
Abstract
Deuterated alcohols are valuable synthetic targets due to their roles in pharmaceuticals, materials, and mechanistic studies. Conventional homogeneous strategies for their synthesis, while effective, often require expensive ligands and offer limited catalyst recovery. Heterogeneous catalysis, by contrast, provides a robust and recyclable alternative with enhanced scalability. Recent advances in supported metal nanoparticles and single-atom catalysts (SACs) have enabled high-efficiency and site-selective deuteration of alcohols. This Perspective presents heterogeneous catalytic systems as evolving into scalable and efficient platforms for deuterated alcohol synthesis, opening new directions for sustainable isotope incorporation.
Similar content being viewed by others


Introduction
Deuterium incorporation technique has become one of the powerful means for organic molecular design, providing unique opportunities to explore reaction mechanisms as well as modulate biological processes1,2,3,4,5,6. The distinct physicochemical properties of carbon-deuterium (C–D) bonds arise from deuterium’s two-fold greater mass compared to hydrogen, resulting in reduced vibrational frequencies and enhanced bond stability (Fig. 1a)7,8. These characteristics give rise to a profound kinetic isotope effect (KIE) that significantly alters molecular behavior9,10,11,12. Leveraging these properties, deuterated compounds play an important role in analytical chemistry as their defined mass shift and spectral distinguishability render them ideal internal standards in mass spectrometry and powerful tools for structure elucidation in NMR spectroscopy (Fig. 1b)13,14. Beyond analytical applications, deuterium incorporation has emerged as a powerful strategy for modulating drug metabolism15,16. By attenuating oxidative degradation, minimizing the formation of toxic metabolites, and improving pharmacokinetic properties, this approach has yielded significantly enhanced drug candidates17. The FDA approval of deutetrabenazine, the first deuterated drug, along with the growing pipeline of deuterium-containing clinical candidates, for example, allosteric inhibitor BMS-986165, highlights the transformative potential of this strategy (Fig. 1c)18,19,20.

a Structural difference between protium (1H) and deuterium (2H)7,8. b Mass spectrometry application: isotopic labeling enables clear distinction between unlabeled and labeled species13,14. c Representative deuterated drugs: Deutetrabenazine and Allosteric inhibitor BMS-98616518,19,20. d Hydroxyl groups in marketed drugs: examples include Advair22 and Jardiance23. e Alcohols as versatile building blocks: representative cases24,25,26,27,28.
Among various deuterated compounds, deuterated alcohols hold a particularly prominent position, as hydroxyl (OH) groups are present in a wide range of marketed drugs and bioactive molecules. Notably, around 30% of the top 200 best-selling drugs worldwide are associated with hydroxyl groups21. Representative examples include therapeutics such as Advair, an inhalation medication for asthma and chronic obstructive pulmonary disease (COPD)22, and Jardiance, an oral agent used to control blood glucose levels in type 2 diabetes and reduce cardiovascular risk23 (Fig. 1d). Moreover, small-molecule alcohol synthons, such as phenylethanol, menthol, and benzyl alcohol, serve as versatile intermediates in drug synthesis, enabling the construction of diverse active pharmaceutical ingredients through selective functional group transformations (Fig. 1e)24,25,26,27,28. Physiological studies have demonstrated that deuterated alcohols offer unique advantages over their non-deuterated counterparts. For instance, the α-deuteration of alcohols can substantially impede cytochrome P450 (CYPs)-mediated oxidation, as exemplified by deuterated losartan derivatives, which exhibit markedly enhanced metabolic stability 29. Furthermore, deuterated alcohols serve as precursors for specialized polymers with tailored optoelectronic properties and enhanced stability 30.
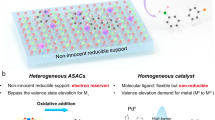
The significance of deuterated alcohols in the aforementioned applications has spurred continuous innovation in their synthetic methodologies, leading to the emergence of increasingly efficient and straightforward approaches29,31,32,33,34,35,36,37,38,39. Conventional strategies relying on stoichiometric reducing-type metal deuterides (e.g., LiAlD4, NaBD4) often encounter substantial limitations (Fig. 2a), including high cost, handling hazards, and poor compatibility with sensitive functional groups36,37,38,39. In contrast, homogeneous transition-metal catalyzed circumvents these challenges by avoiding the use of such sensitive deuterium reagents, thereby offering simpler operations and improved economic viability. Accordingly, Mo31,32 Ru33,34,35, Fe or Mn40, Ir29 complexes have been widely investigated for the synthesis of deuterated alcohols. However, these systems continue to face inherent limitations that hinder their broader applicability (Fig. 2b). In particular, they often depend on costly and air-sensitive ligands, require external base additives for effective deuterium incorporation, and present challenges in catalyst separation and reuse35,40.

Given these constraints, attention has increasingly shifted toward heterogeneous catalysis, including supported metal nanoparticles (e.g., Ru/C, Pd/C and ligand-modified Ru/C)41,42,43,44,45 and SACs46 which offer more practical and scalable platforms for the deuteration of alcohols. To the best of our knowledge, no comprehensive Perspective/Review has yet systematically summarized the methodologies and recent advances in the synthesis of deuterated alcohols. This Perspective aims to fill that gap by providing a focused discussion of recent advances in heterogeneous catalytic strategies for alcohol deuteration, while also comparing heterogeneous and homogeneous catalysts in terms of activity, chemo selectivity, and site selectivity, thereby highlighting the unique advantages of heterogeneous systems. In line with the Perspective format, we further outline key challenges and future directions for heterogeneous deuteration catalysis, with the goal of stimulating the development of next-generation catalytic systems in this area.
Supported nanoparticles for the synthesis of deuterated alcohols
The use of heterogeneous catalysts for the deuteration of alcohols can be traced back to 200247. At that time, Sajiki and co-workers showed that a Pd/C–D2O–H2 system could promote benzylic H/D exchange at room temperature (Fig. 3). Under these mild conditions, deuteration was confined to benzylic positions and no α-deuteration of the hydroxyl-adjacent C–H bonds were observed. For example, in the case of 3-phenylpropan-1-ol (1), deuterium incorporation occurred exclusively at the benzylic position, while the α-C–H bond next to the hydroxyl group remained untouched. In their subsequent 2004 study, they discovered that increasing the temperature to 110–160 °C not only maintained efficient benzylic deuteration but also enabled H/D exchange on the alkyl side chains, including aryl-substituted alkyl carboxylic acids, alkylbenzenes, and aryl-substituted alkyl alcohols41. This work therefore established one of the earliest heterogeneous protocols for the deuteration of aryl-substituted alkyl alcohols. For example, at 110 °C, deuteration of 4-phenylbutan-2-ol (2) was no longer restricted to the benzylic position and deuterium incorporation at the α- and β-positions relative to the hydroxyl group was also observed. The strong influence of temperature on both the deuterium incorporation and the position is further illustrated by 5-phenyl-1-pentanol (3). For example, at 110 °C, no deuteration occurred at the α-position adjacent to the hydroxyl group, whereas at 160 °C, deuterium incorporation was detected at this site and the deuterium incorporation at other alkyl positions increased markedly (Fig. 3).

Temperature-dependent, Pd/C-catalysed H/D exchange of aryl-substituted alkyl alcohols in D2O under H241.
Although the authors recognized that hydrogen gas is essential for the Pd/C–D2O system, its specific role was not clarified in the initial reports. To address this question, Sajiki and co-workers subsequently examined the function of H2 more carefully. They found that when Pd/C was pretreated with H2 in D2O and the H/D exchange reaction was then performed under an argon atmosphere, deuteration still occurred only with reduced efficiency. This result suggests that hydrogen gas is not strictly required for the H/D exchange itself and may instead serve mainly to activate the catalyst. Based on these observations, the authors proposed that a small amount of H2 functions primarily to activate Pd0 on carbon, most likely through the formation of surface Pd–H species that facilitate oxidative insertion into C–H bonds and promote catalytic turnover48.
However, the above preliminary study did not provide a sufficient elucidation of the reaction mechanism and of the Pd/C–D2O–H2 system, which prompted subsequent research to further investigate its mechanism and expand its synthetic applications. In 2007, they developed a Pd/C-catalyzed, redox-mediated H/D exchange strategy that enabled the efficient synthesis of deuterated alcohols from either secondary alcohols or their corresponding ketones and provided detailed mechanistic validation for the deuteration process42. In these studies, they found that 4-phenyl-2-butanol (2) afforded not only the expected deuterated alcohol but also the corresponding deuterated ketone in approximately a 60:40 ratio under the Pd/C–D2O–H2 conditions, and that using the 4-phenyl-2-butanone (4) as the starting material generated a similar mixture, with both the deuterated ketone and the reduced, deuterated alcohol formed in 61:39 proportion (Fig. 4a). This reciprocal interconversion demonstrated that oxidation of the alcohol to the ketone and reduction of the ketone back to the alcohol proceed concurrently under hydrogenation conditions in the absence of any external oxidant.

a Reversible redox-mediated H/D exchange between 2 and its corresponding ketone. b Redox-mediated H/D exchange of linear and cyclic aliphatic alcohols and ketones. c Racemization of a chiral secondary alcohol supporting a ketone-mediated H/D exchange pathway. d Proposed redox-mediated mechanism for Pd/C-catalyzed H/D exchange of secondary alcohols.
Similar redox behavior and efficient deuterium incorporation were observed for a variety of substrates, including linear aliphatic alcohol (5) and ketone (6) as well as cyclic systems such as cyclooctanol (7) and cyclooctenone (8) (Fig. 4b). These results collectively indicated that the H/D exchange of secondary alcohols proceeds through initial oxidation to the corresponding ketone, followed by deuteration of the ketone and subsequent reduction to regenerate the deuterated alcohol. This mechanistic picture was reinforced by the greatly diminished deuterium incorporation observed for substrates that cannot undergo oxidation to a ketone, such as methyl ethers, silyl ethers, and tertiary alcohols. It was further supported by experiments with optically enriched secondary alcohol (5), which afforded a deuterated alcohol with complete loss of enantiomeric excess (Fig. 4c). Taken together, the ether experiments and the results from the chiral alcohol are consistent with a reversible interconversion through a ketone intermediate.
On the basis of these results, the authors proposed the reaction mechanism illustrated in Fig. 4d. The alcohol substrate A first undergoes Pd-catalyzed O–H activation to give the Pd alkoxide B, which then delivers the ketone C through β-hydride elimination. The ketone is reduced in the presence of hydrogen and D2O to form the Pd–alkoxide species D, and repeated sequences of reduction and H/D exchange convert D into the deuterated alcohol G through intermediates F and H. For substrates that can form conjugated or allylic species, further dehydrogenation of F to H, followed by coordination to palladium to give I and J, allows additional H/D exchange at non-activated C–H sites. In this way, multiple cycles of oxidation, H/D exchange, and reduction lead to deeper deuterium incorporation along the carbon chain.
Although the Pd/C system enables deuteration of C–H bonds of alcohols, it fails to achieve site selective deuteration, particularly mono-site-selective transformations. In 2008, Sajiki and coworkers developed a ruthenium-on-carbon (Ru/C) catalyzed protocol for site-selective α-deuteration of alkyl alcohols that lack an aryl substituent in the presence of H2 and D2O (Fig. 5)43. Compared to conventional approaches that rely on stoichiometric metal deuterides or high-pressure D249,50, this protocol offers a practical and scalable alternative route to site-selective deuterated such alkyl alcohols across diverse substrates (Fig. 5a). Within this Ru/C catalytic system, the substrate scope was extensively explored. Secondary alcohols such as 2-decanol (9), 2-hexadecanol (10), 7-ethyl-2-methylundecan-4-ol (11), and cyclooctanol (6) underwent efficient α-deuteration under mild conditions. Primary alcohols including 1-octadecanol (12), 2-isopropyl-5-methylhexan-1-ol (13), 3,7-dimethyloctan-1-ol (14), and cyclohexyl methanol (15) also reacted smoothly under higher temperatures. Moreover, the method enabled site-selective multiple deuteration in diols and triols such as 1,2-ethanediol (16), 1,6-hexanediol (17), 1,10-decanediol (18), and 1,2,3-propanetriol (19), demonstrating broad compatibility with polyhydroxylated substrates. Furthermore, the observed racemization of (R)-2-decanol (20) together with 2-decanone (21) formation provides strong evidence for a redox-mediated mechanism, even though the possibility of direct C–H activation remains unresolved (Fig. 5b).

a Scope of Ru/C-catalyzed α-deuteration of non-aryl alkyl alcohols. b Mechanistic probe experiments supporting a redox-mediated pathway.
Building on their success with Ru/C-catalyzed α-deuteration of alcohols, in 2010, Sajiki and co-workers extended this strategy to carbohydrates, reporting an efficient and highly chemo- and site-selective approach to sugar deuteration enabled by a Ru/C–H2–D2O catalytic system (Fig. 6)44. The protocol achieved nearly quantitative α-deuteration at positions adjacent to hydroxyl groups in a variety of glycosides and six-membered ring sugars (22–25) under mild aqueous conditions, whereas other heterogeneous catalysts (such as Pd/C, Rh/C, Pt/C, Au/C, Ir/C, Ni/C) showed no comparable activity (Fig. 6a). This study further broadened the synthetic utility of heterogeneous deuteration to carbohydrate scaffolds.

a Scope of Ru/C-catalyzed α-deuteration of glycosides and six-membered sugars. b Proposed mechanism for Ru/C-mediated α-deuteration of carbohydrate substrates.
The authors proposed that the reaction likely proceeds through a C–H activation pathway, although no direct experimental evidence was provided. The process is initiated by coordination of D2O and H2 to Ru(0) to generate the active species K. The sugar substrate then coordinates to K through its hydroxyl oxygen, affording intermediate L. Oxidative addition at the α-C–H bond leads to formation of the Ru(II)–hydride complex M, which undergoes intramolecular H–D exchange to produce intermediate N. Reductive elimination from N, followed by aqueous workup, furnishes the α-deuterated product and regenerates the active catalyst (Fig. 6b). It is worth noting that, the frequently proposed direct C–H activation pathways for Pd/C and Ru/C, as exemplified in this sugar-deuteration study, remain largely hypothetical and, to the best of our knowledge, have not yet been corroborated by direct experimental evidence.
A subsequent 2012 study by Sajiki and co-workers further advanced this Ru/C–H2–D2O platform by demonstrating that the α-deuteration of carbohydrates can proceed with high levels of stereocontrol. Building on their earlier protocol for glycosides and six-membered sugars, they showed that a wide range of mono- and disaccharide derivatives undergo efficient H/D exchange at the positions adjacent to hydroxyl groups while largely preserving the original stereochemistry at the anomeric and neighboring centers. This work therefore established Ru/C as a powerful and practical heterogeneous catalyst for regio- and stereoselective deuteration of complex carbohydrate frameworks, and it significantly broadened the synthetic utility of heterogeneous sugar deuteration51.
In 2020, Pieters and co-workers developed a strategy to improve both the chemoselectivity and site selectivity of Ru/C-catalyzed deuteration of alcohols by modifying commercially available Ru/C with N-heterocyclic carbene (NHC) ligands (Fig. 7)45. Surface functionalization with NHCs significantly enhanced chemoselectivity and α-site selectivity in the deuteration of aliphatic alcohols, and thus provided the first general method for selective labeling of the α-position of aliphatic alcohols (Fig. 7a). For instance, aromatic alcohols (26–28) that contain readily reducible aryl groups underwent clean H/D exchange under NHC-modified Ru/C, while undesired aromatic reduction was largely suppressed. Similarly, heterocycles 29–31 such as quinoline and chloroxine, which were fully reduced by native Ru/C, could be deuterated at specific positions with high selectivity when the NHC-modified catalyst was used (Fig. 7b). This advance not only offered a practical route to deuterium-labeled drug-like molecules, but also revealed the potential of this ligand-modification strategy to uncover new C–H activation reactions. For example, electron-rich benzaldehydes were efficiently labeled at the formyl position by NHC-modified Ru/C in good yields, whereas commercial Ru/C promoted both reductive deuteration of the C=O group and additional deuteration at the α-position of the resulting alcohols (32, 33) (Fig. 7).

a Preparation of NHC-modified Ru/C and switch from reductive deuteration to H/D exchange. b Representative examples of α-deuteration and selective C1-deuteration using NHC–Ru/C. Reproduced with permission from ref. 45, © 2020 John Wiley and Sons.
Based on solid-state 13C NMR and XPS analyses, the authors proposed that the observed switch in site- and chemoselectivity arises from strong coordination of bulky NHC ligands on the Ru surface. Aromatic ring reduction usually requires flat π-coordination of the arene on relatively open Ru facets, which demands a large number of contiguous free metal sites. Surface-bound NHCs block these sites and thereby disfavor such π-coordination of aromatic rings, while still allowing a side-on approach and coordination through directing heteroatoms such as nitrogen or oxygen. As a result, reduction pathways are suppressed, and heteroatom-directed C–H activation and deuteration are favored, which accounts for the enhanced α-selectivity in aliphatic alcohols and the selective C1-deuteration of aldehydes observed with the NHC-modified Ru/C catalysts45.
Single-atom catalyst for the synthesis of deuterated alcohols
In existing heterogeneous systems, benzylic alcohol H/D exchange proceeds predominantly through a borrowing-hydrogen mechanism involving reversible oxidation to a ketone, enolization, and subsequent reduction. While this pathway is highly efficient for deuterium incorporation, the keto–enol tautomerization step intrinsically promotes H/D exchange at multiple sites (e.g., both α- and β-positions), leading to undesired multisite labeling and loss of precise site control. As a result, even the ligand-modified Ru/C-based protocols often struggle to achieve truly α-selective deuteration of alcohols, especially at metabolically critical positions in complex molecules.
To address these limitations, we recently developed an iron oxide–supported palladium single-atom catalysts (Pd SACs) that enables highly efficient, α-site-selective H/D exchange of benzylic alcohols (Fig. 8a)46. This system directly targets the α-C(sp3)–H bond adjacent to the hydroxyl group, while strongly suppressing undesired α,β-multisite deuteration. Our Pd SACs delivers up to 95% deuterium incorporation at the α-position together with high α/β selectivity (>20:1), illustrating the substantial performance leap that single-atom can achieve over conventional heterogeneous designs. Accordingly, only a slight loss in H/D exchange activity was observed after five consecutive cycles (Fig. 8b), which showed good recyclability, consistent with its exceptional structural stability. The substrate scope spans a broad range of secondary benzylic alcohols, including both linear and cyclic frameworks (34–39), as well as structurally complex, alcohol-based drug molecules (40, 41), all labeled using D2O as an economical and readily available deuterium source (Fig. 8c).

a Pd SAC for site-selective deuteration of benzylic alcohols. b Catalytic cycling experiment of Pd SAC. c Representative examples of Pd SAC catalyzed site-selective deuteration of benzylic alcohols. d H/D exchange experiments of chiral (S)-34 catalyzed by Pd1/FeOx. e Direct C-H bond activation process. Reproduced with permission from ref. 46, © 2025 John Wiley and Sons.
Next, we investigated the reaction mechanism by examining the racemization behavior of a chiral alcohol (Fig. 8d). Based on the conventional understanding that direct C–H bond activation can, in principle, preserve chirality (Fig. 8e), whereas the borrowing-hydrogen pathway typically leads to complete racemization, we selected (S)-34 as a model substrate. After 10 h of reaction, 81% Dα-incorporation was obtained and the deuterated product was isolated with an enantiomeric ratio (er) of 72:28. This outcome indicates that approximately 37.2% of the (S)-34-d product is formed via a direct C–H activation pathway, while the remainder arises from a borrowing-hydrogen process. In addition, to the best of our knowledge, all previously reported catalytic methods for the synthesis of deuterated chiral alcohols result in complete racemization, presumably because they proceed exclusively through borrowing-hydrogen pathways. In contrast, our results reveal a significant contribution from direct C–H activation in the Pd SACs system and thereby demonstrate the distinctive selectivity of this catalyst in H/D exchange.
Summary and outlook
Recent advances in heterogeneous catalysis have substantially reshaped the field of alcohol deuteration, enabling increasingly selective, scalable, and operationally simple routes for isotopic labeling. Early Pd/C–D2O–H2 systems demonstrated that solid catalysts could mediate both activated and non-activated H/D exchange, thereby establishing the feasibility of heterogeneous approaches. Subsequent studies introduced mechanistic and structural refinements: redox-mediated pathways were elucidated for Pd/C and Ru/C catalysts; Ru/C systems enabled α-selective deuteration across diverse alcohol substrates; and NHC-modified Ru/C platforms further enhanced chemoselectivity and site control by steering reactivity away from reduction-prone surfaces. Most recently, single-atom catalysts (SACs) have delivered a major leap in precision, with Pd SACs achieving up to 95% α-deuterium incorporation in benzylic alcohols through the cooperation of borrowing-hydrogen and direct C–H activation pathways. Collectively, these developments highlight how heterogeneous catalyst design—spanning nanoparticles, ligand-modified surfaces, and atomically dispersed metals—has overcome many of the limitations historically associated with homogeneous systems.
Looking forward, however, several important challenges remain unresolved and present valuable opportunities for future research.
-
(1)
Stereoretentive H/D exchange for chiral secondary alcohols. Although Pd SACs provide the first definitive evidence that direct C–H activation can occur on isolated metal sites, the simultaneous operation of a borrowing-hydrogen pathway inevitably leads to racemization of chiral secondary alcohols. To date, no heterogeneous or homogeneous catalyst can achieve stereoretentive H/D exchange at stereogenic alcohol centers. The development of catalysts capable of suppressing racemization while enabling efficient α-deuteration represents a high-priority goal.
-
(2)
Site-selective deuteration of carbohydrate scaffolds, especially bridgehead positions. While Ru/C systems have enabled α-deuteration adjacent to hydroxyl groups in sugars, achieving true site-selective deuteration at more challenging positions—such as bridgehead carbons in glucose or related saccharide remaining beyond reach. The ability to access such isotopologues would greatly expand the utility of deuterated carbohydrates in metabolic and mechanistic studies.
-
(3)
Selective deuteration of non-benzylic aliphatic alcohols at the hydroxyl-adjacent site. For aryl-substituted aliphatic alcohols in which the hydroxyl group is remote from the aromatic ring (i.e., non-benzylic alcohols), current heterogeneous catalysts cannot achieve α-site-selective deuteration. Methods capable of differentiating between electronically unbiased aliphatic C–H bonds are urgently needed to extend the reach of heterogeneous H/D exchange.
-
(4)
Harnessing enol intermediates for α,β-bis-deuteration of non-benzylic alcohols. Given that borrowing-hydrogen pathways necessarily generate transient enol or enolate-like intermediates, leveraging these species to achieve controlled α,β-multisite deuteration of non-benzylic substrates represents an unexplored but promising direction. Designing catalysts and reaction environments that can selectively intercept these intermediates would open new synthetic possibilities.
Addressing these challenges will require deeper mechanistic understanding of surface-mediated hydrogen transfer, improved control over the balance between borrowing-hydrogen and direct C–H activation pathways, and the design of next-generation heterogeneous catalysts with tunable metal nuclearity and support interactions. With continued advances in single-atom engineering, ligand modification, and operando spectroscopic analysis, heterogeneous catalysis is poised to become a central and broadly applicable platform for precision deuterium incorporation across pharmaceuticals, biochemical probes, and functional materials.
References
Atzrodt, J. et al. The renaissance of H/D exchange. Angew. Chem. Int. Ed. 46, 7744–7765 (2007).
Atzrodt, J. et al. C-H functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. 57, 3022–3047 (2018).
Lepron, M. et al. Nanocatalyzed hydrogen isotope exchange. Acc. Chem. Res. 54, 1465–1480 (2021).
Kopf, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Li, N. et al. Radical deuteration. Chem. Soc. Rev. 51, 6291–6306 (2022).
Prakash, G. et al. C–H deuteration of organic compounds and potential drug candidates. Chem. Soc. Rev. 51, 3123–3163 (2022).
Wiberg, K. B. The deuterium isotope effect. Chem. Rev. 55, 713–743 (1955).
Meanwell, N. A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 54, 2529–2591 (2011).
Perrin, C. L. et al. Stereochemistry of β-deuterium isotope effects on amine basicity. J. Am. Chem. Soc. 127, 9641–9647 (2005).
Charles, L. & Perrin, Y. D. Secondary deuterium isotope effects on the acidity of carboxylic acids and phenols. J. Am. Chem. Soc. 129, 4490–4497 (2007).
Prechtl, M. H. G. et al. Catalytic C-H bond activation at nanoscale lewis acidic aluminium fluorides: H/D exchange reactions at aromatic and aliphatic hydrocarbons. Chem. Eur. J. 17, 14385–14388 (2011).
Zhan, M. et al. A simple and cost-effective method for the regioselective deuteration of phenols. Eur. J. Org. Chem. 2015, 3370–3373 (2015).
Atzrodt, J. et al. Deuterium- and tritium-labelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758–1784 (2018).
Lehmann, W. D. A timeline of stable isotopes and mass spectrometry in the life sciences. Mass Spectrom. Rev. 36, 58–85 (2017).
Katsnelson, A. Heavy drugs draw heavy interest from pharma backers. Nat. Med. 19, 656–656 (2013).
Timmins, G. S. Deuterated drugs; updates and obviousness analysis. Expert Opin. Ther. Pat. 27, 1353–1361 (2017).
Pirali, T. et al. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 35, 493–494 (2017).
Garay, R. P. & Grossberg, G. T. AVP-786 for the treatment of agitation in dementia of the Alzheimer’s type. Expert Opin. Investig. Drugs 26, 121–132 (2017).
Wrobleski, S. T. et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J. Med. Chem. 62, 8973–8995 (2019).
McGrath, N. A., Brichacek, M. & Njardarson, J. T. A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ. 87, 1348–1349 (2010).
Demkowicz, B. J. et al. The comparative effectiveness and safety of fluticasone-salmeterol via metered-dose versus dry powder inhalers for COPD: a new user cohort study. PLoS. Med. 22, e1004596 (2025).
Zinman, B. et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 2iabetes. N. Engl. J. Med. 373, 2117–2128 (2015).
Flick, A. C. et al. Synthetic approaches to the new drugs approved during 2019. J. Med. Chem. 64, 3604–3657 (2021).
Flick, A. C. et al. Synthetic approaches to the new drugs approved during 2020. J. Med. Chem. 65, 9607–9661 (2022).
McInturff, E. L. et al. Synthetic approaches to the new drugs approved during 2021. J. Med. Chem. 66, 10150–10201 (2023).
France, S. P. et al. Synthetic approaches to the new drugs approved during 2022. J. Med. Chem. 67, 4376–4418 (2024).
France, S. P. et al. Synthetic approaches to the new drugs approved during 2023. J. Med. Chem. 68, 2147–2182 (2025).
Itoga, M. et al. Iridium-catalyzed α-selective deuteration of alcohols. Chem. Sci. 13, 8744–8751 (2022).
Shao, M. et al. The isotopic effects of deuteration on optoelectronic properties of conducting polymers. Nat. Commun 5, 3180–3191 (2014).
Balzarek, C. & Tyler, D. R. Intra-and intermolecular H/D exchange in aqueous solution catalyzed by molybdocenes. Angew. Chem. Int. Ed 38, 2406–2408 (1999).
Balzarek, C., Weakley, T. J. R. & Tyler, D. R. C−H bond activation in aqueous solution: kinetics and mechanism of H/D exchange in alcohols catalyzed by molybdocenes. J. Am. Chem. Soc. 122, 9427–9434 (2000).
Tse, S. K. et al. Ruthenium-catalyzed regioselective deuteration of alcohols at the β-carbon position with deuterium oxide. Chem. Eur. J. 17, 13918–13925 (2011).
Khaskin, E. & Milstein, D. Simple and efficient catalytic reaction for the selective deuteration of alcohols. ACS Catal. 3, 448–452 (2013).
Chatterjee, B. & Gunanathan, C. Ruthenium catalyzed selective α-and α,β-deuteration of alcohols using D2O. Org. Lett. 17, 4794–4797 (2015).
Wang, M. et al. Oxidative C(OH)−C bond cleavage of secondary alcohols to acids over a copper catalyst with molecular oxygen as the oxidant. J. Catal. 348, 160–167 (2017).
Tanaka, S. et al. Mixed picolinate and quinaldinate iron(III) complexes for the catalytic oxidation of alcohols with hydrogen peroxide. ChemCatChem 8, 2930–2938 (2016).
Yang, C. et al. Mechanism of electrochemical generation and decomposition of phthalimide-N-oxyl. J. Am. Chem. Soc. 143, 10324–10332 (2021).
Talvitie, J. et al. Electron-deficient phenanthrenequinone derivative for photoactivated hydrogen atom transfer mediated oxidation of secondary alcohols. ChemPhotoChem 7, e202300107 (2023).
Kar, S. et al. Regioselective deuteration of alcohols in D2O catalysed by homogeneous manganese and iron pincer complexes. Green Chem. 20, 2706–2710 (2018).
Sajiki, H. et al. Efficient C−H/C−D exchange reaction on the alkyl side chain of aromatic compounds using heterogeneous Pd/C in D2O. Org. Lett. 6, 1485–1487 (2004).
Esaki, H. et al. Novel Pd/C-catalyzed redox reactions between aliphatic secondary alcohols and ketones under hydrogenation conditions: application to H−D exchange reaction and the mechanistic study. J. Org. Chem. 72, 2143–2150 (2007).
Maegawa, T. et al. A convenient and effective method for the regioselective deuteration of alcohols. Adv. Synth. Catal. 350, 2215–2218 (2008).
Fujiwara, Y. et al. Method for regio-, chemo- and stereoselective deuterium labeling of sugars based on ruthenium-catalyzed C–H bond activation. Chem. Commun. 46, 4977–4979 (2010).
Palazzolo, A. et al. Tuning the reactivity of a heterogeneous catalyst using N-heterocyclic carbene ligands for C−H activation reactions. Angew. Chem. Int. Ed. 59, 20879–20884 (2020).
Li, S. X. et al. Catalytic α-site-selective hydrogen-deuterium exchange of benzylic alcohols by palladium single-atom catalyst. Angew. Chem. Int. Ed. 64, e202507338 (2025).
Sajiki, H. et al. Pd/C-H-catalysed deuterium exchange reaction of the benzylic site in D2O. Synlett 7, 1149–1151 (2002).
Esaki, H. et al. Efficient H/D exchange reactions of alkyl-substituted benzene derivatives by means of the Pd/C–H2–D2O system. Chem. Eur. J. 13, 4052–4063 (2007).
Sreekumar, C. & Pillai, C. N. A convenient method for the synthesis of 2-propanol-2-d. Synthesis 7, 498–499 (1974).
Corey, E. J. & Link, J. O. A new chiral catalyst for the enantioselective synthesis of secondary alcohols and deuterated primary alcohols by carbonyl reduction. Tetrahedron Lett. 30, 6275–6278 (1989).
Sawama, Y. et al. Stereo- and regioselective direct multi-deuterium-labeling methods forsugars. Chem. Eur. J 18, 16436–16442 (2012).
Acknowledgements
This research was funded by National Natural Science Foundation of China (22302199, 22310802000), the NSFC Centre for Single-Atom Catalysis (22388102), the Financial Support from the Natural Science Foundation of Liaoning Province (2024-BS-196), the Educational Department of Liaoning Province (JYTQN2023113), and Startup Fund of Scientific Research of Dalian Polytechnic University for High-Level Talents.
Author information
Authors and Affiliations
Contributions
J.-T.J. and S.-X.L. prepared the original draft of the manuscript. W.-X.Z. prepared the revised manuscript. X.-T.M. and B.Q. contributed to review, resources, and supervision. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, JJ., Li, SX., Zhou, WX. et al. Supported nanoparticles and single-atom catalysts for the synthesis of deuterated alcohols. Commun Chem 9, 53 (2026). https://doi.org/10.1038/s42004-026-01907-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-026-01907-w
