
Abstract
Among the human E3 ubiquitin ligases, NEDD4 (Neural precursor cell expressed developmentally down-regulated 4) plays a critical role in development and cancer, making it a compelling therapeutic target. However, no specific NEDD4 inhibitors have advanced in drug development. In this study, we reveal the inhibitory mechanism of Norclomipramine, a tricyclic antidepressant, which inhibits NEDD4-mediated ubiquitin chain elongation by binding to a hydrophobic pocket in the Ub exosite of the N-lobe. Building on this mechanism, we conducted a focused medicinal chemistry campaign, resulting in the development of covalent inhibitors that specifically target the non-catalytic cysteine C627. These compounds exhibit selective binding to NEDD4 over other family members, effectively inhibiting NEDD4-mediated polyubiquitination while leaving monoubiquitinated substrates unaffected. Among these, compound 32 emerged as a potent lead (IC50 = 0.12 µM) with favorable pharmacokinetic properties, including oral bioavailability, paving the way for future in vivo efficacy studies.

Similar content being viewed by others

Introduction
Targeting the ubiquitin-proteasome system (UPS) has proven to be an effective strategy in cancer therapy, as evidenced by the current clinal use of 26S proteasome inhibitors like Bortezomib and Carfilzomib, as well as NEDD8-activating enzyme inhibitor Pevonedistat1,2,3,4,5. Recent advances in targeted protein degradation have introduced Proteolysis Targeting Chimeras (PROTACs) and molecular glue degraders, which leverage the function of E3 ligases to selectively degrade proteins that are difficult to inhibit with conventional small molecules6,7,8,9.
Among the approximately 600 human E3 ubiquitin ligases, HECT (Homologous to E6AP C Terminus) and RBR (RING between RING) E3s mediate ubiquitination by forming one last transient thioester bond to ubiquitin (Ub) before it is conjugated to the substrate, providing high specificity and selectivity towards targets and Ub chains. Among them, NEDD4 (Neural precursor cell expressed developmentally down-regulated 4) is of particular interest for drug discovery due to its significant role in developmental and disease processes as well as its characterized catalytic mechanism. Indeed, NEDD4 is overexpressed in several cancer types, and its downregulation appears to reduce the proliferation, migration, and invasion of cancer cells10. NEDD4 is also known to negatively regulate the tumor suppressor PTEN11; however, this pro-oncogenic role of NEDD4 in PTEN regulation appears to be context-specific12.
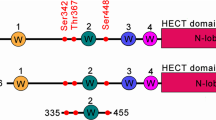
Structurally, NEDD4 is the prototype of the C2-WW-HECT family that shared an evolutionary conserved domain architecture. The N-terminal extension includes a calcium-dependent lipid-binding and regulatory C2 domain and four WW domains, which are crucial for substrate specificity as they recognize PPxY or proline-rich motifs in substrates, adaptors, and regulatory proteins13,14. The C-terminal HECT domain is both necessary and sufficient for mediating the covalent transfer of Ub onto the E3 ligase itself (self-ubiquitination), the substrate or onto Ub to form free chains. The HECT domain presents a bilobal organization: a bulkier N-terminal lobe that contains the E2 binding domain and a non-covalent Ub binding domain, named Ub exosite, which is key for enzyme processivity15,16,17,18, and a C-terminal lobe that carries the catalytic cysteine19,20.
Despite its biomedical importance21, only a few low affinity compounds capable of modulating NEDD4 activity have been disclosed22,23,24,25. None of them have advanced further in the drug development process. A decade ago, through high throughput screening for ITCH inhibitors, Rossi and colleagues identified Clomipramine, an FDA-approved tricyclic antidepressant that acts as a reuptake inhibitor of serotonin and norepinephrine receptors, used in clinics to treat psychiatric disorders. Clomipramine and its analogues, including Norclomipramine, appear to specifically block the HECT catalytic activity of NEDD4 family member ITCH but not the RING ligase Ring1B26.
In this study, we elucidated the mode of action of this class of drugs as HECT inhibitors and exploited the structure of Norclomipramine in complex with the HECT domain of NEDD4 to guide the rational design of novel, cell-active covalent NEDD4 inhibitors. These first-in-class NEDD4 inhibitors demonstrated exceptional specificity and selectivity, leaving the other family members unaffected. Moreover, pharmacokinetic profile of one compound resulted compatible with oral dosing. These findings offer great premises for the development of new therapeutics targeting tumors sensitive to NEDD4 expression and activity.
Results
Norclomipramine inhibits Ub-chain elongation
Rossi and co-workers identified Norclomipramine (compound 1), a tricyclic antidepressant (Fig. 1A), as general inhibitor of HECT-type E3 ligases26 although with limited affinity (Supplementary Fig. 1A). To elucidate its mechanism of action, we tested the effect of this drug on NEDD4 using the isolated catalytic HECT domain and increasing concentration of the compound in a series of biochemical assays. Results indicate that Norclomipramine does not affect the trans-thioesterification reaction from E2 to the E3, as observed in a single-turnover kinetic assay (Fig. 1B) while it specifically inhibits the Ub chain elongation activity of NEDD4, leaving Ub dimer formation unaltered (Fig. 1C). This behavior is reminiscent of the Ub exosite mutants15. Indeed, the binding of Norclomipramine appears to compete with K63-linked Ub chains for the exosite in the N-lobe of the HECT domain (Fig. 1D). Contrary to previous report26, the inhibitory effect of Norclomipramine appears to be reversible as the interaction with HECT was restored upon drug washout (Fig. 1D).

A Chemical structure of Norclomipramine. B Trans-thioesterification reaction performed with bacterially expressed HECT of NEDD4 in presence or absence of Norclomipramine (Norc). HECT thioester formation was monitored after quenching the reaction at different time points by addition of Laemmli buffer without reducing agent. Immunoblot (IB) and Coomassie as indicated. C In vitro ubiquitination assays of the HECT of NEDD4 pre-incubated with the indicated concentration of Norclomipramine (Norc). Reactions were carried out for the indicated time points. IB and Coomassie as indicated. D GST pull-down assays with the isolated NEDD4 HECT and K63-polyUb chains. GST-fusion proteins pre-incubated with Norclomipramine (Norc) at 0.2 mM for 1 hour at 4 °C were either washed or directly incubated with K63-polyUb chains as indicated. IB and Coomassie as indicated.
These results support the idea that NEDD4 adopts distinct catalytic mechanisms for the initial and the subsequent ubiquitination reactions19 and indicate that the mechanism of action of Norclomipramine differs from that of inhibitors targeting the catalytic cysteine, possibly by affecting the requirements for enzyme processivity.
Norclomipramine binds to the Ub exosite
To obtain high-resolution structural insights into the binding of Norclomipramine to the HECT domain of NEDD4, we soaked Norclomipramine into HECT crystals and solved the X-ray structure of the complex. The structure was solved at at 2.12 Å resolution by molecular replacement (Table 1). The structure was refined to Rwork and Rfree of 18.6% and 23.7%, respectively. The overall conformation of HECT is identical to the previously reported structure of NEDD4 (PDB 2XBB)15. Norclomipramine binds in a hydrophobic pocket which overlaps to the Ub exosite (Fig. 2A). A close-up of this region shows that Ub C-terminal tail (residues 71 to 76) expands into a hydrophobic pocket of the HECT domain, shaped by residues L553, Y605, L607, N628, and Y634 (Fig. 2B). These same NEDD4 residues are also involved in the binding of Norclomipramine (Fig. 2C) which, however, inserts its tricyclic moiety deeply into the hydrophobic pocket. This establishes additional interactions with residues M600, F637, and I638, while displacing the side chain of E554, which tilts towards the solvent upon ligand binding. Norclomipramine three-carbon tail also extends towards the solvent and interacts with N628 (Fig. 2C).

A Superposition of the HECT of NEDD4 in complex with Norclomipramine (Norc) or ubiquitin (Ub) in the exosite (from PBD 2XBB). The HECT of NEDD4 is depicted in surface representation, with N-lobe in blue and C-lobe in green. Norclomipramine, in yellow sticks, sits on a hydrophobic pocket which is also exploited by the C-terminal tail of Ub (gold ribbon representation, from PBD 2XBB). B Close-up view of HECTNEDD4:ubiquitin interaction (PBD 2XBB, N-lobe in blue, Ub in yellow). C Close-up view of HECTNEDD4:Norclomipramine interaction (PBD 9H9O, N-lobe in blue, Norclomipramine, in yellow sticks).
The structural data align with the pull-down assay outcomes, indicating that Norclomipramine mechanism of inhibition involves displacing ubiquitin from the Ub exosite located in the N-lobe of the HECT domain.
Generation of covalent inhibitor: identification and characterization of the lead compound 15
Closer inspection of the Norclomipramine binding pocket revealed the presence of a specific cysteine residue in closed proximity (C627, Fig. 2C) that we exploited to obtain covalent inhibitors. We focused our efforts on α,β-unsaturated carbonyl warheads such as acrylate and acrylamide whose low electrophilicity requires proximity to interact with cysteine27, in order to limit the reaction with other thiol groups and possible off target reactions.
To enable real-time monitoring of E3 activity during polyUb chain elongation and quantitatively evaluate the designed molecules, we set-out a ubiquitination assay based on time-resolved fluorescence resonance energy transfer (Ub-TR-FRET, see Methods). This assay measures ubiquitin-chain formation by the Ub machinery (E1, E2, E3) using a mix of fluorescently labelled and unlabelled monoUb, allowing for selective detection of polyubiquitin chains through a FRET signal that increases as the chains extend. After determining the optimal enzyme concentration to achieve the highest signal-to-background ratio, as sanity check we evaluated the behavior of the Ub exosite mutant (F707A) and the catalytically inactive mutant (C867A). As expected, the F707A mutant known to generate short chains28 showed a notable decrease in FRET signal compared to the wild-type enzyme (Supplementary Fig. 1B).
Using this assay, newly synthesized Norclomipramine analogues with acrylamides, methylacrylamides, and methylacrylesters warheads were tested and compared with Norclomipramine (compound 1) (Supplementary Table 1). IC50 values were determined by correlating the area under the curve (AUC) for each tested dose of the inhibitors with the corresponding % signal relative to background. The remarkable activity of compound 5 (IC50 1.18 μM), depicted in Fig. 3A, prompted us to investigate the influence of specific functional groups on NEDD4 inhibitory activity (Table 2). The reduced activity of compounds 11 and 13, featuring a hydrogen and a nitro group in place of the chlorine atom, as well as the partially reduced activity of compound 12 with a bromine substitution, underscore the critical role of chlorine in the hydrophobic pocket. This halogen establishes key chlorine/aliphatic interactions, which are among the most common hydrophobic contacts in protein-ligand interactions29.

A The modification of Norclomipramine’s nitrogen methyl groups with various electrophilic warheads led to the discovery of compound (cmpd) 5, which exhibits low micromolar potency. B Close-up view of HECT NEDD4 bound to compound 15 (PDB 9H9T). Side chains of residues involved in inhibitor binding as well as residue Cys627, covalently bound to the inhibitor are highlighted. C Ub-TR-FRET assay performed with full-length protein NEDD4 pre-incubated for 1 hour with increasing concentrations of compound 15 before ATP addition and measurement. Plots show single time-point measurements of FRET signal to background ratio (% Signal/Background) as function of time (minutes). Mean ± SEM (n = 4). D Ub-TR-FRET assay as in C using the HECT domain of NEDD4. Mean ± SEM (n = 4).
Additionally, we found that the spacer length significantly modulates the proximity of Michael acceptors to C627 (Table 2). Notably, the four-carbon spacer in compound 15 resulted in a substantial increase in activity (IC50 = 0.69 μM) compared to the three-carbon chain in compound 5, while a two-carbon spacer (compound 14) completely abolished inhibition. Further extension of the carbon chain (compound 16) led to a decrease in activity (IC50 = 2.05 μM). These results led us to further characterize compound 15.
Covalent binding of compound 15 to C627 was experimentally tested. Lack of inhibitory activity on the C627A mutant substantiated our assumption (Supplementary Fig. 1C). Additionally, its selective binding to C627, rather than other reactive cysteines, was validated by the efficient formation of Ub-thioester adducts across the enzymes in the ubiquitin cascade (Supplementary Fig. 1D-F). Finally, the X-ray crystal structure of the NEDD4 HECT domain in complex with compound 15 was solved at 2.17 Å, with Rwork = 21.9% and Rfree = 25.2% (Table 1, Fig. 3B). The structure revealed that all interactions established by the tricyclic ring of Norclomipramine are conserved, and the covalent bond with C627 is indeed formed. We hypothesized that compound 15 may also be effective in the context of the full-length enzyme. Consistent with this, incubation of compound 15 exhibited dose-response inhibitory activity on full-length NEDD4 protein (Fig. 3C) similar to the one obtained with the isolated HECT domain (Fig. 3D), with an IC50 value of 1.8 ± 0.182 μM. Thus, compound 15 was further characterized in terms of biochemical selectivity profile.
Selectivity profile of compound 15
Members of the NEDD4 family share a similar structural design, with the HECT domain being particularly conserved also in the inhibitor-binding pocket located in the N-lobe (Fig. 4A). However, only NEDD4-Like has a cysteine residue equivalent to C627 in NEDD4, while ITCH, WWP1 and WWP2 possess a cysteine located at a different position within the pocket (Fig. 4B). To test the activity of compound 15 on other family members, we used optimized Ub-TR-FRET assays with purified HECT domains from NEDD4-Like, ITCH, and HECW2 as representative of the various inhibitor-binding pocket composition (Fig. 4A, B). Compound 15 showed no inhibition for any of these proteins, including NEDD4-Like, even at high concentrations (Fig. 4C, Supplementary Fig. 2A, B and Table 3). A structural comparison explains this exquisite selectivity. Overlaying the compound 15 structure with that of NEDD4-Like HECT (PDB 2ONI, Fig. 4D) revealed that, while C682 in NEDD4-Like aligns with C627, the bulky F608 in NEDD4-Like, replacing L553 in NEDD4, clashes with one of the phenyl rings of compound 15. This was confirmed by testing compound 15 against a NEDD4 L553F mutant, which resulted in no inhibition (Fig. 4E), demonstrating that a phenylalanine at this position is sufficient to prevent the covalent binding of the inhibitor.

A Amino acids alignment, based on 3D structures, of NEDD4 and NEDD4 family members indicating the conserved and variable residues of the inhibitor binding pocket. Cys627, covalently bound to the inhibitor is highlighted in yellow, Phe residues corresponding to amino acid 553 in NEDD4 are highlighted in orange, Leu residues corresponding to the same position are in red. B Close-up view of the inhibitor binding pocket of ITCH (PDB 3TUG). Residue Cys610, positioned differently within the pocket, is highlighted. C Ub-TR-FRET assay performed using the HECT of NEDD4 (left panel) or NEDD4-Like (right panel) in absence or presence of compound 15 at the indicated concentrations. Plots show time-point measurements of FRET signal to background ratio (% S/B) as function of time (minutes). Mean ± SEM (n = 2). D Superposition of the N-lobes of HECT NEDD4 in complex with compound 15 (PDB 9H9T) and HECT NEDD4-Like structures (PDB 2ONI). The presence of Phe608 (orange) in NEDD4-Like hinders inhibitor binding. Leu residue corresponding to the same position in NEDD4 is in green. The respective Cys residues are highlighted using the same colour scheme. E Ub chain formation assay with the indicated HECT domains treated or not with compound 15 at 10 µM concentration. Reactions were stopped at 30 min by quenching the reaction with Laemmli buffer with reducing agent. IB as indicated. Mono-, di- and polyUb chains are labeled on the right. Bottom, Coomassie staining showing comparable loading of proteins. F Schematic representation of the target validation workflow using a mass spectrometry-based approach. A549 wild-type (WT) and NEDD4 knockout (KO) cell lines were treated with 1 µM compound 17B for 4 h. A total of 2 mg of urea lysates were subjected to streptavidin-based immunoprecipitation. The bound proteins were eluted, separated by gel electrophoresis, and analysed by LC-MS/MS. G One-tenth of the protein complexes bound to streptavidin beads were analysed by IB, as indicated. For input controls, 30 µg of total cell lysates were loaded.
To assess the in vivo binding specificity of our inhibitors, we generated a biotinylated derivative, compound 17B, which retained comparable activity in vitro (Supplementary Fig. 3A–D). As an internal control, we used CRISPR-Cas9 to generate NEDD4 knockout A549 cells, which are relatively insensitive to NEDD4 depletion. After cell lysis and streptavidin-based immunoprecipitation, we performed label-free quantitative proteomics, alongside immunoblot analysis of NEDD4 family members expressed at robust levels in A549 cell line (Supplementary Data File 1 and Fig. 4F, G). These experiments demonstrated that compound 17B selectively engages NEDD4, but not other family members (Fig. 4G), nor other E3 ligases (Supplementary Data File 1) also in cellulo.
The NEDD4 enzyme is fully capable of catalyzing both mono- and polyubiquitination of its specific substrates30,31. Since the mechanism of inhibition involves displacing ubiquitin from the Ub exosite located in the N-lobe of the HECT domain—a region known to variably influence catalysis15,16,17,18—we aimed to assess the effect of compound 15 on different types of NEDD4 substrates. We monitor the activity towards polyubiquitinated substrates such as WBP2 (Fig. 5A) and γ-ENaC (Fig. 5B) or towards the endocytic adaptor Eps15, a previously characterized monoubiquitinated substrate (Fig. 5C, D)30,32. WBP2 and γ-ENaC ubiquitination is severely affected when NEDD4 is incubated in the presence of compound 15 (Fig. 5A, B). In contrast, no impairment in Eps15 monoubiquitination was observed either in vitro or in vivo (Fig. 5C, D), confirming that the inhibitor impairs Ub-chain elongation while leaving monoubiquitination unaffected. Notably, in cells treated with compound 15, pAKT levels were markedly reduced, similar to previous findings obtained after NEDD4 depletion33. As further validation of target specificity, NEDD4-Like is completely insensitive to the compound 15 treatment when tested on all the substrates (Fig. 5A–C).

A Substrate ubiquitination assay with the indicated E3s treated or not with compound 15 (10 µM). The complete reaction was loaded onto a gel. IB as indicated. Arrowhead indicates the unmodified WBP2 protein. B Same as in A, using GST-γENAC as substrate. At the end of the reaction, substrate was separated by centrifugation and loaded onto a gel. IB as indicated. Bottom, Coomassie staining showing comparable loading of proteins. C Same as in A, using GST-Eps15 as substrate. Top panel, experiment performed with NEDD4, bottom panel with NEDD4-Like. Coomassie staining showing comparable loading of proteins are reported. D B82L cells were serum starved overnight, treated with compound 15 (1 µM) for 4 h and then stimulated with EGF (100 ng/ml) for the indicated time points. Lysates were analyzed by IB with the indicated antibodies.
Structure Activity Relationship (SAR) around compound 15
Before exploring the structure-activity relationship (SAR) around compound 15, its biochemical selectivity was evaluated against Norclomipramine’s original targets, specifically the 5HT transporter (5HT) and Norepinephrine transporter (NET). As shown in Table 3, compound 15 retained high affinity for the 5HT transporter measured by transporter binding assay, prompting the design and synthesis of additional analogs (Supplementary Table 2).
The initial modifications focused on functionalizing the aliphatic nitrogen with various alkyl and phenylalkyl groups. Compounds such as N-methyl (17), N-ethyl (18), and N-isopropyl (19) retained activity, although they were slightly less potent than compound 15, with IC50 values of 1.26 µM, 1.77 µM, and 1.83 µM, respectively (Supplementary Table 2). More sterically hindered substitutions, such as N-benzyl in compound 20, led to a significant loss of activity.
Subsequent medicinal chemistry efforts targeted the alkylating chain’s ester group, aiming to reduce susceptibility to metabolic degradation. Replacing the ester with an amide in compound 21 led to a 23-fold decrease in potency (IC50 = 16.30 µM), consistent with earlier observations (Supplementary Table 1). Other modifications, such as methyl (22) and isopropyl (24) hydroxamates, failed to enhance potency, while the ethyl hydroxamate derivative (23) displayed similar activity to compound 15. The most promising bioisosteric replacement was the cyano group, with derivative 25 exhibiting the best activity (IC50 = 0.34 µM).
The final SAR exploration focused on variations of the tricyclic scaffold. Introducing an amide function into the 7-membered azepinic ring (compounds 26 and 27) reduced activity compared to compound 15. In contrast, oxazepine analogs (28 and 29) maintained similar activity levels. Further modification of the aromatic ring, replacing it with a N-methyl pyrazole, yielded compound 30, which demonstrated improved potency (IC50 = 0.16 µM). Building on these findings, two additional derivatives (31 and 32) were synthesized, incorporating the most effective moieties identified from earlier dibenzotricycle modifications. Compound 32 emerged as the most potent (IC50 = 0.12 µM).
The four most potent inhibitors (25, 30, 31, and 32) were then subjected to further characterization, focusing on their selectivity profiles across the NEDD4 ligase family as well as their activity against 5HT and NET (Table 4). Interestingly, all four compounds displayed high selectivity for NEDD4 over other HECT E3 ligases in the NEDD4 family. Notably, replacing the benzene ring with pyrazole completely abolished binding to the 5HT transporter, likely due to the loss of π-stacking interactions typically facilitated by the benzene ring with the conserved phenyl residue of the 5HT receptor34. Finally, the antiproliferative activity and in vitro ADME (Absorption, Distribution, Metabolism and Excretion) properties of compounds 25, 30, 31, and 32 were assessed (Supplementary Table 3). All four compounds showed significant and comparable antiproliferative activity when tested on H292 cell line and high plasma protein binding and medium-to-high in vitro clearance in both human and mouse models. Importantly, the nitrile-containing compounds (25 and 32) demonstrated superior stability in mouse plasma compared to the other derivatives.
Overall, the derivatives featuring the N-methyl pyrazole group (30, 31, and 32) exhibited excellent potency, selectivity, and favorable in vitro ADME properties. Among them, compound 32 was selected for further evaluation as an early representative for pharmacokinetic studies. The primary plasma pharmacokinetic parameters are summarized in Table 5. Following a single intravenous (iv) dose of 5 mg/kg, compound 32 exhibited a systemic plasma clearance exceeding the hepatic blood flow (90 ml/min/kg in mice), with an elimination half-life of 44 minutes and a peak concentration (Cmax) of 2.4 µM within 5 min. The high volume of distribution at steady state (Vss = 4 L/kg) indicated extensive tissue distribution. After oral administration (os), in a single dose of 10 mg/kg, compound 32 was rapidly absorbed, reaching a Cmax of 0.1 µM at 30 min, with an oral bioavailability (F%) of 37.5% and a mean residence time (MRT) of approximately 6.5 hours. This pharmacokinetic profile suggests that compound 32 is well-suited for oral administration in future in vivo efficacy studies.
Discussion
Previous efforts to develop NEDD4 inhibitors have largely been hindered by poor selectivity, weak binding affinities, and the inability to differentiate between closely related ligases, such as NEDD4L and other HECT family members22,23,24,25.
In this study, we have successfully identified and characterized a novel class of compounds that demonstrate high specificity and potency in inhibiting NEDD4. Through a series of structural and biochemical studies, we have gained insights into the mechanism of action of these inhibitors, allowing us to fine-tune their selectivity and pharmacokinetic properties.
Norclomipramine was initially recognized as a broad-spectrum inhibitor of HECT-type E3 ligase, with limited specificity26. However, in the present study, we have demonstrated that Norclomipramine and its covalent derivatives offer a more refined and selective mechanism of inhibition, which is distinct from the catalytic cysteine-targeting inhibitors originally hypothesized26. This finding is supported by our structural and biochemical data, showing that the lead compound 15 displaces Ub from the exosite, impairing Ub-chain elongation without affecting the enzyme’s monoubiquitination activity.
Importantly, our compounds exhibited excellent selectivity for NEDD4 over other members of the NEDD4 family, including the closest related NEDD4-Like. The subtle structural differences in the Ub exosite pockets underscore the crucial role of the hydrophobic cleft in shaping inhibitor binding, even before covalent attachment to the non-catalytic C627 residue. This highly specific binding mode enhances both the potency and selectivity of our compounds compared to previously characterized inhibitors23 (Supplementary Fig. 2C and Supplementary Table 1). Moreover, this specificity paves the way for the development of additional inhibitors tailored for other NEDD4 family members that feature distinct cysteine residues in the same pocket (e.g., cysteine 610 in ITCH, Fig. 4B).
Targeted covalent inhibitors have recently revamped in drug discovery, and our compounds well fit in this scenario, as they are able to target a non-catalytic nucleophile that is uniquely conserved across the target protein family. Our medicinal chemistry campaign led to the development of several analogs with improved potency and selectivity. Of particular interest is compound 32, which combines an N-methyl pyrazole moiety with a nitrile group, resulting in the highest potency (IC50 = 0.12 μM) among the tested compounds. In addition to its biochemical efficacy, compound 32 showed favorable in vitro ADME properties, including high protein binding and stability in mouse plasma. Its pharmacokinetic profile, characterized by rapid absorption, extensive tissue distribution, and moderate oral bioavailability, makes it an attractive candidate for oral dosing in future in vivo efficacy studies.
This selective inhibition of polyubiquitination is particularly relevant, as it preserves NEDD4’s physiological function in monoubiquitinating substrates—a process essential for various cellular signaling pathways such as endocytosis, trafficking and other non-degradative processes35,36. By allowing NEDD4 to retain its monoubiquitination capacity while blocking polyubiquitination often associated with the oncogenic activity of NEDD4 in cancer37, these inhibitors strike a balance between maintaining normal cellular functions and inhibiting the oncogenic mechanisms driven by NEDD4.
This unique mechanism of inhibition also holds promise for the development of a novel class of chemical probes. These probes could be designed to modulate NEDD4 activity selectively, bringing it into proximity with specific proteins of interest (POI) to force their monoubiquitination. Such targeted tools would enable researchers to manipulate monoubiquitination events, and their consequences in cells, with precision.
In summary, the selective inhibition of polyubiquitination, combined with favorable pharmacokinetics, positions compound 32 as a leading candidate for preclinical development aimed at targeting NEDD4’s oncogenic functions. Our work represents a significant advancement in the field, overcoming the challenges of specificity and selectivity that have historically limited the development of NEDD4 inhibitors. Further studies will focus on evaluating the in vivo efficacy of compound 32 and assessing its potential as a targeted therapy in NEDD4-driven cancers.
Methods
Reagents
Antibodies and their suppliers were: mouse monoclonal anti-Ub (clone ZTA10, generated in house, dilution 1:5, ref. 38), mouse monoclonal anti-WBP2 (D12, Santa Cruz Biotechnology, sc-514247), rabbit polyclonal anti-NEDD4 (raised against the second WW2 domain of NEDD4 aa 348-382, generated by Eurogentec, dilution 1:5000); rabbit monoclonal anti NEDD4-Like (kindly provided by Pietro De Camilli, dilution 1:1000); mouse monoclonal anti AIP4/ITCH (BD Transduction Laboratories 611199, dilution 1:1000); rabbit polyclonal anti HECW1 (Sigma-Aldrich HPA007593, dilution 1:500); rabbit polyclonal anti HECW2 (raised against aa 596-749 of human HECW2, generated by Eurogentec, dilution 1:500), HRP-conjugated goat anti-mouse IgG (IB 1:10000; Bio-Rad 1721011), HRP-conjugated goat anti-rabbit IgG (IB 1:10000; Bio-Rad 1706515). Biotinylated Ub was from Enzo Life Sciences. Bovine Ubiquitin, Biotin and ATP were from Sigma-Aldrich. Streptavidin HRP and Imperial Protein Stain (Thermo Scientific). Ub TR-FRET mix from South Bay Bio (SBB-TR0051). EGF was from PeproTech. ATP was from JenaBioscience (NU-1010).
Cell lines
A549 (from NCI-60) cells were maintained in DMEM (Euroclone, ECB7501L), supplemented with 10% FBS (Euroclone, ECS0180L) and 2 mM L-glutamine (Euroclone, LOBE17605F). NCI-H292 cells (from ATCC) were growth in RPMI 1640 (Euroclone, ECB9006L), supplemented with FBS 10%, 2 mM L-glutamine, and Pen/Strep 1% (EuroClone, ECB3001L).
To generate the A549 NEDD4 knockout (KO) cell line using CRISPR/Cas9, A549 cells stably expressing Cas9 were electroporated with a guide RNA (gRNA) targeting exon 9 of the NEDD4 gene (gRNA sequence: 5’-ATGAATCTAGAAGAACACAG-3’). Single-cell clones were screened for NEDD4 deletion by immunoblotting, immunofluorescence, and immunoprecipitation. Genomic alterations were further characterized by PCR amplification and sequencing of the targeted region. Clone 2.10, selected for subsequent experiments, harbours two distinct mutant alleles: one with a 2-nucleotide deletion and the other with a 14-nucleotide deletion. Both mutations result in frameshifts that introduce premature stop codons.
Cell Growth Assay
The antiproliferative effect of the inhibitors on cell proliferation was evaluated against NCI-H292 (Non-Small Cell Lung cancer) tumor cell lines using the CellTiter-Glo® 2.0 luminescent cell viability assay (CTG; G9243, Promega, Madison, WI) according to the manufacturer’s instructions. Exponentially growing NCI-H292 cells were seeded at a density of 525 cells/well in 35 µL of media using 384-well plates (Cat# 781080, Greiner Bio-One). Twenty-four hours post-seeding, cells were treated with serial dilutions of NEDD4 inhibitors and incubated for 72 hours. NEDD4 inhibitors were initially dissolved in 100% DMSO at 10 mM and further diluted 1:10 to generate five concentrations ranging from 0.005 to 50 µM. Following compound addition, the final well volume was adjusted to 40 µL. After the 72-hour incubation, 40 µL of CellTiter-Glo (CTG), diluted 1:2 in water, was added to each well. Plates were incubated for 15 minutes at room temperature before measuring luminescence using the Infinite F200 Reader (Tecan). IC50 values were calculated using custom MATLAB scripts. All steps including cell seeding, compound treatment, and CTG addition were performed using a Hamilton MicroLab STAR liquid handling system.
DNA constructs and site-directed mutagenesis
E1, UbeE2D3, NEDD4 HECT and full length, γENAC and Eps15 constructs were previously described15,30. pET43-HIS-MBP-HECW2 HECT domain was amplified from cDNA clone (MGC:150803, IMAGE:40125745) with the following oligos:
5’- GGAAGATCTCCTTACAAGCGGGATTTC -3’‘ and
5’- CCGCTCGAGCGGTCACTCAAGTCCAAAAGTAC -3’, and cloned into pET43.1a_HIS-MBP vector using BamHI/XhoI sites. pGEX6P1-NEDD4-Like HECT was provided by J. Chelly39. pGEX6P1-WBP2-C-K222 was provided by J. Huibregtse40. NEDD4 L553A construct was engineered by site-directed mutagenesis, all the other NEDD4 mutants (F707A and C867A) were previously described15,30. All constructs were sequence-verified. Details are available upon request.
Protein expression and purification
The isolated HECT domain (NEDD4 WT and mutants, NEDD4-Like, ITCH), FL proteins (NEDD4 and NEDD4-Like) of the E3 ligases and substrates (Eps15, γENaC and WBP2) used in this study are produced as Glutathione S-transferase (GST) fusion proteins as reported in Refs. 15,30,41. Briefly, constructs were expressed in E. coli Rosetta cells at 18 °C for 16 hours after induction with 500 μM IPTG at an OD600 of 0.6. Cell pellets were resuspended in lysis buffer (50 mM Na-HEPES, pH 7.5, 200 mM NaCl, 1 mM EDTA, 0.1% NP40, 5% glycerol). Sonicated lysates were cleared by centrifugation at 45,000 × g for 45 min. Supernatants were incubated with glutathione-Sepharose beads (Cytiva). After 2 h at 4 °C, beads were washed with lysis buffer followed by a wash with PBS and finally equilibrated in cleavage buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM DTT and 5% glycerol). To cleave off GST, HRV 3 C protease (produced in house) was added in a ratio (w/w) 1:50 (protease:substrate) and incubated for 16 h at 4 °C.
The cleaved proteins were concentrated in Amicon Ultra Centrifugal Filters (MW cut-off 10 kDa) (MerckMillipore) and loaded onto a Superdex 200 size exclusion chromatography column (Cytiva) equilibrated with 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 5% glycerol, 1 mM DTT. Fractions containing purified proteins were collected and concentrated.
The HECT domain of HECW2 was produced as His-MBP fusion protein. It was expressed in Rosetta cells at 18 °C for 16 h after induction with 500 μM IPTG at an OD600 of 0.6. Cell pellets were resuspended in ysis buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5% Glycerol, 5 mM imidazole, 1 mM DTT, 0.1% NP40, 1 mM PMSF). Sonicated lysates were cleared by centrifugation at 45,000 g for 45 min. Supernatants were incubated with Amylose resin (NEB), previously equilibrated with lysis buffer for 3 h at 4 °C. Protein was eluted from beads with elution buffer (25 mM Tris pH 8, 500 mM NaCl, 5% glycerol, 1 mM DTT and 10 mM maltose). Eluted fractions were concentrated in Amicon Ultra Centrifugal Filters (MW cut-off 10 kDa) and loaded onto a Superdex 200 size exclusion chromatography column (Cytiva) equilibrated with 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5% glycerol, 1 mM DTT. Fractions containing purified HECT domain were collected and concentrated.
E1 (UBA1, Addgene clone 34965) enzyme and E2 enzyme (UBE2D3) were expressed as a 6×His fusion protein, produced and purified as previously described28,41. Bovine ubiquitin was purified onto a Superdex 75 size-exclusion chromatography column equilibrated with 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 1 mM DTT and 5% glycerol.
Isothermal Calorimetry (ITC)
ITC measurements were performed by titrating a 127 μM solution of HECT NEDD4 in 20 mM Tris-HCl pH 8.0, 200 mM NaCl, and 5% glycerol, with 2 mM Norclomipramine, dissolved in the same buffer, on a MicroCal PEAQ-ITC instrument (Malvern Panalytical, Malvern, UK) at 25 °C, with 2 μl injections. Raw data were analyzed with the integrated Malvern analysis software, and heat production was fitted to a one-set-of-sites binding model.
Pull-down assay
For pull-down experiments, 2 μM of GST-HECT of NEDD4 immobilized onto glutathione beads were pre-incubated or not with Norclomipramine (0.2 mM) in YY buffer (50 nM Na-HEPES pH 7.5, 150 mM NaCl, 1 mM EDT, 1 mM EGTA, 10% glycerol, 1% triton X-100) for 1 hour at 4 °C and then incubated with 250 ng of K63-linked Ub chains for 2 h at 4 °C. After four washes with YY buffer, samples were run on SDS-PAGE and detection of protein-bound compound was obtained by immunoblotting using anti-ubiquitin antibody. For the wash-out sample, the beads were washed with YY buffer prior to incubation with ubiquitin chains. Coomassie staining of the membrane was done to show the loading of the GST-fusion proteins.
Ubiquitination assay based on time-resolved fluorescence resonance energy transfer (Ub-TR-FRET) assay
TR-FRET reactions were set up in a white OptiPlate-384 well Microplate (Optiplate, Perkin Elmer, #6007290) using an automated liquid-handler (Hamilton STAR, Hamilton Robotics, Switzerland) and EnVision or EnSpire microplate reader with the following setting: Excitation Filter - UV2 (TRF) 320 nm; Emission Filter 1—APC 665 nm; Emission Filter 2—Europium 615 nm; Delay Time 50 μs; Window Time 400 μs. Briefly, enzymes are bacterially expressed, purified and mix at various concentrations (final concentration, 30 nM E1, 350 nM of E2, 50 nM NEDD4, 32 nM NEDD4-Like or 60 nM HECW2 and ITCH) together with TRF-Ubiquitin-mix (mixture of Eu-cryptate ubiquitin, Cy5-ubiquitin and wild-type ubiquitin, 100x SouthBayBio) in TR-FRET buffer (25 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 100 mM NaCl, 0.01% Tween20), generating a master mix.
All compounds were dissolved in 100% DMSO at high concentration and serially diluted in 96-well plates (from 100 µM to 0.005 µM), ensuring that the final DMSO concentration in the reaction mixture never exceeded 2%. 50 µl/well of the master mix is dispensed in a 384 well plate together with 3 µl of compounds or TR-FRET buffer (with the same amount of DMSO) were added and pre-incubated for 1 hour. The reaction was initiated with the addition of 22 µl of ATP in TR-FRET buffer (2 mM final concentration) and was allowed to proceed for 120 minutes at room temperature. Kinetic raw data were analyzed using GraphPad Prism software version 4.0 (GraphPad Software, San Diego, CA). Due to the sigmoidal shape of the curve representing the reaction kinetics, it was challenging to pinpoint the initial velocity. As a result, we evaluated the effect of the inhibitors by calculating the area under the curve (AUC) of the reaction. Data are reported as % signal/background, where the background corresponds to the signal obtained from the same enzymatic mix without ATP.
IC50 values were calculated using GraphPad Prism considering for each tested dose the corresponding AUC. Percentage of inhibition for each concentration is determined by the equation:
% inhibition = 100 x [1 - (X)/(MAX)], where MAX correspond to AUC of the vehicle (DMSO) treated enzyme, and X corresponds to the AUC of the compound treated enzyme.
For experiments shown in Supplementary fig. 2B, the reaction products at the end of the measurement were further analyzed using SDS-PAGE followed by anti-ubiquitin. Coomassie staining of the membrane is used as a loading control.
Ubiquitination assay
Ubiquitination assays were performed with purified enzymes (20 nM E1, 330 nM of E2, 240 nM E3) and 2 µM of ubiquitin in ubiquitination buffer (25 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 100 mM NaCl). For substrate ubiquitination, 2 µg of substrate (γENaC used as GST fusion proteins or WBP2 cleaved with HRV 3 C protease) were added to the reaction. Samples were incubated with the inhibitors for 1 hour at room temperature at the indicated concentrations. DMSO did not exceed the concentration of 2%. Ub-chain formation was started by adding 2 mM ATP to the reactions (50 µl total volume) and run at 30 °C. Reactions were stopped at the indicated time points by adding Laemmli buffer and samples run on SDS-PAGE. GST-Eps15 ubiquitination was performed in two steps as previously reported30. E1, E2, E3, Ub and ATP were incubated for 1 h in presence of the inhibitor at the indicated concentration. GST-Eps15 (2 µM) and Biotinylated Ub (1 µM) were added to the reaction and substrate ubiquitination was monitored using streptavidin HRP.
In case of GST-γENaC and GST-Eps15 ubiquitination, samples were centrifuged at 4000 g for 5 min to separate the pellet—containing the ubiquitinated substrates—and the supernatant – containing the enzymes and the soluble ubiquitin chains, if produced. The pellet was washed four times in RIPA buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1% SDS, 1 mM EDTA, 5% glycerol, 1% triton X-100) before loading on SDS-PAGE.
Detection was performed by immunoblotting using specific antibody. Coomassie-stained membrane was used to show loading of enzymes and GST-fusion protein after immunoblotting.
Immunoblotting
Proteins were separated by SDS-PAGE and blotted onto nitrocellulose membrane. Filters were blocked with 5% non-fat dry milk (or BSA) in TBS-T and incubated with the appropriate primary antibody for 1 hour at RT. The appropriate horseradish peroxidase-conjugated secondary antibody was incubated for 30 min at RT. Detection was performed with the enhanced ECL solution (Amersham). For Ub detection, proteins were blotted onto PVDF membrane. Filters were denatured for 30 min at 4 °C in 6 M Guanidine-HCl pH 7.6, 5 mM ß-mercaptoethanol and 1 mM PMSF prior to blocking in BSA.
E1 and E2 loading assay
The loading reaction was performed using the E1 (100 nM), the E2 (5 μM) and biotinylated Ub (10 μM) in ubiquitination buffer. Samples were incubated either with 100 µM compound 15 or 2% DMSO for 1 hour at room temperature before starting the reaction (50 µl total volume) with 2 mM ATP for 15 min at 37 °C. Thioester formation on E1 and E2 enzymes was monitored by quenching the reaction with Laemmli buffer with or without reducing agent (100 mM DTT). Samples were run on SDS-PAGE and analyzed by immunoblotting and Coomassie staining.
E2 to E3 trans-thioesterification assay
Thioester formation was tested by single-turnover kinetic assay, performed in two steps. First a loading reaction was performed using the E1 (100 nM), the E2 (5 μM) and biotinylated Ub (10 μM) in ubiquitination buffer with 2 mM ATP for 15 min at 37 °C. Reactions were subsequently quenched on ice by a two-fold dilution with 0.5 M EDTA. In a second step, the loaded E2 was mixed with E3 HECT NEDD4 (pre-incubated with 160-fold excess of inhibitor for 1 hour at room temperature) in ubiquitination buffer to the following final concentrations: 1.4 μM E2, 2.8 μM Ub, 1 μM E3 and 160 µM inhibitor or 2% DMSO. Thioester formation on the HECT NEDD4 was monitored by quenching the reaction at different time points with Laemmli buffer with or without reducing agent (100 mM DTT). Samples were run on SDS-PAGE and analyzed by immunoblotting with Streptavidin HRP and Coomassie staining.
In cellulo analysis of compound 17B
Cells were treated with compound 17B at 1 µM concentration for 4 hours and lysed with UREA-containing buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and 1 M UREA) supplemented with 20 mM sodium pyrophosphate, pH 7.5, 250 mM sodium fluoride, 2 mM PMSF, 10 mM sodium orthovanadate, and protease inhibitors (Calbiochem). Lysates were cleared by ultracentrifugation in MLA 130 rotor (Beckman Coulter) for 45 min at 122,000 × g.
2 mg of lysates were incubated with DB MYONE streptavidin T1 beads (ThermoFisher Scientific) for 2 h at 4 °C. After extensive washes with lysis buffer, beads were re-suspended in Laemmli buffer and proteins were run on sodium dodecyl sulfate-polyacrylamide gel (4–20% TGX precast gel, Bio-Rad or Bolt BisTris plus 4-12%, ThermoFischer Scientific). One-tenth of the eluted samples were analysed by IB, while the remaining materials were subjected to LC–MS/MS analysis.
Liquid chromatography–tandem MS (LC–MS/MS) analysis
Entire gel lanes were processed using the STAGE-digestion protocol42. Peptides were resuspended in 10 μl of solvent A (2% acetonitrile, 0.1% formic acid), and 3 μl of each sample were injected into a quadrupole Orbitrap Q-Exactive mass spectrometer (Thermo Scientific) coupled to a UHPLC Easy-nLC 1000 system (Thermo Scientific). The LC system was connected to a 25 cm fused-silica emitter (75μm inner diameter; New Objective), packed in-house with ReproSil-Pur C18-AQ resin (1.9 μm; Dr. Maisch GmbH, Ammerbuch, Germany) using a high-pressure bomb loader (Proxeon, Odense, Denmark). Mass spectrometry analysis was performed using LC–MS/MS on a quadrupole Orbitrap Q-Exactive HF mass spectrometer (Thermo Scientific). Peptide separation was carried out on a linear gradient from 95% solvent A to 50% solvent B (80% acetonitrile, 0.1% formic acid) over 33 min, followed by an increase to 100% solvent B in 2 min, at a constant flow rate of 0.25 μl/min.
MS data were acquired in data-dependent acquisition (DDA) mode using a top-15 method for HCD fragmentation. Survey full-scan MS spectra (m/z 300–1750) were acquired in the Orbitrap at a resolution of 60,000, with an AGC target of 1e6 and a maximum injection time (IT) of 120 ms. For HCD MS/MS scans, resolution was set to 15,000, with an AGC target of 1e5, IT of 120 ms, normalized collision energy of 28, and an isolation window of 3.0 m/z. Each sample was analyzed in two technical replicates.
For protein identification the raw data were processed with MaxQuant version 1.5.2.8. Peptides were identified from the MS–MS spectra searched against the uniprot_cp_Human_2020 using the Andromeda search engine, in which trypsin specificity was used with up to two missed cleavages allowed. Cysteine carbamidomethylation was used as a fixed modification, methionine oxidation and protein amino-terminal acetylation as variable modifications. The mass deviation for MS and MS–MS peaks was set at 5 and 20 ppm respectively. The peptide and protein false discovery rates (FDRs) was set to 0.01; minimum peptides 2, at least 1 unique, minimum length peptide 6 aminoacids. The lists of identified proteins were filtered to eliminate reverse hits and known contaminants.
Label-free quantification (LFQ) was performed using the ‘match between runs’ feature (time window: 5 min), and only proteins with a minimum ratio count of two were retained. LFQ intensity values were used for quantitation after normalization across the dataset. Proteins were manually curated to remove nonspecific binders detected also in biotin-only samples, and proteins not identified in both technical replicates. Statistical analyses were conducted using Perseus (Version 1.5.1.6) within the MaxQuant environment. A p-value threshold of 0.05 was applied. Missing values were imputed with random numbers from a normal distribution using the ‘imputation’ function (width: 0.3; down shift: 1.8, separately for each column).
Crystallization and structure determination
Crystals of the HECT NEDD4:Norclomipramine complex and or HECT NEDD4:compound 15 complex were obtained soaking HECT NEDD4 crystals with the inhibitors. Crystals were grown in hanging drops in 24 wells with the vapor diffusion technique. Drops were set up mixing 1 μl of protein solution at ca. 10 mg/ml with 1 μl of reservoir solution containing 100 mM MES pH 6.0, 6–10% PEG 400, 90 mM MgCl2, 5 mM TCEP. Nicely diffracting crystals were obtained by microseeding the drops after an overnight equilibration. Compounds were bound to HECT NEDD4 by soaking, adding to the crystal drops 1-2 μl of a cryoprotectant solution containing 100 mM MES pH 6.0, 8-10% PEG 400, 20% glycerol, 1–4 mM inhibitor (0.25–1% DMSO). Crystals were harvested and cryo-cooled in liquid nitrogen after few hours or overnight soak and data collections were carried out at X06DA (PXIII) of the Swiss Light Source at the Paul Scherrer Institut, or at the European Synchrotron Radiation Facility (Grenoble) on beamline ID30A-1. Diffraction data were reduced with the autoPROC or xia2 automated data reduction suites, making use of the XDS/XSCALE and the CCP4 programs. Molecular replacement and model refinement were carried out with Phaser and phenix.refine implemented in the Phenix suite, with iterative cycles of manual building in Coot and refinement. HECT NEDD4 N-lobe and C-lobe models, from PDB entry 2xbf, independently, were used as search models for molecular replacement.
Transporters Binding Assays
The binding assay were conducted by Eurofins Cerep. Compound binding was calculated as the % inhibition of the binding of a radioactively labelled ligand, specific for each target. In each experiment the reference compound was tested concurrently with the test compounds, and the data were compared to historical values previously determined at Eurofins. The results are expressed as a percentage of inhibition of specific binding, calculated as 100–[(measured specific binding/control specific binding) * 100], obtained in the presence of the test compounds.
The experiment was accepted in accordance with Eurofins validation Standard Operating Procedure.
For Norepinephrine transporter assay, human Norepinephrine transporter was expressed in CHO cells transfected with a vector expressing cDNA of human protein and subsequently purified. [3H] nisoxetine at the concentration of 1 nM was used as radioactive ligand (Kd 2.9 nM). Compounds were tested in triplicate at the indicated concentrations, and incubated for 120 min at 4 °C temperature. Non-specific binding was determined in presence of 1 mM Desipramine. Bound radioactivity was then measured via scintillation counting. Reference compounds for the assay: Protriptyline IC50 3.7 nM.
For Serotonin transporter assay, human 5HT transporter was expressed in CHO cells with a vector expressing cDNA of human protein and subsequently purified. [3H] imipramine at the concentration of 2 nM (Kd 1.7 nM) was used a radioactive ligand. Compounds were tested in triplicate at the indicated concentrations, and incubated for 60 minutes at room temperature. Non-specific binding was determined in presence of 10 mM Imipramine. Bound radioactivity was then measured via scintillation counting. Reference compounds for the assay: Imipramine IC50 4,7 nM.
Metabolic stability in liver microsomes
Test compounds were dissolved in DMSO at 0.2 mM concentration and pre-incubated at the final concentration of 1 µM for 10 min at 37 °C in potassium phosphate buffer 50 mM pH 7.4, 3 mM MgCl2, with mouse and human liver microsomes (Sigma M9441 and M0317) at 0.5 mg/ml concentration. After the pre-incubation period, reactions were started by adding the cofactors mixture (NADP, G6P and G6P-DH in a solution of 2% sodium bicarbonate); samples (25 µl) were taken at time 0, 10, 20, 30 and 60 min added to 150 µl of acetonitrile to stop the reaction together with Verapamil 0.02 µM as internal standard. After centrifugation at 14,000 g for 10 minutes, the supernatants were analyzed by LC-MS/MS. A control sample without cofactors was taken at 60 minutes in order to check the stability of test compounds in the matrix. 7-Ethoxycoumarin (7-EC) and propranolol were added as reference standards. Samples were analyzed on a Shimadzu HPLC coupled with an AB Sciex API 4500 Triple Quadrupole. Compounds were analyzed either using acidic or basic conditions. The percent of the area of test compound remaining at the various incubation times were calculated with respect to the area of compound at time 0 min. The rate constant, k (min-1) derived for the exponential decay equation (peak area/IS vs time) was used to calculate the rate of intrinsic clearance (Cli) of the compound using the following equation: Cli (µl/min/mg) = k/ microsomal conc. x 1000.
Plasma protein binding
Test compounds and the standard (Diclofenac) were dissolved in DMSO at 1 mM concentration. 3 µl of stock solutions were spiked in 397 µl of mouse or human plasma to obtain a final concentration of 10 µM. 100 µl of the mix was then dialyzed in the RED inserts (Thermo Scientific) against 300 µl of PBS buffer in the RED plate for 2 h at 37 °C, at 300 rpm stirring speed. At time 0 and at the end of the incubation 50 µl of dialyzed plasma were added to 50 µl of blank PBS buffer and 150 µl of ACN with Verapamil as internal standard solution (0.001 µM). Similarly, 50 µl of incubated PBS at time 0 and 2 hours were added to 50 µl of blank plasma and 150 µl of internal standard solution. Samples were centrifuged at 1300 g for 10 min and supernatants were transferred in vials for LC-MS/MS analysis. Results were calculated as follow: \({fu}=\)Acceptor area ratio /donor area ratio T = 0. % binding = (1-Fu) *100. Samples were tested in duplicate and analyzed on a Shimadzu HPLC coupled with an AB Sciex API 4500 Triple Quadrupole.
Plasma stability
Test compounds were dissolved in DMSO at 1 mM concentration. 1 µl of stock solutions were spiked in 199 µl of mouse or human plasma to obtain a final concentration of 5 µM and aliquots of 30 µl were taken at 0, 15, 30, 60 and 120 min and immediately quenched with 200 µl of a solution of ACN with Verapamil as internal standard (0.001 µM). Samples were vortexed and centrifuged for 15 min at 4 °C at 2600 g before analysis. ML10302 (Sigma) was used as positive standard control. Samples were analyzed on a Shimadzu HPLC coupled with an AB Sciex API 4500 Triple Quadrupole. The percentage of the area of the test compound remaining at the various incubation times was calculated in respect to the area of compounds at time 0.
In Vivo Drug Pharmacokinetic Studies
Pharmacokinetic experiments were performed using 4-week-old male nude CD-1 mice (Harlan, Italy), housed under standard conditions with free access to water and standard laboratory rodent diet. Animals were quarantined for approximately 1 week prior to the study. Each experimental group consisted of three animals.
Compound 32 was dissolved in 5% DMSO and 95% of 50:50 PEG400:H2O and administered to mice in single intravenous (iv) or oral (os) doses of 5 or 10 mg/kg, respectively. Blood samples were collected at 5, 30, 60, 180 and 360 min post dosing and plasma was separated by centrifugation at 2000 g for 10 min. Plasma proteins with internal standard were precipitated by adding 150 µl of cold acetonitrile at the final concentration of 50 ng/ml and samples were kept at -80 °C until submission to LC/MS/MS analysis. Samples were analyzed on a Shimadzu HPLC coupled with an AB Sciex API 4500 Triple Quadrupole. Pharmacokinetic parameters were calculated by a noncompartmental method using the software Analyst™ 6.1 (Applied Biosystems). Following PK parameters were evaluated:
Cmax: Maximum plasma concentration, Tmax: Time of maximum plasma concentration, AUC 0 − ∞ : AUC from time zero extrapolated to infinity, MRT 0 − ∞: Mean Residence Time (AUMC/AUC 0 − ∞) where AUMC is the area under the moment curve, CL:Clearance, Vss: Volume of distribution at steady state, T1/2: elimination half time =ln(2)/Ke (where Ke = -2.303 x slope of elimination phase), F%: oral bioavailability calculated as AUC os/AUC iv.
AUCs were calculated by linear trapezoidal rule. Graphical concentration-time curves are produced after Log transformation and data analyzed using the add-in program PK Solver 2.0.
Compound chemistry
Chemical synthesis of the compounds was performed as indicated in the Supplementary Information. For NMR spectra of the compounds see Supplementary Data 2.
Data availability
The mass spectrometry proteomics data are provided as Supplementary Data 1 and have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD062777. Numerical Source data for Graphs and charts and uncropped blot/gel images were provided as Supplementary Data 3-4. The pdb files for the solved crystal structures are provided as Supplementary Data 5-6 and have been deposited in the RCSB Protein Data Bank. HECT NEDD4:compound 15 (PDB 9H9T); HECT NEDD4:Norclomipramine (PBD 9H9O).
References
Stewart, A. K. et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 372, 142–152 (2015).
Soucy, T. A. et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009).
Torka, P. et al. Pevonedistat, a NEDD8-activating enzyme inhibitor, induces apoptosis and augments efficacy of chemotherapy and small molecule inhibitors in pre-clinical models of diffuse large B-cell lymphoma. EJHaem 1, 122–132 (2020).
Utecht, K. N. & Kolesar, J. Bortezomib: a novel chemotherapeutic agent for hematologic malignancies. Am. J. Health Syst. Pharm. 65, 1221–1231 (2008).
LaPlante, G. & Zhang, W. Targeting the ubiquitin-proteasome system for cancer therapeutics by small-molecule inhibitors. Cancers (Basel) 13 (2021).
Ramachandran, S. & Ciulli, A. Building ubiquitination machineries: E3 ligase multi-subunit assembly and substrate targeting by PROTACs and molecular glues. Curr. Opin. Struct. Biol. 67, 110–119 (2021).
Bekes, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Chamberlain, P. P. & Hamann, L. G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 15, 937–944 (2019).
Arnold, C. PROTAC protein degraders to drug the undruggable enter phase 3 trials. Nat. Med. (2024).
Ye, X., Wang, L., Shang, B., Wang, Z. & Wei, W. NEDD4: a promising target for cancer therapy. Curr. Cancer Drug Targets 14, 549–556 (2014).
Wang, X. et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 (2007).
Fouladkou, F. et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc. Natl Acad. Sci. USA 105, 8585–8590 (2008).
Plant, P. J., Yeger, H., Staub, O., Howard, P. & Rotin, D. The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J. Biol. Chem. 272, 32329–32336 (1997).
Staub, O. et al. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 15, 2371–2380 (1996).
Maspero, E. et al. Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 12, 342–349 (2011).
French, M. E., Kretzmann, B. R. & Hicke, L. Regulation of the RSP5 ubiquitin ligase by an intrinsic ubiquitin-binding site. J. Biol. Chem. 284, 12071–12079 (2009).
Ogunjimi, A. A. et al. The ubiquitin binding region of the Smurf HECT domain facilitates polyubiquitylation and binding of ubiquitylated substrates. J. Biol. Chem. 285, 6308–6315 (2010).
Zhang, W. et al. System-wide modulation of HECT E3 ligases with selective ubiquitin variant probes. Mol. Cell 62, 121–136 (2016).
Fajner, V., Maspero, E. & Polo, S. Targeting HECT-type E3 ligases - insights from catalysis, regulation and inhibitors. FEBS Lett. 591, 2636–2647 (2017).
Lorenz, S. Structural mechanisms of HECT-type ubiquitin ligases. Biol. Chem. 399, 127–145 (2018).
Chen, D., Gehringer, M. & Lorenz, S. Developing small-molecule inhibitors of HECT-type ubiquitin ligases for therapeutic applications: challenges and opportunities. Chembiochem 19, 2123–2135 (2018).
Mund, T., Lewis, M. J., Maslen, S. & Pelham, H. R. Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc. Natl. Acad. Sci. USA 111, 16736–16741 (2014).
Kathman, S. G. et al. A Small Molecule That Switches a Ubiquitin Ligase From a Processive to a Distributive Enzymatic Mechanism. J. Am. Chem. Soc. 137, 12442–12445 (2015).
Quirit, J. G. et al. Indole-3-carbinol (I3C) analogues are potent small molecule inhibitors of NEDD4-1 ubiquitin ligase activity that disrupt proliferation of human melanoma cells. Biochem Pharm. 127, 13–27 (2017).
Yong, D., Green, S. R., Ghiabi, P., Santhakumar, V. & Vedadi, M. Discovery of Nedd4 auto-ubiquitination inhibitors. Sci. Rep. 13, 16057 (2023).
Rossi, M. et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis. 5, e1203 (2014).
Boike, L., Henning, N. J. & Nomura, D. K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 21, 881–898 (2022).
Maspero, E. et al. Structure of a ubiquitin-loaded HECT ligase reveals the molecular basis for catalytic priming. Nat. Struct. Mol. Biol. 20, 696–701 (2013).
Ferreira de Freitas, R. & Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 8, 1970–1981 (2017).
Woelk, T. et al. Molecular mechanisms of coupled monoubiquitination. Nat. Cell Biol. 8, 1246–1254 (2006).
Boase, N. A. & Kumar, S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene 557, 113–122 (2015).
Polo, S. et al. A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature 416, 451–455 (2002).
Huang, Z. J., Zhu, J. J., Yang, X. Y. & Biskup, E. NEDD4 promotes cell growth and migration via PTEN/PI3K/AKT signaling in hepatocellular carcinoma. Oncol. Lett. 14, 2649–2656 (2017).
Sinning, S. et al. Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J. Biol. Chem. 285, 8363–8374 (2010).
Acconcia, F., Sigismund, S. & Polo, S. Ubiquitin in trafficking: the network at work. Exp. Cell Res 315, 1610–1618 (2009).
Magits, W. & Sablina, A. A. The regulation of the protein interaction network by monoubiquitination. Curr. Opin. Struct. Biol. 73, 102333 (2022).
Weber, J., Polo, S. & Maspero, E. HECT E3 ligases: a tale with multiple facets. Front Physiol. 10, 370 (2019).
He, F. et al. Myosin VI contains a compact structural motif that binds to ubiquitin chains. Cell Rep. 14, 2683–2694 (2016).
Broix, L. et al. Mutations in the HECT domain of NEDD4L lead to AKT-mTOR pathway deregulation and cause periventricular nodular heterotopia. Nat. Genet 48, 1349–1358 (2016).
Kim, H. C., Steffen, A. M., Oldham, M. L., Chen, J. & Huibregtse, J. M. Structure and function of a HECT domain ubiquitin-binding site. EMBO Rep. 12, 334–341 (2011).
Taibi, V., Polo, S. & Maspero, E. Monitoring HECT ubiquitination activity in vitro. Methods Mol. Biol. 2602, 81–92 (2023).
Soffientini, P. & Bachi, A. STAGE-diging: A novel in-gel digestion processing for proteomics samples. J. Proteom. 140, 48–54 (2016).
Acknowledgements
We thank Luigi Scietti and Giuseppe Ciossani from BSU- Biochemistry and Structural Biology Unit at European Institute of Oncology (IEO) for the support in protein crystallization and the staff at the European Synchrotron Radiation Facility and Swiss Light Source for assistance in data collection. We gratefully acknowledge the Ravelli family for their generous donation in support of the Experimental Therapeutics Program. We thank Michela Robusto for constructive discussions, Chiara Bigogno and Giulio Massimo Dondio (Aphad S.R.L.) for providing in vitro ADME and pharmacokinetic data. This work was supported by grants from Cariplo Foundation (2017-0746 to E.M.) Worldwide Cancer Research (grant 19-0003 to S.P.) and the Associazione Italiana per Ricerca sul Cancro (IG2018 - ID. 21328 to C.M). L.L, V.T. were and S.D. is a PhD student of the European School of Molecular Medicine (SEMM). Figures were made using BioRender.
Author information
Authors and Affiliations
Contributions
Conceptualization, E.M., M.Va., C.M., and S.P.; investigation, E.M., A.C., J.W., P.T., R.A., A.B., G.F., V.C., R.F., B.L., P.S., L.L., V.T., G.M., M.P., A.R., L.S., M.Vi., S.V., M.C., S.D., M.R., S.Pa.; methodology, E.M., G.F., S.V., S.Pa., A.Ba.; formal analysis, E.M., S.Pa., C.M., and S.P.; supervision, E.M., M.Va., C.M., and S.P.; data curation, E.M., M.V., S.Pa., C.M., and S.P.; writing—original draft, E.M. and S.P.; writing—review & editing, E.M., P.T., A.C., M.Va., M.Vi., S.Pa., C.M., and S.P.; funding acquisition, E.M., C.M., and S.P.
Corresponding authors
Ethics declarations
Competing interests
R.A., G.F., E.M., C.M., M.P., S.Pa., S.P., L.S. and M.Va. are patent co-inventors for “Hect E3 Ligase Nedd4 Inhibitors and uses thereof” (EP3976185B1). The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Danette Daniels and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Maspero, E., Cappa, A., Weber, J. et al. Structure-based design of potent and selective inhibitors of the HECT ligase NEDD4. Commun Chem 8, 164 (2025). https://doi.org/10.1038/s42004-025-01557-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01557-4
This article is cited by
-
Selective ubiquitination of drug-like small molecules by the ubiquitin ligase HUWE1
Nature Communications (2025)
