
Abstract
Accurate prediction of antigen presentation to CD4+ T cells and subsequent induction of immune response are fundamentally important for vaccine development, autoimmune disease treatment and cancer neoepitope discovery. In immunopeptidomics, single-allelic data offer high specificity but limited allele coverage, whereas multi-allelic data provide broader representation at the expense of weak labelling. Current computational approaches either overlook the abundance of multi-allelic data or suffer from label ambiguity due to inadequate modelling strategies. To address these limitations, we present ImmuScope, a weakly supervised deep learning framework that integrates major histocompatibility complex class II (MHC-II) antigen presentation, CD4+ T cell epitopes and immunogenicity assessment. ImmuScope leverages self-iterative multiple-instance learning with positive-anchor triplet loss to decipher peptide-MHC-II binding from weakly labelled multi-allelic data and high-confidence single-allelic data. The training dataset comprises over 600,000 ligands across 142 alleles. Additionally, ImmuScope enables the interpretation of MHC-II binding specificity and motif deconvolution of immunopeptidomics data. We successfully applied ImmuScope to identify melanoma neoantigens, uncovering mutation-driven variations in peptide-MHC-II binding and immunogenicity. Furthermore, we employed ImmuScope to evaluate the effects of SARS-CoV-2 epitope mutations associated with immune escape, with predictions well aligned with experimentally observed immune escape dynamics. Overall, by offering a unified solution for CD4+ T cell antigen recognition and immunogenicity assessment, ImmuScope holds substantial promise for accelerating vaccine design and advancing personalized immunotherapy.
Similar content being viewed by others

Main
T cell-mediated adaptive immunity is crucial for protection against pathogens and diseases1,2,3,4,5,6,7. Antigen presentation by major histocompatibility complex class II (MHC-II) molecules to CD4+ T cells is essential in initiating and coordinating a wide range of immune responses8. The experimental identification of CD4+ epitopes and characterization of MHC-II binding specificities are time-consuming and costly due to the complex nature of antigen processing and the extensive polymorphism of MHC-II molecules9. Consequently, the effective high-throughput prediction of CD4+ T cell epitopes, understanding MHC-II binding specificity and assessing epitope immunogenicity are vital in developing vaccines and immunotherapies10,11,12,13,14,15.
Large-scale immunopeptidome datasets derived from liquid chromatography and mass spectrometry16 have greatly enhanced our understanding of MHC-II antigen presentation. These datasets, also known as eluted ligands (EL), are categorized into single-allelic (SA) data and multi-allelic (MA) data17,18, depending on whether allele-specific or pan-allelic antibodies are used during affinity purification. SA data provide precise, one-to-one peptide-MHC-II (pMHC-II) binding information. By contrast, MA data are weakly labelled, encompassing peptide interactions with multiple MHC-II alleles, where positive samples represent peptides that bind to at least one allele and negative samples represent non-binders. MA data offer broader allele coverage, over twice the coverage of SA data, especially for human leucocyte antigens (HLA)-DQ and HLA-DP loci. Recent findings underscore the clinical relevance of previously underexplored molecules (for example, HLA-DR3/4/5, HLA-DQ and HLA-DP) in autoimmune diseases19,20,21 and transplantation22, highlighting the necessity for the integrated analysis of SA and MA data. Ultimately, incorporating weakly labelled MA data alongside SA data mitigates SA-only biases and enables a comprehensive view of allele-specific binding patterns.
However, the weak labelling inherent in MA data (that is, peptides are not directly assigned to specific allomorphs) presents unique challenges for model design and training. Most studies (for example, HLAIImaster23 and BigMHC24) rely solely on SA data, which restricts their ability to cover broader allele sets. Although Graph-pMHC25 and MixMHC2pred-2.0 (ref. 26) incorporate all possible pMHC-II pairs from MA data during training, these approaches often yield high false-positive rates. NNAlign_MA27 and NetMHCIIpan17,28 leverage SA-trained neural networks to annotate MA data and are then fine-tuned with the pseudo-labelled data. However, prediction biases from the SA-based annotations can propagate through model training, particularly for alleles absent in SA data. Furthermore, naive self-training strategies fail to capture the rich allelic diversity within MA data. Therefore, there is an urgent need to develop a highly precise model that effectively integrates both SA and MA data for predicting CD4+ T cell-related immunity.
Besides antigen presentation, numerous computational approaches have demonstrated impressive potential in predicting epitope characteristics and immunogenicity18,26,29,30,31,32,33. Nonetheless, the complexity of CD4+ T cell activation and differentiation still poses a hurdle11,34,35,36. Most tools target a single facet of the cascaded immune process, such as NetMHCIIpan-4.0 (ref. 17) and NetMHCIIpan-4.2 (ref. 30) for antigen presentation, and DeepNeo37,38 and TLimmuno2 (ref. 39) for epitope immunogenicity. HLAIImaster23, BigMHC24 and Graph-pMHC25 are designed to handle both facets, whereas MoDec29, NNAlign_MA27, MixMHC2pred-2.0 (ref. 26) and NetMHCIIpan-4.3 (ref. 18) can deconvolve MHC-II binding specificity. However, no studies integrate the complete CD4+ T cell immune process—from antigen presentation and T cell recognition to immune response initiation—within one framework8. Transferring cascaded immunological knowledge from the previous stage can enhance CD4+ T cell epitope predictions and help understand how individual components of T cell immunity shape the immune response40,41,42. Furthermore, current algorithms lack fine-grained investigations across diverse immunopathological contexts or disease conditions, which may constrain their application potential in disease diagnosis and therapy43,44.
Here we propose ImmuScope, a weakly supervised deep learning framework for CD4+ T cell immunity prediction, empowered by both SA and MA data. Utilizing self-iterative multiple-instance learning (MIL) approach and quality annotation filtering, ImmuScope pinpoints high-confidence pMHC-II pairs from weakly labelled MA data to broaden allele coverage. It utilizes a positive-anchor triplet loss to uncover discriminative pMHC-II binding patterns. Trained on over 600,000 ligands covering 142 MHC-II alleles, ImmuScope achieves state-of-the-art prediction accuracy with superior robustness and generalizability, expertly designed to navigate the complexities of highly polymorphic alleles in antigen presentation and immune response modelling. Moreover, ImmuScope serves as a comprehensive model that mirrors the cascade of T cell responses through transfer learning on multiple tasks, including antigen presentation prediction, MHC-II binding specificity discovery, CD4+ T cell epitope prediction, immunogenicity prediction and motif deconvolution. We successfully apply ImmuScope to investigate the immunogenicity of melanoma neoantigens and assess the impact of epitope mutations on peptide-MHC-II binding. We further analyse SARS-CoV-2 epitopes to identify key binding cores and explore immune escape mechanisms, particularly in the Omicron variant. These findings underscore ImmuScope’s broad applicability in understanding T cell activation and advancing clinical applications in cancer and viral immunology.
Results
Overview of ImmuScope framework
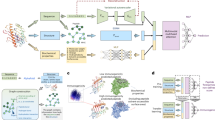
We have developed ImmuScope, a weakly supervised deep learning framework integrating metric learning to robustly predict CD4+ T cell responses. It supports a comprehensive suite of tasks including antigen presentation prediction, MHC-II binding specificity discovery, CD4+ T cell epitope prediction, immunogenicity prediction and motif deconvolution, facilitating a thorough exploration of the cascaded immune process (Fig. 1a). In particular, the weakly labelled MA data span a diverse range of allomorphs and contains massive peptide-binding data, particularly for the HLA-DQ and HLA-DP loci. Specifically, it comprises over 430,000 peptide samples—about 1.75 times that of the SA data—and encompasses about 2.2 times more MHC-II allomorphs (Fig. 1b). ImmuScope employs an MIL module to seamlessly integrate weakly labelled MA data with precisely annotated SA data, thereby harnessing the broad MHC coverage and extensive immunopeptidome of MA data alongside the specificity of SA data. Furthermore, ImmuScope employs a metric loss to capture more nuanced MHC-II binding specificities, thereby enhancing model discrimination.

a, Model backbone of ImmuScope consists of a pMHC-II interaction module (including core-aware convolution and multi-head cross-attention), a residual convolution module and positive-anchor triplet loss. Branch a focuses on learning bag-level features via an attention-based MIL aggregator, whereas branch b utilizes an MLP to learn instance-level features for various application tasks. b, Numbers of MA and SA datasets per MHC-II locus within the EL data, and the ratio of peptide counts for MA and SA data. c, Illustration of the self-iterative boosting strategy to generate high-quality pseudo-labels for MA data and refine the model. d, Downstream tasks of ImmuScope and corresponding data inputs. e, Applying ImmuScope in cohort studies on melanoma neoantigens and SARS-CoV-2. IMM, immunogenicity. Panels a, d and e created with BioRender.com.
The workflow of ImmuScope is as follows: paired pMHC-II sequences are processed through a core-aware convolutional module and a multi-head cross-attention module to extract interaction embeddings (Supplementary Fig. 1). These embeddings are then processed by residual convolutional blocks to construct the final pMHC-II representations. To effectively distinguish positive pairs from negative samples, we apply a positive-anchor triplet loss that minimizes distances among positive pairs and maximizes those from negatives, improving feature discrimination without increasing computational cost. Branch a employs an attention-based MIL aggregator to estimate the contribution of individual alleles within MA samples and compute the bag-level score. Meanwhile, branch b utilizes a multilayer perceptron (MLP) adapted to the specific type of training data for predicting CD4+ T cell epitopes, antigen presentation and immunogenicity. A self-iterative boosting strategy is applied to select high-confidence positive pseudo-labels, which are then combined with SA data to train the final antigen presentation model—ImmuScope-EL (Fig. 1c). Recognizing that MHC-II-mediated antigen presentation is essential for initiating CD4+ T cell activation, ImmuScope-EL is further fine-tuned for downstream tasks, including T cell epitope recognition and immunogenicity assessment (Fig. 1d). To evaluate model interpretability and applicability, we applied ImmuScope to analyse antigen presentation and neoantigen immunogenicity in a melanoma cohort, and to explore SARS-CoV-2 epitope discovery and immune escape mechanisms (Fig. 1e).
ImmuScope achieves state-of-the-art performance on CD4+ epitope benchmark
We evaluated the performance of ImmuScope and other algorithms, including Graph-pMHC25, MixMHC2pred-2.0 (ref. 26), NetMHCIIpan-4.2 (ref. 30) and NetMHCIIpan-4.3 (ref. 18), for identifying CD4+ T cell epitopes on the epitope benchmark. We employed them to predict the binding probability of each peptide to its given MHC-II allomorph and calculated the area under the curve (AUC) for each source protein, epitope and MHC-II allele entry. The AUCs demonstrated that ImmuScope significantly outperformed the current state-of-the-art methods, namely, NetMHCIIpan-4.3 and MixMHC2pred-2.0 (average AUC of 0.825 versus 0.771 and 0.761, respectively; Fig. 2a). In particular, ImmuScope demonstrated greater stability and higher average AUCs across diverse HLA loci (Extended Data Fig. 1a,b). In pairwise comparisons, it surpassed MixMHC2pred-2.0, NetMHCIIpan-4.2 and NetMHCIIpan-4.3 in 71.9%, 77.2%, and 73.7% of alleles, respectively (Fig. 2b). Despite potential statistical noise from few-sample subsets, ImmuScope demonstrated superior performance in most cases. Analysis across peptide lengths (Extended Data Fig. 1c) further confirmed its robustness, with ImmuScope performing best at most lengths, except for 16-mers where it matched NetMHCIIpan-4.3. Overall, the benchmarking results show that ImmuScope accurately predicts CD4+ T cell epitopes.

a, Box plot of AUCs on the CD4+ epitope benchmark. The P values were calculated using a two-sided Wilcoxon signed-rank test to compare ImmuScope with existing methods (NetMHCIIpan-4.3, P = 1.9 × 10−19; NetMHCIIpan-4.2, P = 2.2 × 10−19; MixMHC2pred-2.0, P = 3.3 × 10−22; n = 824). Box centre line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; dashed line, random; ****P < 0.0001. b, Comparisons of ImmuScope with MixMHC2pred-2.0 (left), NetMHCIIpan-4.2 (middle) and NetMHCIIpan-4.3 (right) in terms of AUC. The diagonal line indicates identical AUC values. The percentage in the lower-right corner shows the proportion of alleles for which ImmuScope outperforms the comparator, whereas marker size denotes the number of data points for each allele. Kernel density estimation curves along the top and right margins depict the marginal distributions of AUCs for each method, with the marked numbers indicating the AUC values at the highest density. c, Performance comparison across the SA, SA + MA-MIL and ImmuScope-EL models: AUPR (left) and PPV (right). The P values were calculated by the two-sided Wilcoxon signed-rank test (n = 58): SA versus SA + MA-MIL (AUPR, P = 5.3 × 10−11; PPV, P = 4.3 × 10−11) and SA + MA-MIL versus ImmuScope-EL (AUPR, P = 3.5 × 10−11; PPV, P = 3.5 × 10−11). Box centre line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; ****P < 0.0001. d, AUPR and PPV of ImmuScope-EL by stratifying different HLA loci. The bars represent the mean by 1,000 bootstrap iterations, and the error bars indicate the 95% CIs. e, UMAP visualization of instance embeddings from the SA and ImmuScope-EL models on the test set. In c and d, each data point represents the performance of the corresponding MHC allele.
Triplet loss and high-confidence pseudo-labels boost antigen presentation prediction
To evaluate the contribution of triplet loss and high-confidence pseudo-labels, we performed three ablation experiments using fivefold cross-validation: (1) a baseline SA model trained solely on SA data via branch b; (2) an SA + MA-MIL model incorporating both SA and MA data via MIL and triplet loss, without pseudo-label refinement; and (3) ImmuScope-EL, which additionally employed high-confidence positive pseudo-labels for iterative model refinement.
Integrating weakly labelled MA data through MIL substantially improved the predictive performance, whereas the auxiliary triplet loss helped refine feature-space boundaries, as shown by comparisons between SA + MA-MIL and the SA model in area under the precision–recall curve (AUPR), positive predictive value (PPV) and AUC0.1 (Fig. 2c and Extended Data Fig. 2a). The addition of high-confidence pseudo-labels further improved ImmuScope-EL’s performance over SA + MA-MIL (AUPR, 0.856 versus 0.836; PPV, 0.796 versus 0.779; AUC0.1, 0.823 versus 0.805). Pairwise allele-level analysis demonstrated consistent improvements across almost all MHC-II alleles (Extended Data Fig. 2b and Supplementary Fig. 2), and the performance gain was robust across different HLA loci and evaluation metrics (Fig. 2d and Extended Data Fig. 2c).
To better understand the impact of these components on feature learning, we visualized pMHC-II embeddings generated by the SA and ImmuScope-EL models using uniform manifold approximation and projection (UMAP)45 for representative alleles, including HLA-DRB1*01:01, HLA-DPA1*02:01/DPB1*09:01, H2-IAb, BoLA-DRB3*010:01 and BoLA-DRB3*020:02 (Fig. 2e and Extended Data Fig. 2d). In the ImmuScope-EL model, positive samples (red and blue) formed tight clusters, clearly separated from negatives. This improved clustering is attributed to the positive-anchor triplet loss, where only positives served as anchors due to the heterogeneity and noise in negatives derived from random natural peptides18. The auxiliary loss not only promotes a bottleneck-like transition zone between positives and negatives but also enhances the discriminative capacity of the learned pMHC representations, providing a stronger foundation for high-precision epitope identification and allele binding specificity analysis.
Motif deconvolution on MA data with attention-based MIL module
We applied ImmuScope’s attention-based MIL module to perform motif deconvolution on MA data, integrating attention outputs with antigen presentation scores (Fig. 3a). Due to the absence of precise labels in experimental MA datasets, we first validated this method via fivefold cross-validation on simulated MA data, achieving an average AUPR of 0.884 (95% confidence interval (CI), 0.882–0.885), AUC0.1 of 0.815 (95% CI, 0.814–0.817) and PPV of 0.819 (95% CI, 0.818–0.820; Fig. 3b). Clustering patterns based on the predicted attention scores revealed that ImmuScope accurately identified positives under weak supervision (Fig. 3c). Consequently, leveraging high-confidence positives from MA data via motif deconvolution may enhance antigen presentation prediction by enriching training data and broadening allele coverage.

a, Schematic of MHC-II motif deconvolution pipeline. b, Performance (AUPR, AUC0.1 and PPV) of the attention-based MIL module across different MHC-II alleles in the simulated data. Box centre line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range. Each data point represents the performance of the corresponding MHC allele (n = 58). c, Heat map of MIL attention weights and actual labels of the simulated data. d, Motif deconvolution logos on Racle__4037_DC heterozygous dataset and KLD analysis of PSFMs from the deconvoluted peptides and MHC-II immunopeptidomics. Panel a created with BioRender.com.
We further assessed the attention-based MIL module on several subsets of heterozygous MA datasets comprising varying numbers of HLA alleles. Specifically, subsets Racle__4037_DC29, Racle__RA957 (ref. 29) and Racle__3830_NJF_DQP29 include 4, 9 and 12 HLA alleles, respectively. Motifs derived from the predicted binding peptides were visualized using Seq2Logo46 for different MHC-II allomorphs (Fig. 3d and Extended Data Fig. 3a–c), with a focus on under-represented HLA-DQ and HLA-DP molecules. Comparison with the MHC Motif Atlas47 revealed high similarity at conserved positions (Supplementary Fig. 3a). We computed Kullback–Leibler divergence (KLD) between position-specific frequency matrices (PSFMs) from deconvoluted peptides and those from immunopeptidomics datasets. Most alleles, except HLA-DQA1*01:01/DQB1*05:01, demonstrated high similarity in the Racle__4037_DC dataset (Fig. 3d). Limited peptide ligands may explain the less-defined motif for HLA-DQA1*01:01/DQB1*05:01. Additionally, ImmuScope-EL successfully inferred motifs for MHC-II alleles absent from the database, with performance comparable with NetMHCIIpan-4.3, for example, HLA-DQA1*03:03/DQB1*04:02 and HLA-DQA1*05:05/DQB1*03:02 (Extended Data Fig. 3a,b and Supplementary Fig. 3b). These results highlight ImmuScope-EL’s capability as a robust tool for deciphering MHC-II binding motifs from MA datasets.
ImmuScope quantifies MHC-II binding specificities for allomorphs without known ligands
MHC-II polymorphism may hinder the prediction of pan-allelic binding specificity. To evaluate ImmuScope-EL on unseen alleles, we compared it with state-of-the-art methods, including NetMHCIIpan-4.3 and MixMHC2pred, in predicting binding specificity across different MHC-II loci. A leave-one-allele-out cross-validation strategy was used, excluding the target allele from training. We generated PSFMs for each allele by predicting 100,000 random human peptides using ImmuScope-EL and selecting the top 1% with the highest scores (Fig. 4a). Consistency with immunopeptidomics-derived PSFMs was assessed using KLD distance. ImmuScope-EL showed superior performance in inferring binding specificity for allomorphs without known ligands (Fig. 4b), particularly capturing the multiple specificities of HLA-DRB1*08:02 and the bidirectional specificity of HLA-DPA1*02:01/DPB1*09:01.

a, Workflow of MHC-II binding specificity evaluation. b, Comparison of ImmuScope with other algorithms in predicting MHC-II binding specificity. Bar plots of KLD distance against the PSFMs from the MHC-II immunopeptidomics data (top); peptide-binding motifs obtained by different methods (bottom). Motif of HLA-DQA1*01:02/DQB1*05:02 is derived from peptide ligands in the MHC Motif Atlas Database (http://mhcmotifatlas.org/). Other MHC-II peptide ligands were derived from the corresponding positive samples in the SA data. Panel a created with BioRender.com.
Despite the constraint of a limited dataset, which included only two SA HLA-DQ sets, ImmuScope-EL successfully predicted the binding specificity of HLA-DQA1*01:02/DQB1*05:02. The prediction was supported by MHC Motif Atlas data, reinforcing the reliability of our approach. The binding motifs highlighted substantial differences in binding preferences between HLA-DQA1*01:02/DQB1*05:02 and HLA-DQ allomorphs in the training set, for example, HLA-DQA1*01:02/DQB1*06:02 and HLA-DQA1*01:02/DQB1*06:04 (Supplementary Fig. 4). These findings illustrate ImmuScope-EL’s capability to elucidate local binding patterns across alleles, adeptly addressing the extensive polymorphism characteristic of MHC-II alleles.
ImmuScope enhances the accuracy of epitope immunogenicity prediction
An accurate prediction of immunogenic peptides that activate CD4+ T cells is essential for both vaccine development48 and immunotherapy49. To demonstrate the effectiveness of ImmuScope-IM in predicting immunogenicity, we compared it with five existing algorithms, including DeepNeo37, MixMHC2pred-2.0 (ref. 26), NetMHCIIpan-4.3 (ref. 18), TLimmuno2 (ref. 39) and HLAIImaster23. In particular, ImmuScope-IM exhibited superior performance on the immunogenicity benchmark with an overall AUC of 0.909 (95% CI, 0.901–0.918; Fig. 5a). We further assessed the performance across different MHC-II alleles. For MHC-II alleles with a sample size greater than ten and at least one immunogenic epitope, ImmuScope-IM consistently showed statistically higher AUCs than TLimmuno2 and HLAIImaster, with P values of 1.4 × 10−7 and 2.2 × 10−7, respectively (Fig. 5b). ImmuScope-IM outperformed HLAIImaster in 89.1% of the MHC-II alleles (Fig. 5c). Given the prevalence of fewer positive samples in real-world scenarios, we adopted a positive to negative ratio of 1:10 in building the immunogenicity dataset. Considering data imbalance, the precision–recall curve was utilized for a more accurate assessment of the model performance. The AUPRs highlighted substantial improvements in ImmuScope-IM over existing methods (Extended Data Fig. 4a,b). Predicted AUPRs for ImmuScope-IM and the leading existing model, HLAIImaster, were compared across different MHC-II alleles (Extended Data Fig. 4c). ImmuScope-IM demonstrated superior or comparable performance compared with HLAIImaster in 92.7% of the 55 MHC-II alleles.

a, Receiver operating characteristic curves of ImmuScope-IM and other methods on the immunogenicity benchmark. b, AUCs of ImmuScope-IM and other methods on the immunogenicity benchmark. The P values were calculated by the two-sided Wilcoxon signed-rank test to compare ImmuScope-IM with existing methods (HLAIImaster, P = 2.2 × 10−7; TLimmuno2, P = 1.4 × 10−7, n = 62). Box centre line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; points, data points; ****P < 0.0001. c, Pairwise comparison of AUCs between HLAIImaster and ImmuScope across different MHC-II alleles. d, Predictive analysis of melanoma neoantigen presentation based on ImmuScope-EL. e, Structural conformation of EDIL3290-304 epitopes bound to HLA-DPA1*01:03/DPB1*02:01 on the mutation predicted by AlphaFold3 (average predicted local distance difference test = 92.5; interface-predicted template modelling score = 0.92). The mutated residue is highlighted in red. The dashed lines indicate the hydrogen bonds, and interaction sites within 4 Å are displayed in dark blue. f, SHAP interpretation of p.P298F impact on EDIL3290-304 presentation by HLA-DPA1*01:03/DPB1*02:01. g, Predictive analysis of the immunogenicity of melanoma neoantigens based on ImmuScope-IM. h, Bar plots of AUCs on the SARS-CoV-2 immunogenic epitope benchmark. The bars represent the mean AUCs by 1,000 bootstrap iterations, and the error bars indicate the 95% CIs. i, Predicted binding peptide motifs for HLA-DRB1*01:01 as determined by the antigen presentation (ImmuScope-EL) and immunogenicity (ImmuScope-IM) models.
ImmuScope precisely reveals immunogenic neoantigens in melanoma
To investigate the practicality of ImmuScope for neoantigen identification, we applied it to a cutaneous melanoma cohort50 (Supplementary Tables 1 and 2). ImmuScope effectively detected HLA class II-presented immunogenic neoantigens within the tumour microenvironment and facilitated the evaluation of clinical outcomes. Using ImmuScope-EL, we predicted the binding probabilities of neoantigens to various HLA class II allomorphs in Pt-C and Pt-D, determining the most likely HLA class II restrictions and binding cores (Extended Data Fig. 4d,e and Supplementary Fig. 5). Except for EDIL3, predictions closely aligned with those from ref. 51 using NetMHCIIpan-4.0. Both HLA-DPA1*01:03/DPB1*02:01 and HLA-DPA1*01:03/DPB1*04:02 were predicted to present EDIL3, probably due to their high sequence similarity and comparable presentation capabilities. Further analysis revealed distinct differences in antigen presentation between mutant and wild-type antigens, especially concerning their structural organizations around the mutation site (Fig. 5d). In particular, mutations at key MHC-II anchor positions resulted in substantial changes in antigen presentation probability compared with wild-type antigens, as observed in LAMC1, EDIL3, ATXN2L II and PLXNB2. The corresponding wild-type epitopes failed to elicit immune responses, probably due to inadequate biophysical conditions for MHC-II binding.
Moreover, we employed AlphaFold3 to predict interaction conformations between EDIL3290-304 and HLA-DPA1*01:03/DPB1*02:01 before and after mutation (Fig. 5e and Supplementary Table 3). The proline-to-phenylalanine substitution probably enhanced peptide-MHC-II binding by increasing hydrophobic contacts and steric complementarity. An additional hydrogen bond further stabilized the complex and may have contributed to increased conformational flexibility. Structural analysis indicated that the mutated peptide fit better within the MHC binding groove, potentially enhancing presentation efficiency. SHapley Additive exPlanations (SHAP) analysis highlighted the mutated residue as critical for both peptide-MHC binding and T cell activation (Fig. 5f and Supplementary Fig. 6). Consistently, ImmuScope-IM demonstrated that most mutant neoantigens elicited stronger T cell responses than wild-type counterparts, except for RPS9 (Fig. 5g). A comparison with other methods is provided in Supplementary Note 6. These findings support the robustness of ImmuScope in predicting antigen presentation, binding core structure and immunogenicity, reinforcing its utility in neoantigen discovery for cancer immunotherapy.
ImmuScope predictions align closely with SARS-CoV-2 epitope discovery results
To rigorously evaluate ImmuScope’s performance in predicting SARS-CoV-2-specific T cell immunogenicity, we conducted comprehensive benchmarking using curated SARS-CoV-2 epitope data. Our analysis showed that ImmuScope-IM consistently outperformed existing methods in immunogenicity prediction (Fig. 5h). We further validated its clinical potential via a longitudinal study52, which systematically characterized CD4+ T cell epitopes derived from SARS-CoV-2 and restricted by the prevalent HLA-DR1 (DRB1*01:01) allotype. Their experimental study measured the maximal immune responses across eight donors against 29 candidate SARS-CoV-2 epitopes (Extended Data Fig. 5a), using HLA-DR4 (DRB1*04:01) as a negative control. This study design enabled the direct comparison of peptide immunogenicity between DR1+ and DR4+ donors via in vitro assays. Applying ImmuScope-IM to this clinical dataset, we predicted the immunogenicity of all peptides in both DR1+ and DR4+ donors (Extended Data Fig. 5b). Statistical validation via a paired Wilcoxon signed-rank test yielded a significant P value of 0.002, confirming that HLA-DR1-restricted peptides were consistently more immunogenic. In particular, this finding precisely matches the experimental observations from ref. 52. This strong concordance between computational predictions and clinical measurements highlights ImmuScope’s ability to accurately capture HLA-restricted immunogenicity patterns.
We also predicted the antigen presentation and immunogenicity-related binding motifs of peptides bound to HLA-DRB1*01:01 (Fig. 5i). We accurately identified the peptide-binding groove of HLA-DR1 epitopes derived from SARS-CoV-2, as defined structurally in ref. 52, through computational alignment scores (Fig. 6a,b and Extended Data Fig. 5c–f). In particular, we identified a more closely matching core binding site on the spike protein epitope S486-505. The complex structure of the binding peptide LQSYGFQPTNGVGY with HLA-DRB1*01:01 was predicted by AlphaFold3 with a predicted local distance difference test score above 90 and an interface-predicted template modelling score of 0.94 (Supplementary Table 3). The structure exhibited high local accuracy and interface alignment quality, rendering it highly reliable and instrumental in identifying potential immunogenic epitopes.

a,b, Predicted binding positions and alignment scores by ImmuScope-EL for SARS-CoV-2 spike protein epitope S486-505 (a) and S761-775 (b). The residues of the binding core within each peptide are labelled according to their positions. The interactions between the peptides and HLA-DR1 are displayed by dashed lines. pLDDT, predicted local distance difference test; ipTM, interface-predicted template modelling score. c, Structural comparison of HLA-DR1-S486-505Omicron (BA.1) aligned on the HLA-DR1-S486-505Wuhan HU-1 structure. The HLA-DR1 peptide-binding groove is depicted as a grey cartoon, whereas the S486-505Omicron (BA.1) and S486-505Wuhan HU-1 peptide are displayed as pink and lavender sticks, respectively. The mutant AAs are also highlighted. d, Structural comparison of HLA-DR1-S761-775Omicron (BA.1) aligned on the HLA-DR1-S761-775Wuhan HU-1 structure in two registers. Left: a +1 register shift in asymmetric unit (ASU) copies 1 and 3. Right: the same register as seen in ASU copy 2 (ref. 52). e, Alignment scores for the binding core of HLA-DR1-S486-505Wuhan HU-1 and HLA-DR1-S486-505Omicron (BA.1) predicted by ImmuScope-EL and the corresponding immunogenicity scores predicted by ImmuScope-IM. f, Alignment scores for the binding core of HLA-DR1-S761-775Wuhan HU-1 and various binding positions of Omicron (BA.1) predicted by ImmuScope-EL. g, Immunogenicity scores of HLA-DR1-S761-775Wuhan HU-1 and HLA-DR1-S761-775Omicron (BA.1) predicted by ImmuScope-IM.
ImmuScope facilitates understanding SARS-CoV-2 immune escape dynamics
Building on the work of ref. 52, who employed crystallographic analyses to elucidate how SARS-CoV-2 variant mutations affect epitope presentation and enable immune escape, we further investigated these mechanisms using ImmuScope-IM. Their structural studies of HLA-peptide complexes revealed that although Omicron (BA.1) variants S486–505 and S761–775 exhibited enhanced binding to HLA-DR1, they effectively escaped T cell recognition. We utilized ImmuScope-IM to investigate changes in the binding cores and immunogenicity of SARS-CoV-2 epitopes following mutation.
Structural analysis of HLA-DR1-S486-505Wuhan HU-1 revealed that S486-505Omicron (BA.1) induced mutations located within both the binding core and the peptide-flanking region. The S486-505Omicron (BA.1) epitope was bound by HLA-DR1 using the same register (Fig. 6c), consistent with core binding alignment scores predicted by ImmuScope-EL (Fig. 6e). The core sequence YFPLRSYSF exhibited a slight reduction in the post-mutation binding score, indicating that S486-505Omicron (BA.1) maintained favourable binding affinity (BA) with HLA-DR1. All core-positioned mutations occurred at potential T cell receptor (TCR) contact positions, particularly non-anchor residues Q493R (P5) and G496S (P8). Q493R (P5) introduced the most evident conformational change, with positively charged P5-Arg positioned centrally in the binding core. G496S added a polar hydroxyl at P8-Ser. The immunogenicity scores predicted by ImmuScope-IM decreased from 0.248 to 0.188 after mutation, consistent with the immune escape mechanisms of S486-505Omicron (BA.1), as revealed by structural analysis and further supported by SHAP analysis (Fig. 6e and Supplementary Fig. 7a). We also analysed a register shift caused by a single mutation in S761-775Omicron (BA.1) by ImmuScope. N764K, positioned at archetypal P1 anchor position for HLA-DR1, resulted in two distinct peptide conformations of HLA-DR1-S761-775Omicron (BA.1) (Fig. 6d). In the first conformation, the HLA molecule contacted the neighbouring Leu at the P1 pocket, generating a +1 register shift (that is, TQLKRALTGIAVEQD to TQLKRALTGIAVEQD). This new peptide conformation bound to HLA-DR1 via P1-Leu, P4-Ala and P9-Ala, with an unfavourable Thr at P6 due to its large polar hydroxyl side chain52. The second conformation accommodated the N764K mutation at P1 to bind S761-775Omicron (BA.1), aligning with the binding register of S761-775Wuhan HU-1.
We also calculated the alignment scores of binding regions after mutation. KRALTGIAV achieved the highest score, corresponding to the second peptide conformation. The alignment score of LKRALTGIA was –0.758 (Fig. 6f) and 1.574 when excluding P6 (Supplementary Fig. 8). LKRALTGIA still exhibited suboptimal binding probability relative to other regions, consistent with structural analysis52. In particular, ImmuScope-IM predicted a 72.8% reduction in immunogenicity score for HLA-DR1-S761-775Omicron (BA.1) compared with its HLA-DR1-S761-775Wuhan HU-1 counterpart (Fig. 6g), probably due to sequence alterations and structural rearrangements introduced by the mutation. SHAP analysis (Supplementary Fig. 7b) supported this reduction, suggesting impaired T cell recognition. Additional analyses of epitope mutations across diverse SARS-CoV-2 variants and HLA-II alleles are provided in Supplementary Note 7, offering broader insights into immune escape strategies.
Discussion
ImmuScope represents a significant advance in computational immunology by integrating weakly supervised learning and metric learning to predict CD4+ T cell-mediated immune responses with unprecedented accuracy. Our framework overcomes key limitations of existing approaches through its self-iterative MIL architecture, which effectively combines weakly labelled MA data with highly specific SA datasets to achieve a twofold improvement in allele coverage. Incorporating positive-anchor triplet loss further enhances ImmuScope’s ability to resolve challenging pMHC-II interactions, enabling state-of-the-art performance in antigen presentation prediction, T cell epitope recognition and immunogenicity assessment. Beyond these core capabilities, ImmuScope provides robust tools for motif deconvolution and binding specificity analysis, demonstrating particular value in two clinically important applications: evaluating melanoma neoepitope immunogenicity and deciphering SARS-CoV-2 immune escape mechanisms associated with mutations. Different from the established CD4+ T cell epitope predictors, ImmuScope uniquely integrates SA and MA immunopeptidomics data within a unified framework that mirrors the biological MHC-II antigen processing and CD4+ T cell recognition cascade. This design broadens allele coverage and boosts accuracy across key tasks, addressing long-standing limitations in the field. A key feature of ImmuScope is its residue-level attribution module, which enables molecular insight into CD4+ T cell immunity by elucidating binding specificity and immune escape dynamics at residue-level resolution. As immunopeptidomics resources continue to grow in scale and resolution, ImmuScope’s biologically grounded design and analytical capabilities position it as a powerful AI platform for next-generation vaccine development and precision immunotherapy, bridging the gap between epitope prediction and mechanistic understanding of CD4+ T cell responses.
Although these advances are substantial, several challenges must be addressed to fully realize ImmuScope’s clinical potential. Our current reliance on IFN-γ production as the primary readout for CD4+ T cell responses, although conventional, provides an incomplete picture of T cell immunity. A comprehensive assessment should incorporate Th2/Th17 cytokine profiles (IL-4, IL-10 and IL-17), T cell proliferation assays and activation markers (CD25 and CD69) to better capture the full spectrum of T cell functionality53,54,55. Another critical limitation is ImmuScope’s current inability to systematically evaluate how mutations affect epitope immunogenicity, despite the profound impact such alterations can have on MHC-II binding and T cell recognition. This gap could be addressed by incorporating deep mutational scanning data or in silico mutagenesis approaches to enable the quantitative prediction of mutation effects56. Furthermore, although ImmuScope effectively leverages large-scale pMHC-II sequence datasets to predict CD4+ T cell epitopes, the limited availability of structural data for pMHC-II and TCR-pMHC-II complexes constrains its ability to capture peptide-MHC binding preferences. With only approximately 300 experimentally resolved complex structures currently available in the TCR3d57 database, our understanding of the structural determinants of T cell recognition remains incomplete.
Looking ahead, several strategic improvements will enhance ImmuScope’s capabilities and applications. First, integrating multidimensional immune datasets encompassing cytokine profiles, proliferation measurements, activation markers and TCR repertoire information will enable more comprehensive epitope characterization. Second, advances in structural prediction algorithms like AlphaFold3, coupled with emerging high-resolution structural biology techniques, promise to dramatically expand the database of TCR-pMHC-II complexes58,59, facilitating the atomic-level modelling of immune recognition. Third, developing mutation-aware modelling will improve our ability to predict and optimize epitope immunogenicity. Finally, extending ImmuScope to model HLA-dependent immunogenicity across genetically diverse populations will be crucial for developing globally effective vaccines and personalized immunotherapies. By incorporating both CD4+ and CD8+ T cell response prediction capabilities, future versions of ImmuScope could become a unified platform for vaccine design, cancer immunotherapy development and infectious disease research60,61, ultimately bridging the gap between computational immunology and clinical practice.
Methods
Datasets
The statistics of the datasets used for training and validation for different tasks are shown in Supplementary Table 4. The following is a detailed dataset description.
MHC-II antigen presentation data
To train the antigen presentation model, we used the large-scale antigen presentation data collected in refs. 17,18,30,62, comprising three data types: BA, SA and MA EL datasets (Fig. 1b and Supplementary Fig. 9a,b). All data were filtered to remove possible contaminants and MHC class I-restricted peptides, retaining peptides of 12–21 amino acids (AAs) in length26. The EL datasets were then enriched by uniformly sampling five times of 12–21-AA random natural peptides as negative samples. The datasets were divided into five subsets for cross-validation using the common-motif method, ensuring that peptides sharing a subsequence of nine or more AAs were grouped into the same subset63. The final SA dataset contains 246,590 positive and 2,448,316 negative samples, whereas the MA dataset includes 432,255 positive and 4,467,755 negative samples, covering 142 MHC-II molecules. Additionally, the BA dataset comprises 129,110 data points across 80 class II molecules.
CD4+ epitope benchmark
The CD4+ epitope benchmark18, compiled by Nilsson et al. in 2023, was assembled following a specific protocol. Initially, positive CD4+ T cell epitopes ranging from 12 AAs to 21 AAs, without post-translational modifications and with complete four-digit MHC-II typing, were selected from the Immune Epitope Database (IEDB; https://www.iedb.org/)64. Only epitopes associated with well-documented source proteins were considered. Subsequently, the corresponding negative samples were generated based on the source protein sequences retrieved from the UniProt database (https://www.uniprot.org/)65. Each {epitope, allele, protein} triplet was then segregated into a distinct test subset. Within each subset, using a sliding window of the same length as the epitope, overlapping peptides were generated from the source protein sequence and designated as negative samples, excluding the epitope itself. Furthermore, it was ensured that none of the samples in the test set had previously appeared in the MHC-II antigen presentation training data. Ultimately, the test set comprised 842 {epitope, allele, protein} triplets, encompassing 40 HLA-DR, 13 HLA-DQ and 4 HLA-DP molecules.
Immunogenicity data
We curated immunogenicity assay data from IEDB64 and integrated it with the MHCBN66 dataset, following the methodology described in DeepNeo37,38. This dataset contains records up to 14 May 2024. Specifically, we selected the data of T cell reactivity based on IFN-γ secretion. Furthermore, we refined the dataset to include only entries with full MHC-II restriction and peptide lengths ranging from 12 AAs to 21 AAs. Given the variable nature of pMHC-II immunogenicity experiments, we followed the method in ref. 37 to classify pMHC-II with contradictory results as binding pairs. Moreover, we identified proteins with sequence similarity below 0.5 in the RCSB Protein Data Bank67 and generated ten times as many negative samples by randomly splitting peptides of the same length as the positive samples. The strategy aligns with the approaches used in IEPAPI68 and MHCflurry 2.0 (ref. 69). Subsequently, we randomly divided the data into training/validation and test sets at an 8:2 ratio. Consequently, the training/validation set comprised 71,584 data points, and the test set included 17,897 data points for our immunogenicity analysis.
Simulated MA dataset
Due to the absence of precise labels in the MA data, we constructed a simulated MA dataset using the SA dataset, which has been divided into a fivefold cross-validation set, to evaluate the capability of the MIL module in detecting positive pMHC-II samples within bags. The process was as follows: we selected four out of the five folds as the training set. These data were then randomly shuffled and organized into bags, each containing ten samples. Subsequently, we randomly sampled negative instances to achieve a 1:3 ratio of positive to negative bags.
Melanoma neoantigen data
The melanoma neoantigen data were obtained from ref. 51, who identified and functionally characterized 13 HLA class II-restricted neoantigens in two melanoma patients (Pt-C and Pt-D). The corresponding epitope information is available in the IEDB (reference IRI: http://www.iedb.org/reference/1042469). We included only neoantigens with reported TCR reactivity and quantitative avidity measurements. Each selected neoantigen was directly compared with its wild-type counterpart, enabling reliable immunogenicity assessment based on EC50 values.
SARS-CoV-2 immunogenic epitope data
The SARS-CoV-2 immunogenic epitope data were curated from the IEDB database64 (accessed 2 April 2025) and relevant primary literature (Supplementary Tables 4 and 5). Data from IEDB were retrieved using the following query parameters: disease set to COVID-19, full MHC class II restriction, source limited to peer-reviewed journal articles and T cell reactivity measured by IFN-γ secretion. Additional epitopes were manually extracted from selected primary publications. The resulting dataset comprises immunogenic epitopes derived from SARS-CoV-2 structural proteins (S, E, M and N) and the non-structural protein nsp12. All epitopes were clustered using MMseqs2 (ref. 70) with a sequence identity threshold of 0.5. The resulting clusters were then split into training and test sets at a 6:4 ratio, yielding 6,181 and 3,763 samples, respectively. To improve model generalizability, the training set was augmented with a general immunogenicity dataset containing non-SARS-CoV-2-derived epitopes, resulting in a total of 95,237 training samples.
ImmuScope architecture
MA and SA data representation
In this study, our model processed two predominant forms of mass spectrometry immunopeptidomics data: MA and SA data. Following the paradigm of MIL, we treated each MA sample as a ‘bag’ containing multiple instances, specifically pMHC-II pairs (Supplementary Fig. 9c). A positive bag suggests that the peptides are presented by at least one of the MHC molecules expressed in that sample. Conversely, a negative bag indicates that all pMHC-II pairs are negative instances. Similarly, for SA data, we defined each pMHC-II sample as either a positive bag with a single positive instance or a negative bag with a single negative instance. This consistent representation of MA and SA data enabled our framework to simultaneously learn from both data types and make predictions, thereby facilitating its application across diverse immunopeptidomics datasets.
Attention-based MIL aggregator
In branch a of Fig. 1a, we employed an attention-based MIL pooling mechanism71,72 to aggregate instance features within each bag. This mechanism not only enhances interpretability for predicting bag labels but also enables the identification and prioritization of the most critical instances crucial for the final prediction. Let Z = ℇ(X; θ) represent the embedding of pMHC-II instance obtained from the backbone of ImmuScope ℇ parameterized by θ. zk denotes the kth instance in the bag Z = {z1…zk}. We implemented the following gated attention aggregator:
where w, V and U denote the model parameters and ⊙ represents an element-wise multiplication. The function tanh(·) refers to the hyperbolic tangent activation function and sigm(·) denotes the sigmoid nonlinearity.
High-confidence positive pseudo-labels selection module
We introduced high-confidence positive pseudo-labels to improve the accuracy of antigen presentation prediction. The number of positive samples in MA data is approximately twice that in SA data, and allele coverage is 2.2 times larger. This difference is particularly evident at the HLA-DP and HLA-DQ loci, where MA data substantially supplements coverage gaps in SA data. In particular, these weakly annotated positive MA samples contain multiple pMHC-II pairs, with at least one pair exhibiting positive signals. Such characteristics pose challenges for directly incorporating MA data into model training. To address this, we have developed a high-confidence positive pseudo-label selection module, which self-iteratively incorporates pseudo-labels from MA data to refine our predictive model (Fig. 1c).
High-confidence sample selection is performed using the trained backbone of ImmuScope, with the training process detailed in the ‘Antigen presentation prediction’ section. MA data are first input into the ImmuScope backbone, which incorporates Monte Carlo dropout73 to assess variability and enhance reliability. An attention-based MIL aggregation module is then used to estimate the uncertainty distribution of the MA samples, enabling the identification of high-confidence positive samples. Specifically, we iteratively select high-confidence samples by controlling the confidence ratio (Top R%) based on the attention scores. Samples already showing high confidence within the antigen presentation prediction branch are excluded. The selected samples are then integrated into the SA data for model fine tuning. Throughout this iterative process, we progressively adjust confidence thresholds to incorporate a broader range of positive MA samples, thereby improving model generalization. The optimal ratio of positive pseudo-labelled samples is determined based on validation performance.
Positive-anchor triplet loss
MHC-II molecules exhibit extensive diversity, exemplified by the human HLA-DR, HLA-DQ and HLA-DP loci, which collectively comprise 11,674 allelic variants according to the IPD-IMGT/HLA database74. Additionally, the peptides themselves show notable variability in sequence and length. The peptide-binding groove of MHC-II is highly specific for binding AAs in peptides75, determining which peptides can be bound and presented. Triplet loss76 enhances the model’s ability to perceive these subtle differences by minimizing the distance between similar samples (positive samples) and maximizing the distance between dissimilar samples (negative samples). This loss is particularly suitable for predicting pMHC-II BA and antigen presentation, as it improves learning on challenging-to-discriminate pMHC-II samples and facilitates the discovery of nuanced binding patterns between peptides and specific MHC-II molecules.
In the experimental setup, triplet loss was calculated using only positive samples as anchors. This strategy enabled the model to better distinguish crucial binding features within pMHC-II complexes. The positive-to-negative sample ratio in the antigen presentation dataset was set to 1:10. Using negative samples as anchors increased computational costs and might distract from the model’s primary goals by unnecessarily optimizing distances between negative samples. Such optimization failed to enhance discrimination and reduced the learning efficiency. To address these challenges and align with critical learning objectives, we have formulated the triplet loss for each mini-batch as follows:
where d(xi, yi) = ||xi – yi||p, we used Euclidean distance as the metric function, setting p = 2. In this context, i represents a mini-batch, N is the batch size and a exclusively denotes all the positive samples used as anchors; pos and neg indicate the positive and negative samples within the mini-batch, respectively; margin is a threshold defining the minimum distance that the negative sample must exceed beyond the positive sample from the anchor to avoid incurring a loss.
ImmuScope training process
ImmuScope backbone training process
The backbone of ImmuScope is a pretrained model for other downstream tasks. Initially, we loaded the SA and MA data, and then we computed the positive-anchor triplet loss for the embeddings of pMHC-II instances, denoted as \({{\mathcal{L}}}_{{{\rm{triplet\_loss}}}}\). In branch a, the bag labels for SA and MA data were optimized using the binary cross-entropy loss function, represented as \({{\mathcal{L}}}_{{{\rm{MIL\_SA}}}}\) and \({{\mathcal{L}}}_{{{\rm{MIL\_MA}}}}\), respectively. Concurrently, in branch b, the SA data were optimized using the binary cross-entropy loss \({{\mathcal{L}}}_{{{\rm{instance\_SA}}}}\). The composite loss function for the backbone is defined as
where τ represents the weighting factor for the triplet loss, setting τ = 0.1. Throughout the training process, the parameters of the ImmuScope backbone network were refined by synergistically combining individual instance learning, aggregated label optimization and metric learning strategies. This integrative approach ensured a robust optimization of model parameters, effectively capturing both micro- and macro-level data characteristics. The Adam optimizer with a learning rate of 1 × 10−3 was used to train the backbone of ImmuScope for up to 20 epochs, with the final model being selected based on the best performance on the validation set.
Antigen presentation prediction
On the basis of the backbone of ImmuScope, we gradually introduced high-quality positive pseudo-labels from MA data to construct an antigen presentation prediction model. In each epoch, we first obtained the predicted antigen presentation probability on branch b, the attention score in the MIL aggregator and the corresponding bag score through forward calculations. To ensure prediction stability and accurately gauge model uncertainty, we employed an architecture with Monte Carlo dropout to perform ten forward passes and analysed both mean and variance of these predictions. Initially, we selected the top 8% of samples with high attention weights and whose variances ranked in the top 80% (from lowest to highest). These thresholds (8% and 80%) were determined through preliminary experiments and an examination of the distribution of attention scores, ensuring that we focused on high-confidence, relatively low-variance samples. We also excluded samples with predicted antigen presentation probabilities exceeding 0.95 and those whose variances ranked in the top 40% (from lowest to highest), as they were already reliably identified by the model.
As the iterations progressed and the model’s internal representations became more stable, we gradually relaxed the threshold on attention weights from the top 8% to 12%. This step—commonly employed in self-training approaches—aims to broaden the scope of positive pseudo-labelled samples, thereby enriching the training dataset with more diverse pMHC-II binding candidates and further enhancing the model’s learning capacity. At the same time, we utilized this expanded dataset, SA-extend (EL), for incremental fine tuning of the backbone model. Finally, we fine-tuned the ImmuScope backbone with the final SA-extend (EL) dataset over ten additional epochs using the Adam optimizer (learning rate = 3 × 10−5), yielding the optimized ImmuScope-EL model for antigen presentation prediction.
CD4+ T cell epitope prediction
Antigen presentation is a prerequisite for the CD4+ T cell immune response. In line with the methodology of NetMHCIIpan-4.3, our CD4+ T cell epitope prediction model, ImmuScope, similarly incorporated both BA and EL data. Specifically, the antigen presentation model ImmuScope-EL was fine-tuned using BA data, employing a learning rate of 2 × 10−5, and leveraging the Adam optimizer to minimize the mean squared error loss over 20 epochs. To balance the influence of BA and EL data on CD4+ T cell epitope prediction, we set an 8:2 weighting ratio for BA and SA data in the validation set. This ratio was determined based on preliminary experiments and data correlation: although BA data provide precise BA information, SA data capture actual antigen presentation events in vivo. The final validation metrics were calculated as follows:
where AUPRBA and AUPRSA denote the AUPR values of the BA and SA subsets, respectively, within the validation set. Finally, we evaluated the performance of CD4+ T cell epitope prediction on the CD4+ epitope benchmark.
MHC-II epitope immunogenicity prediction
Immunogenicity is crucial as it determines the efficacy and safety of vaccines and therapies by triggering immune responses. We refined ImmuScope-EL further with immunogenicity data to develop the ImmuScope-IM model, tailored to immunogenicity prediction. The ImmuScope-IM model was optimized by an Adam optimizer with a learning rate of 1 × 10−3 and binary cross-entropy loss, for a maximum of 20 epochs. For the application of the ImmuScope-IM model in SARS-CoV-2 epitope discovery and dynamic escape mechanism studies, we excluded the epitope binding data pertaining to SARS-CoV-2 from our initial immunogenicity dataset to construct a dedicated SARS-CoV-2 immunogenicity benchmark dataset, ensuring unbiased benchmarking. This benchmark dataset was then used to train the ImmuScope-IM model for assessing the immunogenicity of SARS-CoV-2 epitopes.
All deep learning models were developed using PyTorch v. 1.12.1 and trained on an NVIDIA GeForce RTX 4090 GPU. Details of the algorithm and model hyperparameters are provided in Supplementary Tables 6 and 7, respectively. Computational efficiency and scalability are described in Supplementary Note 8.
Analysis of motif deconvolution
We employed the trained ImmuScope-EL model to perform motif deconvolution and obtain the binding peptide sequence set for different MHC-II allomorphs. Specifically, a subset of MA data was fed into ImmuScope-EL, and the attention weights from the attention-based MIL aggregator, along with the antigen presentation probabilities from branch a and branch b, were obtained, respectively. To ensure high-quality deconvolution, we selected the antigen presentation peptides with an antigen presentation probability greater than 0.8 and an attention weight exceeding the reciprocal of the number of MHC-II categories in the bag. We then employed Seq2Logo to visualize the motif logo of different MHC-II allomorphs based on the sequences of selected peptides.
Quantification of MHC-II binding specificity
We first calculated the antigen presentation score by inputting 100,000 random human peptide sequences and the alleles to be assessed into ImmuScope-EL. Then, the samples with the top 1% of the predicted scores were selected for cluster analysis using GibbsCluster77, and the optimal number of clusters, that is, binding specificity, was determined based on the lowest average KLD. Finally, we evaluated the MHC binding specificity quantified by ImmuScope-EL by comparing the KLD with the PSFM matrix based on the peptidomics data. The prediction results of NetMHCIIpan were obtained by predicting the top 1% of random human peptides using the NetMHCIIpan-4.3 software package, whereas MixMHC2pred was obtained by predicting using the MixMHC2pred-2.0 web server.
Measuring the similarity of MHC binding motifs
To evaluate the similarity between sequence motifs generated by various algorithms and those obtained from peptidomics data, we first represented each set of peptide-binding cores with PSFMs. Each PSFM was then converted into a single vector by concatenating the frequency values at its nine positions, with each position containing 20 values corresponding to the 20 standard AAs. Finally, we calculated the symmetric KLD18 for any two PSFMs, denoted as a and b, using the following formula:
where ε is employed as an exceedingly small positive number, typically set at 1 × 10−10, to prevent division by zero.
Calculation of binding core alignment scores for epitopes
In our analysis of the melanoma neoepitope and SARS-CoV-2 epitope binding cores, we employed the ImmuScope-EL model to analyse the binding cores of various epitopes and to examine changes on mutations. Initially, we used ImmuScope-EL to predict 100,000 random human peptides and selected the top 1% based on the highest binding scores to create a position-specific scoring matrix for specific alleles (like HLA-DRB1*01:01 in SARS-CoV-2 epitope analysis). Subsequently, we calculated the matching degree for each 9-mer window of the candidate peptides against the position-specific scoring matrix. The alignment score for each window was then computed to assess how well it matched the binding pattern defined by the position-specific scoring matrix.
Statistical analyses
Error bars depicted in the bar plots indicate 95% CIs, unless specified otherwise. Performance benchmarks such as AUC and AUPR were computed using the scikit-learn Python package (v. 1.3.0). UMAP analysis was conducted with the umap-learn Python package (v. 0.5.3). The predicted binding peptide ligands were further clustered using the GibbsCluster tool (v. 2.0). Sequence motifs were generated and visualized using the Seq2Logo tool (v. 2.0). Additionally, the three-dimensional structures of pMHC-II complexes were visualized using PyMOL (v. 2.5.7).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets used for training and testing the algorithm are available via Zenodo at https://doi.org/10.5281/zenodo.14184201 (ref. 78). The MHC-II antigen presentation data and CD4+ epitope benchmark are sourced from NetMHCIIpan-4.318 (https://services.healthtech.dtu.dk/services/NetMHCIIpan-4.3/). Immunogenicity datasets are derived from IEDB64 (https://www.iedb.org/). The motifs of MHC-II alleles are obtained from the MHC Motif Atlas Database47 (http://mhcmotifatlas.org/) and NetMHCIIpan-4.3. Melanoma neoepitope immunogenicity data are derived from original study51 (https://doi.org/10.1038/s41586-022-04682-5). The benchmark dataset for SARS-CoV-2 immunogenicity prediction is systematically curated from the IEDB and relevant primary literature (Supplementary Tables 4 and 5 provide details). The core dataset of SARS-CoV-2-derived CD4+ T cell epitopes, including those analysed for HLA-DR1- and HLA-DR4-restricted immune responses, along with the structural data of HLA class II-presented SARS-CoV-2 epitopes and associated immune escape metadata, is obtained from the foundational study by Chen et al.52 (https://doi.org/10.1016/j.celrep.2023.112827). To enable extended epitope mutation analysis (Supplementary Information), we incorporated additional datasets from two key studies examining SARS-CoV-2 immune escape79,80 (https://doi.org/10.1126/sciadv.abl5394 and https://doi.org/10.1038/s41590-022-01351-7). All HLA class II sequence data are retrieved from the IPD-IMGT/HLA database74 (https://www.ebi.ac.uk/ipd/imgt/hla/), whereas SARS-CoV-2 variant sequence evolution data are derived from ViralZone81 (https://viralzone.expasy.org/). Source data are provided with this paper.
Code availability
The source code and model weights of ImmuScope are available via GitHub at https://github.com/shenlongchen/immuscope, Zenodo at https://doi.org/10.5281/zenodo.14184201 (ref. 78) and as a reproducible CodeOcean capsule (https://codeocean.com/capsule/4332973/tree/v1 (ref. 82)).
References
Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 23, 186–193 (2022).
Deets, K. A. & Vance, R. E. Inflammasomes and adaptive immune responses. Nat. Immunol. 22, 412–422 (2021).
Roy, P., Orecchioni, M. & Ley, K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat. Rev. Immunol. 22, 251–265 (2022).
Chi, H., Pepper, M. & Thomas, P. G. Principles and therapeutic applications of adaptive immunity. Cell 187, 2052–2078 (2024).
Dikiy, S. & Rudensky, A. Y. Principles of regulatory T cell function. Immunity 56, 240–255 (2023).
Sultan, H. et al. Neoantigen-specific cytotoxic Tr1 CD4 T cells suppress cancer immunotherapy. Nature 632, 182–191 (2024).
Dendrou, C. A., Petersen, J., Rossjohn, J. & Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 18, 325–339 (2018).
Pishesha, N., Harmand, T. J. & Ploegh, H. L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 22, 751–764 (2022).
Logunova, N. N. et al. MHC-II alleles shape the CDR3 repertoires of conventional and regulatory naïve CD4+ T cells. Proc. Natl Acad. Sci. USA 117, 13659–13669 (2020).
Brightman, S. E. et al. Neoantigen-specific stem cell memory-like CD4+T cells mediate CD8+T cell-dependent immunotherapy of MHC class II-negative solid tumors. Nat. Immunol. 24, 1345–1357 (2023).
Joglekar, A. V. & Li, G. T cell antigen discovery. Nat. Methods 18, 873–880 (2021).
Wells, D. K. et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183, 818–834. e813 (2020).
Hudson, D., Fernandes, R. A., Basham, M., Ogg, G. & Koohy, H. Can we predict T cell specificity with digital biology and machine learning? Nat. Rev. Immunol. 23, 511–521 (2023).
Vegesana, K. & Thomas, P. G. Cracking the code of adaptive immunity: the role of computational tools. Cell Syst. 15, 1156–1167 (2024).
Sidhom, J.-W., Larman, H. B., Pardoll, D. M. & Baras, A. S. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat. Commun. 12, 1605 (2021).
Purcell, A. W., Ramarathinam, S. H. & Ternette, N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 14, 1687–1707 (2019).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020).
Nilsson, J. B. et al. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning. Sci. Adv. 9, eadj6367 (2023).
Hu, X. et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet. 47, 898–905 (2015).
Sollid, L. M. Tolerance-inducing therapies in coeliac disease—mechanisms, progress and future directions. Nat. Rev. Gastroenterol. Hepatol. 21, 335–347 (2024).
Busch, R., Kollnberger, S. & Mellins, E. D. HLA associations in inflammatory arthritis: emerging mechanisms and clinical implications. Nat. Rev. Rheumatol. 15, 364–381 (2019).
Fernández-Viña, M. A. et al. Multiple mismatches at the low expression HLA loci DP, DQ, and DRB3/4/5 associate with adverse outcomes in hematopoietic stem cell transplantation. Blood 121, 4603–4610 (2013).
Yang, Q. et al. HLAIImaster: a deep learning method with adaptive domain knowledge predicts HLA II neoepitope immunogenic responses. Brief. Bioinform. 25, bbae302 (2024).
Albert, B. A. et al. Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity. Nat. Mach. Intell. 5, 861–872 (2023).
Thrift, W. J. et al. Graph-pMHC: graph neural network approach to MHC class II peptide presentation and antibody immunogenicity. Brief. Bioinform. 25, bbae123 (2024).
Racle, J. et al. Machine learning predictions of MHC-II specificities reveal alternative binding mode of class II epitopes. Immunity 56, 1359–1375 e1313 (2023).
Alvarez, B. et al. NNAlign_MA; MHC peptidome deconvolution for accurate MHC binding motif characterization and improved T-cell epitope predictions. Mol. Cell. Proteomics 18, 2459–2477 (2019).
Reynisson, B. et al. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J. Proteome Res. 19, 2304–2315 (2020).
Racle, J. et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 37, 1283–1286 (2019).
Nilsson, J. B. et al. Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome. Commun. Biol. 6, 442 (2023).
McMaster, B., Thorpe, C., Ogg, G., Deane, C. M. & Koohy, H. Can AlphaFold’s breakthrough in protein structure help decode the fundamental principles of adaptive cellular immunity? Nat. Methods 21, 766–776 (2024).
Motmaen, A. et al. Peptide-binding specificity prediction using fine-tuned protein structure prediction networks. Proc. Natl Acad. Sci. USA 120, e2216697120 (2023).
Shen, L.-C. et al. Supervised contrastive learning enhances MHC-II peptide binding affinity prediction. Expert Syst. Appl. 269, 126463 (2025).
Yang, Y. et al. MHCII-peptide presentation: an assessment of the state-of-the-art prediction methods. Front. Immunol. 15, 1293706 (2024).
Peters, B., Nielsen, M. & Sette, A. T cell epitope predictions. Annu. Rev. Immunol. 38, 123–145 (2020).
Schaap-Johansen, A.-L., Vujović, M., Borch, A., Hadrup, S. R. & Marcatili, P. T cell epitope prediction and its application to immunotherapy. Front. Immunol. 12, 712488 (2021).
Kim, J. Y. et al. MHC II immunogenicity shapes the neoepitope landscape in human tumors. Nat. Genet. 55, 221–231 (2023).
Kim, J. Y., Bang, H., Noh, S. J. & Choi, J. K. DeepNeo: a webserver for predicting immunogenic neoantigens. Nucleic Acids Res. 51, W134–W140 (2023).
Wang, G. et al. TLimmuno2: predicting MHC class II antigen immunogenicity through transfer learning. Brief. Bioinform. 24, bbad116 (2023).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Oliveira, G. & Wu, C. J. Dynamics and specificities of T cells in cancer immunotherapy. Nat. Rev. Cancer 23, 295–316 (2023).
Xu, H. et al. ImmuneApp for HLA-I epitope prediction and immunopeptidome analysis. Nat. Commun. 15, 8926 (2024).
Yang, K., Halima, A. & Chan, T. A. Antigen presentation in cancer—mechanisms and clinical implications for immunotherapy. Nat. Rev. Clin. Oncol. 20, 604–623 (2023).
Gressier, E. et al. CD4+T cell calibration of antigen-presenting cells optimizes antiviral CD8+T cell immunity. Nat. Immunol. 24, 979–990 (2023).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2019).
Thomsen, M. C. & Nielsen, M. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 40, W281–287 (2012).
Tadros, D. M., Eggenschwiler, S., Racle, J. & Gfeller, D. The MHC Motif Atlas: a database of MHC binding specificities and ligands. Nucleic Acids Res. 51, D428–D437 (2023).
Heitmann, J. S. et al. A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity. Nature 601, 617–622 (2022).
Speiser, D. E., Chijioke, O., Schaeuble, K. & Münz, C. CD4+T cells in cancer. Nat. Cancer 4, 317–329 (2023).
Bawden, E. G. et al. CD4+T cell immunity against cutaneous melanoma encompasses multifaceted MHC II–dependent responses. Sci. Immunol. 9, eadi9517 (2024).
Oliveira, G. et al. Landscape of helper and regulatory antitumour CD4+T cells in melanoma. Nature 605, 532–538 (2022).
Chen, Y. et al. Structural definition of HLA class II-presented SARS-CoV-2 epitopes reveals a mechanism to escape pre-existing CD4+T cell immunity. Cell Rep. 42, 112827 (2023).
Ruterbusch, M., Pruner, K. B., Shehata, L. & Pepper, M. In vivo CD4+T cell differentiation and function: revisiting the Th1/Th2 paradigm. Annu. Rev. Immunol. 38, 705–725 (2020).
Künzli, M. & Masopust, D. CD4+T cell memory. Nat. Immunol. 24, 903–914 (2023).
Tarannum, M. et al. Engineering innate immune cells for cancer immunotherapy. Nat. Biotechnol. 43, 516–533 (2025).
Frank, F. et al. Deep mutational scanning identifies SARS-CoV-2 nucleocapsid escape mutations of currently available rapid antigen tests. Cell 185, 3603–3616. e3613 (2022).
Lin, V. et al. TCR3d 2.0: expanding the T cell receptor structure database with new structures, tools and interactions. Nucleic Acids Res. 53, D604–D608 (2025).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Ellis, G. I., Sheppard, N. C. & Riley, J. L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 22, 427–447 (2021).
Oh, D. Y. & Fong, L. Cytotoxic CD4+T cells in cancer: expanding the immune effector toolbox. Immunity 54, 2701–2711 (2021).
Jensen, K. K. et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406 (2018).
Nielsen, M., Lundegaard, C. & Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinf. 8, 238 (2007).
Vita, R. et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Lata, S., Bhasin, M. & Raghava, G. P. MHCBN 4.0: a database of MHC/TAP binding peptides and T-cell epitopes. BMC Res. Notes 2, 61 (2009).
Burley, S. K. et al. RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 51, D488–D508 (2023).
Deng, J. et al. IEPAPI: a method for immune epitope prediction by incorporating antigen presentation and immunogenicity. Brief. Bioinform. 24, bbad171 (2023).
O’Donnell, T. J., Rubinsteyn, A. & Laserson, U. MHCflurry 2.0: improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 11, 42–48. e47 (2020).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Chen, R. J. et al. Whole slide images are 2D point clouds: context-aware survival prediction using patch-based graph convolutional networks. In Proc. Medical Image Computing and Computer Assisted Intervention 339–349 (Springer, 2021).
Ilse, M., Tomczak, J. & Welling, M. Attention-based deep multiple instance learning. In Proc. International Conference on Machine Learning 2127–2136 (PMLR, 2018).
Gal, Y. & Ghahramani, Z. Dropout as a Bayesian approximation: representing model uncertainty in deep learning. In Proc. International Conference on Machine Learning 1050–1059 (PMLR, 2016).
Barker, D. J. et al. The IPD-IMGT/HLA Database. Nucleic Acids Res. 51, D1053–D1060 (2022).
You, R., Qu, W., Mamitsuka, H. & Zhu, S. DeepMHCII: a novel binding core-aware deep interaction model for accurate MHC-II peptide binding affinity prediction. Bioinformatics 38, i220–i228 (2022).
Schroff, F., Kalenichenko, D. & Philbin, J. Facenet: a unified embedding for face recognition and clustering. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 815–823 (IEEE, 2015).
Andreatta, M., Alvarez, B. & Nielsen, M. GibbsCluster: unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 45, W458–W463 (2017).
Shen, L.-C. shenlongchen/ImmuScope: ImmuScope v0.1.0. Zenodo https://doi.org/10.5281/zenodo.14810445 (2025).
Obermair, F.-J. et al. High-resolution profiling of MHC II peptide presentation capacity reveals SARS-CoV-2 CD4 T cell targets and mechanisms of immune escape. Sci. Adv. 8, eabl5394 (2022).
Tye, E. X. et al. Mutations in SARS-CoV-2 spike protein impair epitope-specific CD4+T cell recognition. Nat. Immunol. 23, 1726–1734 (2022).
De Castro, E. et al. ViralZone 2024 provides higher-resolution images and advanced virus-specific resources. Nucleic Acids Res. 52, D817–D821 (2024).
Shen, L.-C. ImmuScope: ImmuScope v1 CodeOcean https://codeocean.com/capsule/4332973/tree/v1 (2025).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant nos. 62372234 and 62072243 to D.-J.Y.) and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX23_0490 to L.-C.S.). Funding was also provided by the National Health and Medical Research Council of Australia (grant nos. APP1127948, APP1144652 and APP2036864 to J.S.) and by the Major and Seed Inter-Disciplinary Research projects from Monash University (to J.S.).
Funding
Open access funding provided by Monash University.
Author information
Authors and Affiliations
Contributions
L.-C.S., Y.Z., D.-J.Y. and J.S. conceived the concept. L.-C.S. and Z.W. designed the computational architecture. L.-C.S. conducted the computational experiments and developed the downstream tasks. L.-C.S. and Y.Z. performed the bioinformatics analyses. L.-C.S. prepared the figures and wrote the paper. D.R.L., Y.L., J.T. and J.R. provided guidance on data analysis. D.-J.Y. and J.S. supervised the project. All authors contributed ideas to the work and participated in editing and revising the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Leng Han and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 CD4+ T cell epitope prediction by ImmuScope.
a, Bar plots of AUCs at different HLA loci (HLA-DP, HLA-DQ and HLA-DR), the points represent the average AUC of the corresponding MHC molecule. b, Bar plot of AUCs on the CD4+ epitope benchmark. In a, b, the bars represent the mean AUCs by 1,000 bootstrap iterations, the error bars indicate the 95% CIs, and each data point represents the performance of the corresponding HLA loci. c, Comparison of AUC performance between ImmuScope and other methods on the CD4+ epitope benchmark across different peptide lengths. Shaded bands around the lines represent 95% CIs, estimated from 1,000 bootstrap iterations and scaled by a factor of 0.2 for visual clarity.
Extended Data Fig. 2 Ablation analysis of ImmuScope-EL modules.
a, AUC0.1 performance of SA, SA + MA-MIL, and ImmuScope-EL. The P values were calculated by the two-sided Wilcoxon signed rank test (n = 58): SA vs. SA + MA-MIL (\(P=1.3\times {10}^{-10}\)) and SA + MA-MIL vs. ImmuScope-EL (\(P=1.7\times {10}^{-8}\)). Box center line, median; box limits, upper and lower quartiles; whiskers, 1.5×interquartile range; ****P < 0.0001. Each data point represents the performance of the corresponding MHC allele. b, AUPR performance of SA, SA + MA-MIL, and ImmuScope-EL, calculated per MHC-II allele, only including alleles with at least 25 positive samples and a minimum of 30 total samples. c, AUC0.1 performance of the ImmuScope-EL model for different MHC alleles. The bars represent the mean performance values by 1,000 bootstrap iterations, and the error bars indicate the 95% CIs. Each data point represents the performance of the corresponding MHC allele. d, UMAP visualization of instance embeddings from the SA and ImmuScope-EL models on the BoLA-DRB3 subset.
Extended Data Fig. 3 Motif deconvolution of ImmuScope on multi-allelic data.
a, b, Motif deconvolution logo on Racle__RA957 (a) and Racle__3830_NJF_DQP (b) datasets. c, Motif deconvolution peptide counts on the heterozygous subsets.
Extended Data Fig. 4 Benchmarking Immunogenicity prediction and neoantigen analysis.
a, b, Precision-recall curves (a) and bar plots of AUPRs (b) of the candidate methods on the immunogenicity benchmark. The bars represent the mean AUPRs by 1,000 bootstrap iterations, and the error bars indicate the 95% CIs. The gray dashed lines denote the results of random predictions. c, AUPR performance comparison between HLAIImaster and ImmuScope-IM on different MHC alleles. d, Predictive analysis of HLA restriction for melanoma neoantigens. The x-axis corresponds to different neoantigens, and the y-axis represents different HLA alleles. Each position represents the antigen presentation probability of the corresponding antigen and allele predicted by ImmuScope-EL. The upper left matrix corresponds to Pt-C, and the lower right matrix corresponds to Pt-D. The color depth corresponds to the antigen presentation probability. e, Predicted cores and alignment scores for melanoma neoantigen-HLA binding with ImmuScope-EL. Purple represents high alignment scores.
Extended Data Fig. 5 SARS-CoV-2 epitope recognition and HLA binding position prediction.
a, Summary of maximal responses to all peptides (n = 29) across tested donors (n = 8), grouped by DR status. Immune responses were quantified as spot-forming cells (SFCs) per 10,000 PBMCs and plotted on a \({\log }_{10}(\frac{{sfcs}}{\mathrm{10,000}}+1)\) scale. Each point represents the maximal response of a donor to a peptide, based on duplicate ELISpot wells. The dashed line at 1.415 notes the cutoff for donor-peptide responses, representing 25 sfcs/10,000 cells. The response rate is expressed as a percentage. An inset shows P value was calculated using a two-sided Fisher’s exact test, to compare the DR1+/DR4+ statuses (groups) with the positive peptide responses. b, Summary of DR1+/DR4+ status predictions by ImmuScope-IM for all peptides (n = 29). The P value was calculated using two-sided Wilcoxon signed rank test to compare the peptide responses associated with DR1+/DR4+ status. c, d, e, f, Binding positions and alignment scores on HLA-DR1-S511-530 (PDB: 8CMC, c), HLA-DR1-M176-190 (PDB: 8CME, d), HLA-DR1-nsp31350-1364 (PDB: 8CMF, e) and HLA-DR1-nsp146420-6434 (PDB: 8CMG, f), respectively.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–12, Tables 1–8 and Notes 1–8.
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, LC., Zhang, Y., Wang, Z. et al. Self-iterative multiple-instance learning enables the prediction of CD4+ T cell immunogenic epitopes. Nat Mach Intell 7, 1250–1265 (2025). https://doi.org/10.1038/s42256-025-01073-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-01073-z
This article is cited by
-
Mapping T helper cell targets with deep learning
Nature Machine Intelligence (2025)
