. Commun Biol 9, 425 (2026). https://doi.org/10.1038/s42003-026-09679-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09679-8
