
Abstract
At TN ≃ 300K the layered insulator BaCoS2 transitions to a columnar antiferromagnet that signals non-negligible magnetic frustration despite the relatively high TN, all the more surprising given its quasi two-dimensional structure. Here, we show, by combining ab initio and model calculations, that the magnetic transition is an order-from-disorder phenomenon, which not only drives the columnar magnetic order, but also the inter-layer coherence responsible for the finite Néel transition temperature. This uncommon ordering mechanism, actively contributed by orbital degrees of freedom, hints at an abundance of low energy excitations above and across the Néel transition, in agreement with experimental evidence.
Similar content being viewed by others


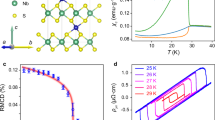
Introduction
Frustrated magnets often display a continuous accidental degeneracy of the classical ground state that leads to the appearance of pseudo-Goldstone modes within the harmonic spin-wave approximation1. Since those modes are not protected by symmetry, they may acquire a mass once anharmonic terms are included in the spin-wave Hamiltonian. This mass, in turn, cuts off the singularities brought about by the pseudo-Goldstone modes, in that way stabilising ordered phases otherwise thwarted by fluctuations. To put it differently, let us imagine that the classical potential has a manifold of degenerate minima generally not invariant under the symmetry group of the Hamiltonian. It follows that the eigenvalues of the Hessian of the potential change from minimum to minimum. Allowing for quantum or thermal fluctuations is therefore expected to favour the minima with lowest Hessian determinant, although the two kinds of fluctuations not necessarily select the same ones2. Moreover, it is reasonable to assume that the minima with lowest Hessian determinant are those that form subsets invariant under a symmetry transformation of the Hamiltonian so that choosing any of them corresponds to a spontaneous symmetry breaking. Such a phenomenon, also known as order from disorder3, emerges in many different contexts4, from particle physics5,6 to condensed matter physics7,8, even though frustrated magnets still provide the largest variety of physical realisations1,2,3,9,10,11,12,13,14,15.
The layered insulator BaCoS2 might be legitimately included in the class of frustrated magnets. Below a critical temperature TN, BaCoS2 becomes an antiferromagnet characterised by columnar spin-ordered planes, which we hereafter refer to as antiferromagnetic striped (AFS) order, a classic symptom of frustration. The planes are in turn stacked ferromagnetically along the c-axis, so called C-type stacking as opposed to the antiferromagnetic G-type one. Inelastic neutron scattering (INS) experiments show that magnetic excitations below TN have pronounced two-dimensional (2D) character16,17 implying strong quantum and thermal fluctuations that join with magnetic frustration to further hamper magnetic order. In spite of all that, the Néel temperature of BaCoS2 is rather large, between 290K18 and 305K19, which is highly surprising. Indeed, a direct estimate of the spin exchange constants by neutron diffraction has been recently attempted in doped tetragonal BaCo0.9Ni0.1S1.9 subject to an uniaxial strain17. This compound undergoes a Néel transition to the C-type AFS phase at 280 K17, not far from TN of undoped BaCoS2. The neutron data were fitted by a conventional J1 − J2 Heisenberg model13 on each plane plus an inter-plane ferromagnetic exchange Jc, yielding J2 ~ 9.3 meV, J1 ~ −2.3 meV and 0 < ∣Jc∣ < 0.04 meV, with the upper bound due to experimental resolution. The Néel temperature can be overestimated discarding J120 and taking ∣Jc∣ equal to the upper bound. In that way, one obtains21 TN ≃ 200K, which, despite supposedly being an overestimate, is 2/3 smaller than the observed value. This discrepancy is puzzling.
Another startling property is the anomalously broad peak of the magnetic susceptibility at TN18,19, which suggests a transition in the Ising universality class rather than the expected Heisenberg one19. A possible reason of this behaviour might be spin-orbit coupling19. Indeed, a Rashba effect due to the layered structure and the staggered sulfur pyramid orientation, see Fig. 1, has been found to yield sizeable band splittings at specific points within the Brillouin zone, at least in metallic tetragonal BaNiS222. The Rashba-like spin-orbit coupling strength may barely differ in BaCoS2, or be weakened by strong correlations23. In either case, its main effect is to introduce an easy plane anisotropy, as indeed observed experimentally19, which, at most, drives the transition towards the XY universality class. It is well possible that the weak orthorhombic distortion in BaCoS2 may turn the easy plane into an easy axis, but the resulting magnetic anisotropy should be negligibly small and thus unable to convincingly explain the experimental observations.

a Three-dimensional view of tetragonal BaCoS2. Ba atoms are the large green spheres, while S atoms are shown in yellow. The cobalt atoms sit inside the blue square-based pyramids. b Top and lateral view of the structure, respectively. Note that, since the apexes of nearest neighbour pyramids point in opposite directions, there are two inequivalent Co atoms, shown as blue and grey spheres, with opposite vertical displacements from the a − b plane, which are connected by the non-symmorphic symmetry. c Magnetic order in the low-temperature orthorhombic phase. Dots represent Co atoms, arrows their spins and blue triangles indicate the orientation of the surrounding sulfur pyramids. Within each a − b plane the spins form a striped antiferromagnet (AFS) with ferromagnetic chains coupled antiferromagnetically. The ferromagnetic chains can be either along a (AFS-a) or along b (AFS-b). The planes are stacked ferromagnetically, C-type stacking, thus the two equivalent configurations C-AFS-a and C-AFS-b.
Lastly, BaCoS2 shows a very strange semiconducting behaviour above TN, with activated dc conductivity, no evidence of a Drude peak and, yet, an optical conductivity that grows linearly in frequency24.
The relatively high TN despite magnetic frustration and quasi-two dimensional character, as well as the abundance of low energy excitations above and across the Néel transition are pieces of evidence that some kind of order-from-disorder phenomenon takes place in BaCoS2, a scenario that we here support by a thorough analysis combining ab initio and model calculations.
Results
Phase diagram of BaCoS2
BaCoS2 is a metastable layered compound that, quenched from high temperature, crystallises in an orthorhombic structure with space group Cmme, no. 6725, characterised by in-plane primitive lattice vectors a ≠ b. However, we believe physically more significant to consider as reference structure the higher-symmetry non-symmorphic P4/nmm tetragonal one (a = b) of the opposite end member, BaNiS2, and regard the orthorhombic distortion as an instability driven by the substitution of Ni with the more correlated Co. The hypothetical tetragonal phase of BaCoS2 is shown in Fig. 1a. Each CoS a − b plane has two inequivalent cobalt atoms, Co(1) and Co(2), see Fig. 1b, which are related to each other by a non-symmorphic symmetry.
Below TN, an AFS magnetically ordered phase sets in. In the a − b plane it consists of ferromagnetic chains, either along a (AFS-a) or b (AFS-b), coupled antiferromagnetically, see Fig. 1c. The stacking between the planes is C-type, i.e., ferromagnetic, thus the labels C-AFS-a and C-AFS-b that we shall use, as well as G-AFS-a and G-AFS-b whenever we discuss the G-type configurations with antiferromagnetic stacking. We mention that the orthorhombic distortion with b > a (a > b) is associated with C-AFS-a (C-AFS-b), i.e., ferromagnetic bonds along a (b)19, at odds with the expectation that ferromagnetic bonds are longer than antiferromagnetic ones. This counterintuitive behaviour represents a key test for the ab initio calculations that we later present.
Neutron scattering refinement and magnetic structure modelling in the low-temperature phase point to an ordered moment of μCo ~ 2.63 − 2.9μB19,26, suggesting that each Co2+ is in a S = 3/2 spin configuration, in agreement with the high-temperature magnetic susceptibility19. Moreover, the form factor analysis of the neutron diffraction data26 indicates that the three 1/2-spins lie one in the \({d}_{3{z}^{2}-{r}^{2}}\), the other in the \({d}_{{x}^{2}-{y}^{2}}\), and the third either in the dxz or dyz 3d-orbitals of Co. Since dxz and dyz, which we hereafter denote shortly as x and y orbitals, form in the P4/nmm tetragonal structure a degenerate Eg doublet occupied by a single hole, such degeneracy is going to be lifted at low-temperature. That hints at the existence of some kind of orbital order, besides the spin one, in the magnetic orthorhombic phase. Let us try to anticipate by symmetry arguments which kind of order can be stabilised. We observe that in the Cmme orthorhombic structure the cobalt atoms occupy the Wyckoff positions 4g, which, for convenience, we denote as Co(1) ≡ (0, 0, z), Co(2) ≡ (1/2, 0, −z), Co(3) ≡ (0, 1/2, −z), Co(4) ≡ (1/2, 1/2, z), and have symmetry mm2. As a consequence, the hole must occupy either the x orbital or the y one, but not a linear combination, and the chosen orbital must be the same for Co(1) and Co(4), as well as for Co(2) and Co(3). Therefore, we denote as dn, d = x, y, the orbital occupied by the hole on Co(n), n = 1, …, 4, and as d1d2d3d4 a generic orbital configuration. Then, there are only four of them that are symmetry-allowed: xxxx, yyyy, xyyx and yxxy, see Fig. 2.

Co(1), Co(2), Co(3) and Co(4) correspond to the 4g Wyckoff positions occupied by the cobalt atoms. The label x(y) indicates that the hole occupies the dxz(dyz) orbital of the 3/4-filled dxz/dyz doublet of the corresponding cobalt atom.
We remark that xxxx is degenerate with yyyy in the tetragonal phase. The choice of either of them is associated with the same C4 → C2 symmetry breaking that characterises both the AFS-a or AFS-b spin order and the orthorhombic distortion, b > a or a > b. All these three choices can be associated with three Ising variables τ, σ and X such that τ = + 1 corresponds to xxxx, σ = +1 to AFS-a, X = +1 to b > a, and vice versa. Since they all have the same symmetry, odd under C4, they would be coupled to each other should we describe the transition by a Landau-Ginzburg functional. We shall hereafter denote as Z2(C4) the Ising sector that describes the C4 → C2 symmetry breaking.
The other two allowed orbital configurations xyyx and yxxy (see Fig. 2) are instead degenerate both in the tetragonal and orthorhombic phases, but break the non-symmorphic symmetry (NS) that connects, e.g., Co(1) with Co(2) and Co(3). We can therefore associate to those configurations a new Ising sector Z2(NS).
We emphasise that the above conclusions rely on the assumption of a Cmme space group. A mixing between x and y orbitals is instead allowed by the Pba2 space group proposed in ref. 27 as an alternative scenario for BaCoS2 at room temperature. As a matter of fact, the two symmetry-lowering routes, P4/nmm → Cmme and P4/nmm → Pba2, correspond to different Jahn-Teller-like distortions involving the dxz-dyz doublet and the Eg phonon mode of the P4/nmm structure at the M point, which is found to have imaginary frequency by ab initio calculations27. However, latest high-accuracy X-ray diffraction data18 confirm the Cmme orthorhombic structure even at room temperature, thus supporting our assumption.
Ab initio analysis
Using density functional theory (DFT) and DFT + U calculations, in the first place, we checked if the tetragonal phase is unstable towards magnetism, considering both a conventional Néel order (AFM) compatible with the bipartite lattice and the observed AFS. We found, using a Hubbard interaction of U = 2.8 eV and a Hund’s coupling constant J = 0.95 eV for the Co-3d orbitals as motivated by constrained random phase approximation (cRPA)24, that the lowest energy state is indeed the AFS, the AFM and non-magnetic phases lying above by about 0.5 eV and 2.3 eV, respectively. Let us therefore restrict our analysis to AFS and stick to U = 2.8 eV. Note, however, that the ordering of the four configurations with lowest energy within our DFT + U simulations does not change within 2 eV around that value. The main assumptions entering our ab initio modelling are thereby not sensitive to the precise choice of U around the value motivated by cRPA calculations. We use an 8-site unit cell that includes two planes, which allows us to compare C-AFS with G-AFS. In addition, we consider both the tetragonal structure with AFS-a, since AFS-b is degenerate, and the orthorhombic structure with b > a, in which case we analyse both AFS-a and AFS-b. For all cases, we investigate all four symmetry-allowed orbital configurations, xxxx, yyyy, xyyx and yxxy, assuming either a C-type or G-type orbital stacking between the two planes of the unit cell, so that, for instance, G(xxxx) means that one plane is in the xxxx configuration and the other in the yyyy one.
In Table 1, we report the energies per formula unit of several possible configurations in the tetragonal structure, including those that would be forbidden in the orthorhombic one. All energies are measured with respect to the lowest energy state and are expressed in Kelvin. In agreement with experiments, the lowest energy state T0 has spin order C-AFS, a or b being degenerate. In addition, it has C-type antiferro-orbital order, C(xyyx). We note that its G-type spin counterpart T1 is only 2 K above, supporting our observation that TN = 290K is anomalously large if compared to these magnetic excitations. The abundance of nearly degenerate ground states is consistent with the seminal DFT + U study by Zainullina and Korotin28, where the importance of different orbital configurations for a given stripe magnetic phase was studied for a larger value of U.
The energy differences between C-type orbital stacked configurations and their G-type counterparts are too small to allow obtaining a reliable modelling of the inter-plane orbital coupling. On the contrary, the energy differences between in-plane orbital configurations can be accurately reproduced by a rather simple modelling. We assume on each Co-site an Ising variable τ3 equal to the difference between the hole occupations of orbital x and of orbital y. The Ising variable on a given site is coupled only to those of the four nearest neighbour sites in the a − b plane, with exchange constants Γ1a = Γ1 + σ δΓ1 and Γ1b = Γ1 − σ δΓ1 along a and b, respectively. In addition, the Ising variables feel a uniform field Bτ σ. Here, σ is the Ising Z2(C4) order parameter that distinguishes AFS-a, σ = +1 from AFS-b, σ = −1. We find that the spectrum is well reproduced by the parameters in Table 2. It is worth noticing that the in-plane antiferro-orbital order is unexpected in light of the nematic columnar spin order that would rather suggest the ferro-orbital xxxx or yyyy configurations to have lowest energy. The explanation is that the antiferro-orbital order yields within DFT + U a larger insulating gap than the ferro-orbital order, see Fig. 3.

The density of states (DOS) around the Fermi level is shown for the two orbital nematic configurations yyyy (full green line) and xxxx (dashed blue line) as well as for the orbital ordered configuration xyyx (dashed orange line), which is found to be lowest in energy within Hubbard-U corrected density functional theory (DFT + U) calculations.
We now move to the physical orthorhombic structure, assuming b > a with b/a = 1.00825, and recalculate all above energies but considering only the orbital configurations allowed by the Cmme space group. In this case, we have to distinguish between AFS-a and AFS-b, which are no longer degenerate. The results are shown in Table 3.
The calculated magnetic moment per Co atom in the lowest energy state, O0 in Table 3, is μAFS ~ 2.65μB, in quite good agreement with experiments19,26. We remark that the ab initio calculation correctly predicts that the lowest energy state O0 has ferromagnetic bonds along a despite b > a, which, as we mentioned, is an important test for the theory. The energy difference between AFS-a and AFS-b, i.e., O0 and O3, is about 20 K, and gives a measure of the spin-exchange spatial anisotropy in the a and b directions due to the orthorhombic distortion. This small value implies that the Néel transition temperature TN ≃ 290K is largely insensitive to the orthorhombic distortion that exists also above TN18,19. In particular, the energy difference between C-type and G-type stacking, O0 and O1 in Table 3, remains the same tiny value found in the tetragonal phase. Table 3 thus suggests that the spin configurations C-AFS-a, C-AFS-b, G-AFS-a and G-AFS-b are almost equally probable at the Néel transition, and that despite the orthorhombic structure.
The orbital arrangement of the C-AFS-a configuration is also important to describe, e.g., the pressure-induced metal-insulator transition in BaCoS2, see Supplementary Note 1.
Orthorhombic distortion
A further evidence of the marginal role played by the orthorhombic distortion at the Néel transition comes from the total energy as function of the parameter d = 2(a − b)/(a + b) that quantifies the distortion, shown in Fig. 4 for different orbital configurations assuming AFS-a magnetic order. We note that for all orbital configurations the energy gain due to a finite d is tiny with respect to d = 0. For instance, the lowest-energy orbital configuration xyyx reaches a minimum at about \({d}_{\min }^{xyyx} \sim -0.5 \%\), not far from the experimental value \({d}_{\exp } \sim -0.8 \%\)25, which reduces to \({d}_{\exp } \sim -0.4 \%\) under high pressure synthesis18. However, the energy gain with respect to d = 0 is less than 3 K. This result suggests that, despite the hypothetical P4/nmm structure of BaCoS2 being inherently unstable to an orthorhombic Jahn-Teller distortion, the latter plays almost no role in stabilising the AFS magnetic order in contrast to naïve expectations.

Energy ΔE per formula unit as function of the orthorhombic distortion d at fixed unit cell volume and measured with respect to the C(xyyx) phase at d = 0 (T0 in Table 1). We compare C(xyyx) with the two nematic configurations C(xxxx) and C(yyyy) assuming a magnetic order C-AFS-a. Experimental data are taken from refs. 19,25,51. The phases T0, T7, and T17 refer to Table 1, phases O0, O12, and O17 to Table 3.
Wannierisation
To gain further insight into the mechanisms that drive the Néel transition, we generate two tight-binding Hamiltonians with maximally-localised Wannier functions for Co-d-like and Co-dxz/yz-like orbitals, respectively. Both tight-binding models reproduce overall well the DFT band structure of the PM phase in the orthorhombic structure, see Fig. 5. Whereas the fit of the 5-orbital model is nearly perfect, the 2-orbital model shows small deviations along the M − Γ direction due to missing hybridisation with the other Co-d orbitals. Table 4 shows the leading hopping processes of the 5-band model restricted to the \(({d}_{xz},{d}_{yz})\) subspace.

DFT band structure and eigenstates of our Wannier Hamiltonians along the high-symmetry path Γ − X − M − Γ − Y − M: (a) Co-d model and (b) dxz-dyz model with projection onto the dxz-orbital character in red.
We note that, because of the staggered shift of the Co atoms out of the sulfur basal plane, the largest intra-layer hopping is between next-nearest neighbour (NNN) cobalt atoms instead of nearest-neighbour (NN) ones. Moreover, the stacking of the sulfur pyramids and the position of the intercalated Ba atoms makes the inter-layer NN hopping negligible, contrary to the NNN one that is actually larger than the in-plane NN hopping, but still smaller than the in-plane NNN one (\({t}_{NN}^{{{{{{{{\rm{inter}}}}}}}}}\ll {t}_{NN}^{{{{{{{{\rm{intra}}}}}}}}} \, < \, {t}_{NNN}^{{{{{{{{\rm{inter}}}}}}}}} \, < \, {t}_{NNN}^{{{{{{{{\rm{intra}}}}}}}}}\)).
We finally remark that the orthorhombic distortion has a very weak effect on the inter-layer hopping, which is consistent with the tiny energy difference between C-AFS and G-AFS being insensitive to the distortion, compare, e.g., the energies of T1 and O1 in Tables 1 and 3, respectively.
Effective Heisenberg model
Armed with all the above ab initio results, we are now ready to address the main questions of this work, i.e., why TN is so high and why the Néel transition looks Ising-like.
We already mentioned that the largest energy scales that emerge from the ab initio calculations are the magnetic ones separating the lowest-energy AFS configuration from the Néel and non-magnetic states. Therefore, even though BaCoS2 seems not to lie deep inside a Mott insulating regime, we think it is worth discussing qualitatively the spin dynamics in terms of an effective S = 3/2 Heisenberg model. If we assume that the leading contribution to the exchange constants derives from the hopping processes within the dxz − dyz subspace, then Table 4 suggests the Heisenberg model shown in Fig. 6. According to this figure, the exchange constants J1x/y, J2, and J3x/y are related to the hopping terms T(±1, 0, 0)/(0, ±1, 0), T±(1, −1, 0)/±(1, 1, 0), and T(±1, 0, 1)/(0, ±1, 1), reported in Table 4. This model consists of frustrated J1 − J2 planes13,29,30,31,32 coupled to each other by a still frustrating J3 coupling, see Fig. 6b. In order to be consistent with the observed columnar magnetic order, the exchange constants have to satisfy the inequality \(2{J}_{2} > \vert {J}_{3}\vert +\vert {J}_{1}\vert\). Moreover, J3 forces to deal with a two sites unit cell, highlighted in yellow colour in Fig. 6a where the non-equivalent cobalt sites are referred to as Co(1), in blue, and Co(2), in red, respectively. The reason is that Co(1) on a plane is only coupled to Co(2) on the plane above but not below, and vice versa for Co(2).

a shows the J1 − J2 model on the a − b plane, while (b) shows how nearest neighbour planes are coupled to each other by the exchange J3. The latter forces to deal with a two-site unit cell, highlighted in yellow in (a). Blue and red balls indicate the two different Co sites of the unit cell.
To simplify the notation, we write, for a = 1, 3, \({J}_{ax}={J}_{a}\,\left(1-{\delta }_{a}\right)\) and \({J}_{ay}={J}_{a}\,\left(1+{\delta }_{a}\right)\), where, in analogy with the single-layer J1 − J2 model13, δa ≠ 0 are Ising order parameters associated with the C4 → C2 symmetry breaking. Moreover, we define the in-plane Fourier transforms of the spin operators:
where R labels the N unit cells in the a − b plane, ℓ = 1, 2 the two sites (sublattices) within each unit cell, and n = 1, …, L the layer index. With those definitions, the Hamiltonian reads:
where Ha is proportional to Ja, a = 1, 2, 3, and:
The classical ground state corresponds to the three-dimensional modulation wave vector (π, 0, Qz) ≡ (0, π, Qz), which describes an antiferromagnetic order within each sublattice on each layer, and where the inter-plane Qz is the value that minimises the classical energy per site, \(E({Q}_{z})=-2{J}_{2}-2\sqrt{{J}_{1}^{2}{\delta }_{1}^{2}+{J}_{3}^{2}{\delta }_{3}^{2}+2{J}_{1}{J}_{3}{\delta }_{1}{\delta }_{3}\cos {Q}_{z}\,}\). The expression of E(Qz) shows that inter-layer magnetic coherence sets in only when the two Ising-like order parameters, δ1 and δ3, lock together. Specifically, J1J3δ1δ3 > 0 stabilises C-AFS, Qz = 0, otherwise G-AFS, Qz = π. We already know that the former is lower in energy, though by only few Kelvins, see Table 3. Moreover, an orthorhombic distortion b > a favours AFS-a, which implies J1 δ1 + J3 δ3 > 0, even though AFS-b is higher by only 20K according to DFT + U, see O3 in Table 3.
Using our J1 − J2 − J3 model to fit the INS data of ref. 17 at 200 K, we estimate J2 ≃ 9.3 meV, J1 + J3 ≃ −2.34 meV, J1δ1 + J3δ3 ≃ 0.53 meV, and \(0 \, < \, \sqrt{{J}_{1}{J}_{3}{\delta }_{1}{\delta }_{3}\,} < 0.14\,\,{{\mbox{meV}}}\,\), where, we recall, the upper bound is due to experimental resolution. Such small bound suggests that the two order parameters δ1 and δ3 are already formed at 200K, whereas their mutual locking is still suffering from fluctuations. We finally observe that the ferromagnetic sign of J1 and J3 is consistent with the diagonal hopping matrices in the corresponding directions (see Table 4) and the antiferro-orbital order. Estimations of the exchange constants based on the DFT + U energies can be found in Supplementary Note 2.
Spin-wave analysis of the \({{{{{{{{\mathcal{C}}}}}}}}}_{4}\) symmetric model
To better understand the interplay between the \({Z}_{2}\left({C}_{4}\right)\) Ising degrees of freedom and the magnetic order at TN, we investigate in more detail the Hamiltonian (2) with δ1 = δ3 = 0, thus J1x = J1y = J1 and J3x = J3y = J3. Since J2 > 0 is the dominant exchange process, the classical ground state corresponds to the spin configuration:
where S = 3/2 is the spin magnitude, n3,i,n is a unit vector, and \({{{{{{{\bf{Q}}}}}}}}=\left(\pi ,0\right)\equiv (0,\pi )\), the equivalence holding since G = (π, π) is a primitive in-plane lattice vector for the two-site unit cell. In other words, each sublattice on each plane is Néel ordered, and its staggered magnetisation n3,i,n is arbitrary. We therefore expect that quantum and thermal fluctuations may yield a standard order-from-disorder phenomenon3.
Within spin-wave approximation, the spin operators can be written as:
where n1,i,n, n2,i,n and n3,i,n are orthogonal unit vectors, \({x}_{i,n}^{{{{\dagger}}} }({{{{{{{\bf{q}}}}}}}})={x}_{i,n}^{}(-{{{{{{{\bf{q}}}}}}}})\) and \({p}_{i,n}^{{{{\dagger}}} }({{{{{{{\bf{q}}}}}}}})={p}_{i,n}^{}(-{{{{{{{\bf{q}}}}}}}})\) are conjugate variables, i.e.:
and
The three terms of the Hamiltonian (2) thus read, at leading order in quantum fluctuations, i.e., in the harmonic approximation:
where E0 = 2NL S(S + 1) J2(Q), γ(q) = γ(q, δ = 0), and:
We note that H2 does not depend on the choice of n3,i,n, reflecting the classical accidental degeneracy, unlike H1 + H3. We start treating H1 and H3 within perturbation theory. The unperturbed Hamiltonian H2 can be diagonalised and yields the spin-wave dispersion:
Free energy in perturbation theory and quadrupolar coupling
The free energy in perturbation theory can be written as F = ∑ℓ Fℓ, where Fℓ is of ℓth order in H1 + H3, and F0 is the unperturbed free energy of the Hamiltonian H2. Notice that only even-order terms are non vanishing, thus ℓ = 0, 2, 4, … . Given the evolution operator in imaginary time:
where Ha(τ), a = 1, 3, evolves with the Hamiltonian H2, the second order correction to the free energy is readily found to be:
where
with ωλ = 2πλT, \(\lambda \in {\mathbb{Z}}\), bosonic Matsubara frequencies. Even without explicitly evaluating Ξ2, we can conclude that the free-energy gain at second order in H1 + H3 is maximised by n3,1,n ⋅ n3,2,m = ± 1, with m = n, n + 1, which reduces the classical degeneracy to 4L configurations, where L is the total number of layers in the system. Such residual degeneracy is split by a fourth order correction to the free energy proportional to \({J}_{1}^{2}\,{J}_{3}^{2}\) that reads:
where
We remark that, despite ω2(q) vanishes linearly at q = 0, Q, both Ξ2(T) and Ξ4(T) are non-singular.
The fourth order correction F4 in Eq. (14) has a twofold effect: it forces n3,1,n ⋅ n3,2,n to be the same on all layers and, in addition, stabilises a ferromagnetic inter-layer stacking. Therefore, the ground state manifold at fourth order in H1 + H3 is spanned by n3,1,n = n3 and n3,2,n = σ n3, where n3 is an arbitrary unit vector reflecting the spin SU(2) symmetry, and σ = ± 1 is associated with the global C4 → C2 symmetry breaking.
Similarly to the single-plane J1 − J2 model13, the above results imply that an additional term must be added to the semiclassical spin action. Specifically, if we introduce the Ising-like fields σn(R) = n3,1,n(R) ⋅ n3,2,n(R) and σn+1/2(R) = n3,1,n(R) ⋅ n3,2,n+1(R), Eqs. (12) and (14) imply that, at the leading orders in J1 and J3, the effective action in the continuum limit includes the quadrupolar coupling term13:
We expect a 3D Ising transition to occur at a critical temperature Tc, below which 〈σn(R)〉 = m1, 〈σn+1/2(R)〉 = m3, with m1 m3 > 0. In turn, the Ising order should bring along the 3D AFS one below a finite Néel temperature bounded from above by Tc20. To get a rough estimate of the latter, based on Eq. (16) we assume that, upon integrating out the spin degrees of freedom, the classical action describes an anisotropic three-dimensional ferromagnetic Ising model with exchange constants I1 on layers n, I3 on layers n + 1/2, and I⊥ < I1, I3 between layers. Hereafter, we take for simplicity J1 = J3, thus I1 = I3 ≡ I∥.
We then note that, for the J1 − J2 − J3 model with J2 ≃ 9.3 meV and J1 = J3 ≃ −1.17 meV, the 2D Ising critical temperature with S = 3/2 of each layer n and n + 1/2 is about 0.4 (S + 1/2)2J2 ≃ 173K30. This critical temperature corresponds to I∥ ≃ 6.6 meV in the 2D Ising model. The 3D Ising critical temperature Tc grows with I⊥, reaching 280K and 345K at I⊥ = 0.5I∥ and I⊥ = I∥, respectively33, which are reassuringly of the same order of magnitude as TN.
However, BaCoS2 remains orthorhombic above TN, which implies that the structural C4 → C2 symmetry breaking occurs earlier than magnetic ordering upon cooling. Therefore, even though the effects of the orthorhombic distortion on the electronic structure are rather small, see Table 4, it is worth repeating the above discussion assuming from the start that σn(R) = n3,1,n(R) ⋅ n3,2,n(R) = 1 (AFS-a), so that σn+1/2(R) = n3,1,n(R) ⋅ n3,2,n+1(R) = n3,1,n(R) ⋅ n3,1,n+1(R). It follows that the quadrupolar term (16) becomes:
with K(T) ≫ h(T) > 0. The term proportional to K(T) is bi-quadratic in the original spin operators and, alone, it would drive an Ising-like transition either towards C-AFS, n3,1,n ⋅ n3,1,n+1 = 1 or G-AFS, n3,1,n ⋅ n3,1,n+1 = − 1. On the contrary, the term proportional to h(T) is quadratic and yields an inter-layer ferromagnetic coupling that stabilises C-AFS, though by only two kelvins according to our DFT + U calculations. Therefore, (17) corresponds to an unusual model of coupled Heisenberg layers in which the dominant inter-layer coupling is bi-quadratic. We believe that this term is responsible of the higher TN than the estimate obtained assuming just the small ferromagnetic exchange, as earlier discussed, as well as of the pronounced Ising-like character of the Néel transition. We also remark that which among C-AFS and G-AFS is lower in energy does depend on the orbital configurations, see Table 3. Therefore, we expect that the neglected coupling between spins and orbital fluctuations should further reduce the already small energy difference between C-AFS and G-AFS.
Conclusion
BaCoS2 is a frustrated magnet with a pronounced two-dimensional character of the magnetic excitations that, nonetheless, orders magnetically at a Néel temperature of TN ~ 300K18,19 through a second order phase transition more similar to an Ising than a Heisenberg one. We have shown that these puzzling features can be pieced together within an order-from-disorder scenario that we have uncovered by a thorough ab initio analysis demonstrating the critical role of the specific Co d-orbitals involved in magnetism. Although specific to BaCoS2, our results might be relevant to other spin-frustrated transition metal compounds that also crystallise in the non-symmorphic P4/nmm space group, like, e.g., the iron pnictides. In many (mainly electron-doped) iron-based superconductors the disordered phase at high temperature first spontaneously breaks C4 symmetry when cooling below a critical temperature Tnem, thus entering a nematic phase34. Only at a lower temperature TN < Tnem, also spin SU(2) is broken and a stripe-ordered magnetic long-range order emerges34,35. The Néel temperature can be quite large as in BaCoS2 or even vanishing within experimental accuracy as in FeSe, where TN ≠ 0 is observed only under pressure36.
Also from a model point of view, our description of BaCoS2 fits into the modelling of these materials. Indeed, the key role played by several order parameters in BaCoS2 has parallels to the phenomenological Landau free energy description of FeSC34. Besides itinerant multi-orbital Hubbard models37,38,39, also spin-1 Heisenberg models conceptually similar to ours have been proposed for iron-based superconductors and FeSe40,41. There, however, instead of achieving the C4 symmetry breaking via an order-from-disorder phenomenon, it is often assumed from the start42 or by explicitly adding bi-quadratic spin exchanges40 that mimic the quadrupolar terms (16).
Moreover, our ab initio simulations for BaCoS2 predict that the lowest-energy phase has not ferro-orbital order, as often discussed in the context of FeSC, but rather an anti-ferro orbital ordering that breaks the non-symmorphic symmetry instead of C4. Therefore, BaCoS2 seems to realise a situation where collinear magnetism and orthorhombicity do not imply orbital nematicity, unless for specific crystal structures under pressure.
Methods
Ab initio calculations
We carried out ab initio DFT and DFT + U calculations using the Quantum ESPRESSO package43,44. The density functional is of generalised gradient approximation type, namely the Perdew-Burke-Ernzerhof functional45, on which local Hubbard interactions and Hund’s coupling terms were added to the Co atoms in case of the DFT + U within a fully rotational invariant framework46,47. If not stated otherwise, the geometry of the unit cell and the internal coordinates of the atomic positions in the orthorhombic structure were those determined experimentally, taken from ref. 25. For non-magnetic calculations, the relative atomic positions were kept fixed and the in-plane lattice constants a = b chosen such that the unit cell volume matched the one of the orthorhombic structure. Co and S atoms are described by norm-conserving pseudopotentials (PP) with non-linear core corrections, Ba atoms are described by ultrasoft pseudopotentials. The Co PP contains 13 valence electrons (3s2,3p6,3d7), the Ba PP 10 electrons (5s2,5p6,6s2), and S PPs are in a (3s2,3p3) configuration. The plane-waves cutoff has been set to 120Ry and we used a Gaussian smearing of 0.01Ry. The k-point sampling of the electron-momentum grid was at least 8 × 8 × 8 points in the 8 Co atom supercell.
Wannier interpolation
To determine the band structure and derive an effective low-energy model, we performed a Wannier interpolation with maximally localised Wannier functions48,49 using the Wannier90 package50. We constructed Wannier fits based on the non-magnetic DFT + U calculation using a 4 × 4 × 4 k-grid with a doubled in-plane unit cell comprising 4 Co atoms.
Data availability
The data used for generating the figures of the main text and the Supplementary Information are available from the corresponding author on reasonable request.
Code availability
The codes used and described in the “Methods” section are publicly available. The scripts used for generating the figures are available from the corresponding author on reasonable request.
References
Rau, J. G., McClarty, P. A. & Moessner, R. Pseudo-goldstone gaps and order-by-quantum disorder in frustrated magnets. Phys. Rev. Lett. 121, 237201 (2018).
Schick, R., Ziman, T. & Zhitomirsky, M. E. Quantum versus thermal fluctuations in the fcc antiferromagnet: alternative routes to order by disorder. Phys. Rev. B 102, 220405 (2020).
Villain, J., Bidaux, R., Carton, J. P. & Conte, R. Order as an effect of disorder. J. Phys. France 41, 1263–1272 (1980).
Burgess, C. Goldstone and pseudo-Goldstone bosons in nuclear, particle and condensed-matter physics. Phys. Rep. 330, 193–261 (2000).
Weinberg, S. Approximate symmetries and pseudo-goldstone bosons. Phys. Rev. Lett. 29, 1698–1701 (1972).
Coleman, S. & Weinberg, E. Radiative corrections as the origin of spontaneous symmetry breaking. Phys. Rev. D 7, 1888–1910 (1973).
Demler, E., Hanke, W. & Zhang, S.-C. SO(5) theory of antiferromagnetism and superconductivity. Rev. Mod. Phys. 76, 909–974 (2004).
Fernandes, R. M. & Chubukov, A. V. Low-energy microscopic models for iron-based superconductors: a review. Rep. Prog. Phys. 80, 014503 (2016).
Tessman, J. R. Magnetic anisotropy at 0∘K. Phys. Rev. 96, 1192–1195 (1954).
Belorizky, E., Casalegno, R. & Niez, J. J. Calculation of the spin wave energy gap at k = o for a simple cubic ferromagnet with anisotropic exchange interactions. Phys. Status Solidi B 102, 365–372 (1980).
Shender, E. Anti-ferromagnetic garnets with fluctuation-like interacting sub-lattices. Zh. Éksp. Teor. Fiz. 83, 326–337 (1982).
Henley, C. L. Ordering due to disorder in a frustrated vector antiferromagnet. Phys. Rev. Lett. 62, 2056–2059 (1989).
Chandra, P., Coleman, P. & Larkin, A. I. Ising transition in frustrated heisenberg models. Phys. Rev. Lett. 64, 88–91 (1990).
Gvozdikova, M. V. & Zhitomirsky, M. E. A Monte Carlo study of the first-order transition in a Heisenberg FCC antiferromagnet. J. Exp. Theor. Phys. Lett. 81, 236–240 (2005).
Rau, J. G., Lee, E. K.-H. & Kee, H.-Y. Generic spin model for the honeycomb iridates beyond the Kitaev limit. Phys. Rev. Lett. 112, 077204 (2014).
Shamoto, S.-I., Kodama, K., Harashina, H., Sato, M. & Kakurai, K. Neutron scattering study of BaCo0.82Ni0.18S2. J. Phys. Soc. Jpn 66, 1138–1144 (1997).
Shamoto, S.-i, Yamauchi, H., Ikeuchi, K., Kajimoto, R. & Ieda, J. Broken C4 symmetry in the tetragonal state of uniaxial strained BaCo0.9Ni0.1S1.9. Phys. Rev. Res. 3, 013169 (2021).
Abushammala, H. et al. Two-dimensional fluctuations and competing phases in the stripe-like antiferromagnet BaCoS2. arXiv https://doi.org/10.48550/arXiv.2302.12208 (2023).
Mandrus, D. et al. Magnetism in BaCoS2. J. Appl. Phys. 81, 4620–4622 (1997).
Syljuåsen, O. F., Paaske, J. & Schecter, M. Interplay between magnetic and vestigial nematic orders in the layered J1 − J2 classical Heisenberg model. Phys. Rev. B 99, 174404 (2019).
Juhász Junger, I., Ihle, D. & Richter, J. Thermodynamics of layered Heisenberg magnets with arbitrary spin. Phys. Rev. B 80, 064425 (2009).
Santos-Cottin, D. et al. Rashba coupling amplification by a staggered crystal field. Nat. Commun. 7, 11258 (2016).
Brosco, V. & Capone, M. Rashba-metal to Mott-insulator transition. Phys. Rev. B 101, 235149 (2020).
Santos-Cottin, D. et al. Linear behavior of the optical conductivity and incoherent charge transport in \({{{{{{{{\rm{BaCoS}}}}}}}}}_{2}\). Phys. Rev. Mater. 2, 105001 (2018).
Snyder, G., Gelabert, M. C. & DiSalvo, F. Refined structure and properties of the layered Mott insulator BaCoS2. J. Solid State Chem. 113, 355 – 361 (1994).
Kodama, K. et al. Electronic structure of the quasi two-dimensional Mott system BaCo1−xNixS2. J. Phys. Soc. Jpn 65, 1782–1786 (1996).
Schueller, E. C. et al. Structural signatures of the insulator-to-metal transition in BaCo1−xNixS2. Phys. Rev. Mater. 4, 104401 (2020).
Zainullina, V. M. & Korotin, M. A. Ground state of BaCoS2 as a set of energy-degenerate orbital-ordered configurations of Co2+ ions. Phys. Solid State 53, 978–984 (2011).
Moreo, A., Dagotto, E., Jolicoeur, T. & Riera, J. Incommensurate correlations in the t-J and frustrated spin-1/2 Heisenberg models. Phys. Rev. B 42, 6283–6293 (1990).
Capriotti, L., Fubini, A., Roscilde, T. & Tognetti, V. Ising transition in the two-dimensional quantum J1 − J2 Heisenberg model. Phys. Rev. Lett. 92, 157202 (2004).
Weber, C., Becca, F. & Mila, F. Finite-temperature properties of frustrated classical spins coupled to the lattice. Phys. Rev. B 72, 024449 (2005).
Lante, V. & Parola, A. Ising phase in the J1 − J2 Heisenberg model. Phys. Rev. B 73, 094427 (2006).
Oitmaa, J. & Enting, I. G. Critical behaviour of the anisotropic Ising model. J. Phys. C Solid State Phys. 5, 231 (1972).
Fernandes, R. M., Chubukov, A. V. & Schmalian, J. What drives nematic order in iron-based superconductors? Nat. Phys. 10, 97–104 (2014).
Wang, F., Kivelson, S. A. & Lee, D.-H. Nematicity and quantum paramagnetism in FeSe. Nat. Phys. 11, 959–963 (2015).
Kothapalli, K. et al. Strong cooperative coupling of pressure-induced magnetic order and nematicity in FeSe. Nat. Commun. 7, 12728 (2016).
Chubukov, A. V., Efremov, D. V. & Eremin, I. Magnetism, superconductivity, and pairing symmetry in iron-based superconductors. Phys. Rev. B 78, 134512 (2008).
Stanev, V., Kang, J. & Tesanovic, Z. Spin fluctuation dynamics and multiband superconductivity in iron pnictides. Phys. Rev. B 78, 184509 (2008).
Graser, S., Maier, T. A., Hirschfeld, P. J. & Scalapino, D. J. Near-degeneracy of several pairing channels in multiorbital models for the Fe pnictides. New J. Phys. 11, 025016 (2009).
Hu, J., Xu, B., Liu, W., Hao, N.-N. & Wang, Y. Unified minimum effective model of magnetic properties of iron-based superconductors. Phys. Rev. B 85, 144403 (2012).
Glasbrenner, J. K. et al. Effect of magnetic frustration on nematicity and superconductivity in iron chalcogenides. Nat. Phys. 11, 953–958 (2015).
Zhao, J. et al. Spin waves and magnetic exchange interactions in CaFe2As2. Nat. Phys. 5, 555–560 (2009).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Anisimov, V. I., Zaanen, J. & Andersen, O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 44, 943–954 (1991).
Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467–R5470 (1995).
Marzari, N. & Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 56, 12847–12865 (1997).
Souza, I., Marzari, N. & Vanderbilt, D. Maximally localized Wannier functions for entangled energy bands. Phys. Rev. B 65, 035109 (2001).
Mostofi, A. A. et al. An updated version of wannier90: a tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 185, 2309–2310 (2014).
Kanada, M. et al. High-pressure neutron and X-ray studies on the Mott transition of BaCoS2. J. Phys. Chem. Solids 60, 1181–1183 (1999).
Acknowledgements
We are thankful to H. Abushammala, A. Gauzzi, and Y. Klein for fruitful discussions. We acknowledge the allocation for computer resources by the French Grand Équipement National de Calcul Intensif (GENCI) under the project numbers A0110906493 and A0110912043. M.F. acknowledges financial support from the European Research Council (ERC), under the European Union’s Horizon 2020 research and innovation programme, Grant agreement No. 692670 “FIRSTORM”.
Author information
Authors and Affiliations
Contributions
M.C. and B.L. designed the research, B.L. performed the DFT + U calculations and M.F. developed the model. All authors analysed the data, discussed the results and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks Pawel Maksimov, Kyle Miller and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lenz, B., Fabrizio, M. & Casula, M. Order from disorder phenomena in BaCoS2. Commun Phys 7, 35 (2024). https://doi.org/10.1038/s42005-023-01514-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42005-023-01514-4
