
Abstract
Background
Despite significant advancements in cancer immunotherapy, survival outcomes remain poor in patients with pancreatic ductal adenocarcinoma.
Methods
In this early phase II clinical trial, we evaluated the combined effects of immunotherapy with dual immune checkpoint inhibitors durvalumab and tremelimumab, local tumor ablation with microwave energy, and gemcitabine on progression-free survival in twelve patients with non-metastatic unresectable locally advanced pancreatic adenocarcinoma. Single-cell transcriptomics and T cell receptor profiling were used to characterize the tumor microenvironment and peripheral blood immune cell repertoire.
Results
Here we show that from these twelve patients (median progression-free survival = 8.9 months, 95% confidence interval 3.2–18.4), eight patients received the combination therapy (median progression-free survival of 11.2 months, 95% confidence interval 4.0–18.4). One of these eight patients experiences grade 5 toxicity. Using single-cell transcriptomics and T cell receptor profiling, we characterize the tumor microenvironment and peripheral blood immune cell repertoire of six patients, of which three patients contain paired samples before and after the start of immunotherapy. We find substantial overlap in T cell receptors and CD8-Temra cells in the tumor microenvironment and in peripheral blood. Integration of the single-cell dataset with an independent bulk transcriptome cohort reveals that a high CD8-Temra gene signature is associated with improved overall survival. While there are interesting trends, T cell receptor-related metrics do not show statistically significant correlations with progression-free survival in this dataset.
Conclusions
These findings suggest that CD8-Temra cells may serve as potential biomarkers and therapeutic targets for immunotherapy efficacy in pancreatic ductal adenocarcinoma, pending validation in larger cohorts. We hypothesize that local tumor ablation may enhance tumor immunogenicity and systemic anti-tumor responses, supporting their integration into future treatment strategies. Future studies with larger cohorts are needed to validate these findings and optimize treatment protocols for wider clinical applicability.
Plain Language Summary
Pancreatic cancer is one of the deadliest forms of cancer, and current treatments have only limited success. This study aimed to explore whether combining immunotherapy with local tumor ablation and chemotherapy could improve outcomes for patients with advanced pancreatic cancer that cannot be surgically removed. Local tumor ablation is a treatment that destroy cancer cells inside the body by applying heat without surgery. We treated twelve patients and studied how their immune systems responded, using advanced techniques to analyze cancer and blood samples. We found that certain immune cells, called CD8-Temra cells, were linked to better survival, suggesting they could help guide future treatments. Our findings suggest that combining local tumor ablation with immunotherapy may improve the body’s ability to fight cancer, but larger studies are needed to confirm these results.
Similar content being viewed by others


Introduction
Pancreatic ductal adenocarcinoma (PDAC) is characterized by an extremely poor prognosis due to its aggressive biology, late-stage diagnosis, and limited effective treatment options. By the end of this decade, PDAC is projected to be the second most frequent cancer-related cause of death worldwide1. It is one of the most aggressive malignancies with an overall 5-year survival rate of only 9%2. Most patients present with incurable metastatic disease and/or loco-regional invasion of the surrounding major blood vessels. Patients with early-stage localized PDAC are potential candidates for surgical therapy, which, often in combination with chemo(radio)therapy, is the only chance for cure3,4. In the absence of metastases, PDAC with vascular involvement—classified as either borderline resectable or locally advanced (LAPC) unresectable disease—can be treated with combination modalities aimed at converting the tumor to a potentially resectable state. A third of patients present with LAPC without detectable metastases. As these patients might become potentially resectable after effective systemic treatment, there is an increasing focus on improving outcomes with combination treatment modalities. For patients with LAPC, treatment guidelines propose systemic chemotherapy with FOLFIRINOX or gemcitabine in combination with nab-paclitaxel, depending on the patient’s performance status5.
Over the last few years, significant efforts have been made to enhance the survival rate of patients with unresectable non-metastatic LAPC. One emerging approach is local thermal ablation (LTA), which focuses on local tumor control and modulation of the immune response. Although this approach has been successful in treating patients with small liver or renal malignancies6,7, there is only limited and low-level evidence regarding LTA of LAPC. Despite not being the standard treatment option, radiofrequency, microwave, or electroporation energy has also been used as an LTA technique to treat LAPC in carefully selected patients in small studies. Although these studies suggest a survival benefit compared to standard chemotherapy, no randomized trials exist, and high-level evidence is lacking8,9. Moreover, the challenging nature of LTA of LAPC, along with concerns of potential damage to surrounding structures, such as the bile duct, duodenum, and major blood vessels or collateral veins, has limited its use in most medical centers. On the other hand, LTA of LAPC seems promising due to its potential to generate tumor-specific antigens and its ability to prime the immune system to better recognize and kill cancer cells, particularly when combined with immunotherapy.
Monoclonal antibodies targeting immune checkpoint molecules, such as programmed death ligand 1 (PD-L1), death-1 (PD-1), and/or cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), are well-established treatments for several malignancies10. Clinical trials have shown immune checkpoint inhibitors (ICI) to be more effective in cancers with high microsatellite instability (MSI-H) or deficient mismatch repair (dMMR). In PDAC, though, which is MMR-proficient or microsatellite stable (MSS), the response to ICI monotherapy remains limited. Most of these trials were performed in a metastatic setting11,12,13,14,15, and the highly desmoplastic and immunosuppressive tumor microenvironment (TME) was deemed responsible for these poor results. However, the molecular mechanisms underlying the lack of response to ICI treatment remain unclear and could pave the way for novel multi-modality approaches that boost immunogenicity in PDAC.
In this study, we evaluate the combination of dual ICI with durvalumab and tremelimumab, LTA using minimally invasive surgical microwave ablation (MIS-MWA), and gemcitabine in patients with non-metastatic unresectable LAPC. We show that this treatment is feasible and results in a median progression-free survival (PFS) of 8.9 months, with patients receiving the full regimen reaching 11.2 months. We find that CD8-Temra cells and shared T-cell receptor clonotypes between the tumor and blood are associated with improved survival, suggesting that these immune features may serve as biomarkers of immunotherapy response. These findings highlight the potential of combining local tumor ablation with systemic immunotherapy to enhance anti-tumor immunity in pancreatic cancer.
Methods
Study design and patient selection
In total, 20 patients were planned to be included in this mono-center, early phase-2 non-randomized, open-label, prospective trial at the University Hospitals Leuven, Belgium, which is a pancreatic cancer referral center. This sample size calculation was based on the primary endpoint of median PFS. Historical data report a median PFS of approximately 6 months in this patient population (18). Assuming an improvement to 12 months in our cohort and using a one-sided log-rank test with a significance level (alpha) of 0.05 and a statistical power (1-Beta) of 80%, a minimum of 17 patients is required to detect this difference. This calculation was performed using the Schoenfeld approximation. To account for potential drop-out or loss to follow-up, we planned to include 20 patients in total.
This study consisted of two parts. The initial six patients were evaluated in a pilot study as a safety run-in. The pilot study was considered safe if no dose-limiting toxicity (DLT) occurred in more than two out of six patients for the DLT period, which was defined as the time from administration of the first dose of durvalumab and tremelimumab until the planned administration of the second dose. The decision to stop or continue the trial after the initial six patients or after the run-in phase was made by the multidisciplinary team of oncologists, radiologists, and surgeons.
Therapy-naive patients of 18 years or older with histologically proven unresectable non-metastatic LAPC according to the NCCN Guidelines Version 2.2017 were screened for inclusion between April 2020 and November 2022 (Supplementary Table 1). Final patient eligibility was determined intra-operatively, as a substantial number of patients could be excluded due to unexpected intra-abdominal metastases discovered during laparoscopic surgical exploration. Treatment of the first patient started on 15 May 2020. Treatment of the last patient ended on 28 September 2023. Recruitment was stopped on 21 October 2022. The survival data cutoff date was 29 May 2024.
The study was conducted in compliance with the principles of the Declaration of Helsinki (most recent version) and the principles of Good Clinical Practice. Before the start of the trial, ethical and regulatory approvals were obtained from the Clinical Trial Center of the University Hospitals KU Leuven (UZ S61508). This trial was registered as NCT04156087 on clinicaltrials.gov on 6 November 2019 (https://clinicaltrials.gov/study/NCT04156087) and as EudraCT 2018-002852-34 on the European database for clinical trials.
Written informed consent was obtained from all included patients prior to study start.
Treatment schedule
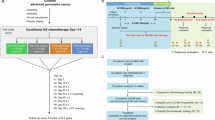
The study scheme is depicted in Fig. 1a. Following screening and baseline endoscopic ultrasound (EUS)-guided fine needle biopsy, the first dose of ICI, comprising a fixed dose of 1500 mg of durvalumab and a fixed dose of 75 mg of tremelimumab, was administered 2 weeks before MIS-MWA. The second dose was administered 2 weeks after the MIS-MWA procedure. The 3rd dose and 4th dose were given 6 and 10 weeks after MIS-MWA, respectively. After week 10, durvalumab in monotherapy was administered every 4 weeks. Gemcitabine (1000 mg/m²) was started 6 weeks after MIS-MWA and was administered once a week for 3 weeks, followed by a week of rest, in combination with continued ICI. This regimen was continued until disease progression was reported.

a Study scheme. b CONSORT diagram, D Durvalumab, T tremelimumab, G gemcitabine, MIS-MWA minimally invasive surgical microwave ablation, pt patient.
The MIS-MWA procedure started with a staging laparoscopy. Any macroscopic disease suspected of metastasis was examined with frozen section histopathology. If metastasis was confirmed, the procedure was ended, and patients were treated according to the latest European Society of Medical Oncology (ESMO) Guidelines on metastatic PDAC. If no metastases were detected, the procedure was continued, and surgical dissection was performed to gain access to the pancreas. Prior to ablation, an ultrasound-guided biopsy using a 14-gauge Tru-Cut needle was taken from the LAPC tumor primary. Pancreatic head tumors were freed from the retroperitoneum to allow cold-water cooling of the duodenum, the peri-ampullary region, including the bile duct, vena cava inferior, and the abdominal aorta. Cold-water cooling irrigation was installed in the duodenal lumen via a nasogastric tube and around the peri-duodenal/peri-tumoral region via direct irrigation with a surgical suction/irrigation device. The MWA probe was positioned under laparoscopic ultrasound guidance, and ablation was performed using the Angiodynamics Solero MWA system (Angio Dynamics, New York, USA). One low-suction drain was placed in the vicinity of the surgical ablation field to allow assessment for pancreatic fistula. After MIS-MWA, patients were submitted to an enhanced recovery after surgery (ERAS) program.
Data and sample collection
Study data were collected and managed using the secure, web-based software platform REDCap (Research Electronic Data Capture)16.
Two biopsies of the LAPC tumor primary were taken at two distinct time points: one at baseline during endoscopy (pre-treatment) and the other immediately prior to MIS-MWA during exploratory laparoscopy (on-treatment). Biopsies were only performed if technically feasible and deemed safe. If the initial samples were of low quality, repeat biopsies were not conducted to avoid unjustifiable complications such as post-puncture pancreatitis. EUS-guided tissue samples were collected using an 18-gauge core needle. Laparoscopic biopsies were obtained using a 14-gauge trucut needle under direct guidance of a laparoscopic ultrasound probe, which was positioned in direct contact with the pancreatic surface. The collected tissue samples from the LAPC primary were utilized for pathological assessment, including PD-L1 status, single-cell transcriptome (scRNA-seq), and T-cell receptor sequencing (scTCR-seq).
At each of the following time points, blood samples were collected for peripheral blood mononuclear cell (PBMC) isolation: at baseline before treatment (time point 1), 2 weeks after the first infusion of immunotherapy, before MIS-MWA (time point 2), before the second immunotherapy infusion, 2 weeks after MIS-MWA (time point 3), and at tumor progression or at the end of treatment for any other reason (time point 4).
Clinical assessments
Primary endpoint
We hypothesized that the combination of dual ICI, MIS-MWA, and gemcitabine would improve median PFS from 6 (historical ref. 17) to 12 months in patients with non-metastatic unresectable LAPC, based on synergistic effects between local ablation and systemic immunotherapy.PFS time was defined as the duration from the start of treatment until disease progression or death from any cause. To distinguish true progression of the LAPC tumor primary from pseudo-progression due to ICI treatment, disease progression was defined by iRECIST criteria version 1.118. Patients were classified as long-term survivors (LTS) or short-term survivors (STS) if their PFS time was higher or lower than the median PFS time, respectively.
Target tumor activity was assessed on a contrast-enhanced CT scan of the abdomen or MRI measurements in 3 dimensions. Oncological evaluations were performed with contrast-enhanced CT scans or MRIs of the abdomen and thorax every 2 months as per standard of care, for a maximum of 2 years after the study drug was last administered.
Survival estimates were calculated using the Kaplan-Meier method (IBM SPSS Statistics version 29 0.2.0.).
Secondary endpoints
The toxicity of ICIs and gemcitabine was coded and graded based on the NCI CTCAE v. 5.0 and presented descriptively. The number and types of postoperative complications were registered. The therapy-oriented severity grading system (TOSGS)19 of complications was used, and complications were allocated to either surgical or non-surgical site complications.
Tertiary endpoint
Overall survival (OS) time was assessed using the Kaplan–Meier method.
Single-cell analyses
Single-cell RNA and T-cell receptor (TCR) sequencing of tumor samples
Tumor tissue biopsies were obtained from six patients at two different time points: before the start of immunotherapy (referred to as “pre-treatment”) and during immunotherapy at the time of surgery (designated as “on-treatment”). Unfortunately, for some patients, no high-quality pre-treatment (Pt 1) or on-treatment (Pt 7, Pt 12) samples could be obtained.
Tumor biopsies were dissociated20, and cells were subsequently sorted according to their CD45 status. This resulted in a total of 15 available single-cell samples, of which 8 were pre-treatment samples (4 CD45− and 4 CD45+), and 7 were on-treatment samples (3 CD45− and 4 CD45+). Single-cell RNA (scRNA) and single-cell TCR (scTCR) sequencing libraries were subsequently generated using the Chromium Single Cell 5’ V(D)J library kit (10x Genomics, Pleasanton, CA, USA) and sequenced on an Illumina NovaSeq 6000 system.
scRNAseq clustering analysis leading to cell (sub)types
All single cells were mapped with Cell Ranger v5.0.0 (10x Genomics) using reference genome GRCh38 (version 2020 A). To filter out ambient RNA, CellBender21 was used with default settings, except for the “total-droplets-included” parameter, which was adjusted to 20,000 following an inspection of the individual sample UMI curves. After this correction, the samples were processed using Seurat version 422, retaining only cells that express >200 and <6000 genes, have at least 400 unique molecular identifiers (UMIs), and have a mitochondrial content below 25%. The presence of doublet cells was assessed using DoubletFinder version 2.0.323. The data were then normalized, highly variable features were calculated, counts were scaled, and a principal component analysis (PCA) was performed for linear dimensional reduction. Regression was done for UMI counts, percentage of mitochondrial genes, and cell cycle (S and G2M scores were calculated using the CellCycleScoring function in Seurat). Twenty principal components (PCs) were used for subsequent clustering and Uniform Manifold Approximation and Projection (UMAP) for dimension reduction. The resulting clusters were annotated as distinct cell types based on the expression of marker genes, as described previously24. This resulted in a total of 50,748 cells for further downstream analysis. A similar approach was used for clustering the major immune cell types (natural killer and T cells, n = 21 538; B cells, n = 3794; and myeloid cells, n = 14,280, including macrophages, monocytes, and dendritic cells) into different cell subtypes, except Harmony (version 1.0.1) was applied with default settings to account for sample-specific variation25. For the natural killer and T-cell subclusters, cells were initially filtered using the CD45 status and regressed using the number of UMI counts, the percentage of mitochondrial genes, the cell cycle scores (including S score, G2M score, interferon score, and stress score) and sample identity, followed by sample-specific correction by Harmony. Myeloid stages were similarly filtered by removing low-quality cells before Harmony was used. For B cells, the immunoglobulin VDJ genes were removed from the variable features, as this can drive sample-specific clustering. These and subsequent downstream analyses were performed in R (version 4.2.2).
scRNAseq and scTCRseq downstream response analysis
To assess response to immunotherapy, the single-cell dataset was restricted to the CD45+ fraction of patient samples, resulting in nine samples (n = 21,141 cells). The samples were stratified according to clinical response, whereby patients with a PFS time above the median PFS time of 8.9 months were considered LTS and patients with a PFS time below the median as short-term survivors (STS). The pre-treatment condition consisted of 3 LTS samples (Pt 9, 12, and 16) and 2 STS samples (Pt 7 and 14). The on-treatment condition included 2 LTS samples (Pt 9 and 16) and 2 STS samples (Pt 1 and 14). Three patients (Pt 9, 14, and 16) had paired data, i.e., both pre-treatment and on-treatment samples were available. No high-quality pre-treatment biopsy was obtained for Pt 1, while no on-treatment samples were available for Pt 7 and 12.
Differential gene expression analysis related to clinical response was performed with the FindAllMarkers function in Seurat using the default Wilcoxon rank sum test settings.
To assess differences in the relative abundance of cell types between LTS and STS, cell fractions were calculated in the CD45+ restricted single-cell dataset, for both pre- and on-treatment conditions. Boxplots were plotted using ggplot2, version 326. To improve clarity and avoid overlap, data points were plotted on top of each boxplot with slight random variation along the horizontal axis. The upper and lower edges of the boxplots represent the first and third quartiles, respectively, while the whiskers extend to the nearest data points within 1.5 times the interquartile range. Significant differences in cell fractions based on responder status were evaluated using Mann-Whitney testing. Spearman’s correlation coefficients between cell type abundances in pre-treatment and on-treatment conditions were computed using the corrplot package in R27.
Samples with available responder status were used for scTCRseq analysis. In total, 9 pre- and on-treatment samples from 6 patients (Pt 1, 7, 9, 12, 14, and 16) were available. TCR clonotypes were defined based on the complementary-determining regions 3 (CDR3) nucleotide sequences. T-cell expansion was assessed by analyzing TCR sequences both pre- and on-treatment for the three patients with paired data. A clonotype was considered expanded if either the frequency (i.e., the number of cells sharing the same TCR clonotype) or proportion (i.e., frequency normalized for the number of cells with TCR detected in a sample) increased upon treatment and had an on-treatment frequency > 2 or > 5 [1].
Correlation with patient PFS time was assessed using Spearman’s correlation analysis.
Single-cell RNA and T-cell receptor (TCR) sequencing of PMBC samples
Pooled blood samples from 12 unique patients (Pt 1, 3, 6, 7, 8, 9, 10, 11, 12, 14, 15 and 16) were processed using 10x Genomics sequencing. Only cells with at least 200 and fewer than 7000 genes, at least 400 UMIs, and less than 25% mitochondrial content were retained. Subsequently, Souporcell was applied to trace the cells back to each original patient sample28. Barcodes with unknown origin and doublets were excluded. The data was normalized, variable features were generated, scaling was performed, and PCA and UMAP representations were generated. Sample-specific variation was adjusted using Harmony (version 0.1.1). A total of 156,782 cells were obtained. The clusters were then annotated using known marker genes and SingleR version 1.10.029 using immune cell expression datasets, including the Database of Immune Cell Expression30, the Monaco Immune database31, and the Novershtern Hematopoietic Dataset32. This pipeline was subsequently repeated for the NK and T cell cluster (n = 62,565 cells) to allow for further clustering of distinct NK and T cell subtypes.
Validation of single-cell insights using deconvolution of bulk RNA sequence data
To validate the results from the single-cell analysis, we utilized publicly available bulk RNA sequence data from a phase 2 colorectal cancer (CRC) and PDAC clinical trial with immunotherapy response data (NCT03104439)33. This dataset comprises samples from metastatic PDAC patients treated with a combined regimen of radiation therapy and immunotherapy using ipilimumab and nivolumab. Out of the 25 PDAC cases in the clinical trial, 10 pre-treatment bulk samples were available. DESeq2 normalized counts for these samples were downloaded from the NCBI website (GSE179351). We then applied Scissor version 2.0.0 in R to identify clinically relevant subpopulations within the single-cell T cell data, based on the expression profiles from bulk data, which were significantly associated with the PFS of the bulk data patients34. Boxplots of the relative abundance of cell types were constructed as described above.
We then focused on the gene expression of CD8-Temra marker genes identified in our study, applied to the bulk dataset. We first performed a differential gene expression analysis by comparing CD8-Temra single cells with the other T cell subtypes. The derived gene list was ranked by the adjusted p value, yielding a list of the top 10 genes. The complete CD8-Temra signature included the following genes: “PLEK”, “GNLY”, “CX3CR1”, “FGFBP2”, “CTSW”, “PRSS23”, “GZMH”, “GZMB”, “ADGRG1” and “S1PR5”. Next, the sample enrichment score of each bulk sample was calculated using the ssGEA method within the GSVA package (version 1.46.0) in R. The bulk samples were categorized into high and low CD8-Temra signature groups based on the median enrichment score. Kaplan-Meier curves for OS and PFS times were generated, stratifying patients by high or low signature scores.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Patient disposition and baseline characteristics
In total, 16 patients were screened for eligibility (Fig. 1b). Four of these patients did not meet the eligibility criteria (Pt 2, 4, 5, and 13) due to metastatic disease detected on additionally performed MRI or laparoscopy. The remaining 12 patients, whose characteristics are shown in Supplementary Table 2, received at least one dose of durvalumab and tremelimumab and were included in the intention-to-treat analysis. Four patients were women, and 8 patients were men, with a median age of 67 (range 34-81). Four of the 12 patients were found to have metastases during the staging laparoscopy before MIS-MWA (pt 7, 8, 10, 14). Median follow-up time was 15.4 months (95% Confidence Interval [CI] 3.9–23.6).
Efficacy
The pilot phase of the study was considered safe. However, due to slow patient recruitment and concerns about treatment efficacy, the study was prematurely terminated after enrolling 12 patients instead of the originally planned 20, thus limiting the statistical power to definitively evaluate the primary endpoint. Indeed, the median PFS time of the eligible 12 patients was 8.9 months (95% CI 3.2–18.4), and the median OS time was 15.4 months (95% CI 3.9–23.6) (Fig. 2a, b). The slow recruitment was believed to be primarily due to the COVID-19 pandemic, which resulted in reduced patient willingness to participate and fewer referrals from other centers. On the other hand, patients who underwent MIS-MWA and continued with the per-protocol treatment (n = 8) had a median PFS time of 11.2 months (95% CI 4.0-18.5) and an OS time of 17.3 months (95% CI 9.2-23.6).

a Progression-free survival. b Overall Survival PFS, progression-free survival.
Safety
Out of 12 patients included in the intention-to-treat analysis, 7 had at least 1 treatment-related adverse event (trAE) (Table 1). Common trAE, occurring in 2 or more patients, were nausea (3/12), fatigue (4/12), diarrhea (3/12), and disguise (2/12). Three patients had grade 3 or higher trAE. Two of these cases qualified as serious adverse events (SAE). One patient (Pt 3) developed severe, refractory immune-related colitis 3 months after the first dose of ICI, requiring hospitalization and intravenous corticosteroids, leading to permanent discontinuation of study treatment. Another patient (Pt 11) developed a postoperative pancreatic fistula and died due to hemorrhagic shock following laparoscopic direct biopsy and MWA; this was classified as a grade 5 SAE and a surgical site complication (TOSGS grade 5). One further patient (pt 16) refused to continue study treatment 6 months after inclusion. In the remaining patients, no postoperative surgery-related complications occurred. Median length of hospital stay after MIS-MWA was 2 days (range 2–17).
Tumor microenvironment
A total of 20 tumor samples were obtained from 6 patients. Low-quality samples and pre-treatment samples of patients (Pt 2, 4, 5, and 13) who did not meet the eligibility criteria to be included in the trial were discarded. Three patients (Pt 9, 14, and 16) had paired data, i.e., both pre-treatment and on-treatment samples. Ultimately, high-quality transcriptomic data of 15 tumor samples from 6 patients were analyzed, including two Pt 1 samples, two Pt 7 samples, four Pt 9 samples, one Pt 12 sample, three Pt 14 samples, and three Pt 16 samples.
Single-cell RNAseq bioinformatics analysis on these samples yielded a dataset of 50,748 single cells. Subsequent analysis involving dimensionality reduction and clustering identified, based on marker gene expression, several clusters including epithelial/cancer cells (n = 5305), T cells and NK cells (n = 21,538), myeloid cells (n = 14,280), fibroblasts (n = 4161), B-cells (n = 3794), endothelial cells (n = 1344), and mast cells (n = 328) (Fig. 3).

a UMAP depicting cell types identified in pre-treatment and on-treatment samples. b Heatmap showing the expression of marker genes for each cell type. scRNA-seq single-cell RNA sequencing, UMAP uniform manifold approximation and projection.
To improve our understanding of the cell types relevant to adaptive immune response, we subclustered the T cells and NK cells of the transcriptomic data separately and identified multiple phenotypes specific for CD4, CD8 T cells and NK cells (Fig. 4a). The most abundant clusters constitute a mixture of CD4 naive T cells and CD4 effector memory cells (CD4-Tn-Tem, CD-Tn and CD-Tem, n = 8988). In addition, a relatively large cluster of cytotoxic NK cells was found (n = 1980), characterized by the expression of typical cytotoxic markers (GZMB, PRF1). We also identified a cluster of CD4 resident memory (CD4-Trm) T cells (n = 1641), annotated using typical markers (CCL4, CD8A, and XCL1). Activated CD8 T cells (n = 554) with an exhaustion-like phenotype were found, characterized by expression of the immune-checkpoint marker PDCD1 and the effector marker IFNG. We also identified a cluster of CD45RA activated effector/memory CD8 T cells (CD8-Temra, n = 982) with a relatively high expression of marker genes GZMH, GZMB and GLNY. This cluster resembled the cytotoxic NK cluster due to the presence of elevated cytotoxic markers, but displayed a different phenotypic profile due to the expression of other markers such as CD8A and ZNF683. Finally, we annotated a cluster of regulatory CD4 T cells (CD4-Treg, n = 1033) with a relatively elevated expression of marker genes FOXP3, CTLA4, IL2RA, TNFRSF4, and DUSP4. We were unable to accurately annotate a CD4 exhausted cluster in the current dataset.

UMAP depicting NK/T cells (a), B-cells (b), and myeloid cells (c). Corresponding heatmaps are shown for NK/T cells (d), B-cells (e) and myeloid cells (f). TME tumor microenvironment, migDC migratory dendritic cell, pDC plasmacytoid dendritic cell, cDC conventional dendritic cell.
In the relatively smaller B-cell compartment, we identified three distinct clusters of memory B cells: Memory B cells expressing TCL1A (n = 70), Memory B cells expressing high levels of CD27 (n = 1082), and Memory B cells expressing low levels of CD27 (n = 99) (Fig. 4b). In addition, we annotated a cluster of B naive cells (n = 499) and plasma cells (n = 271). Finally, we subclustered the myeloid cells, constituting macrophages (n = 3597), monocytes (n = 5248), and dendritic cells (n = 963) (Fig. 4c).
From the 15 samples used for initial clustering, we selected 9 samples from the CD45+ fraction (n = 21,141 cells) for further analysis, stratifying them by treatment (pre- versus on-treatment) and clinical response (LTS versus STS). While the sample size was too small to draw definitive conclusions, we did observe proportional differences between the clinical response groups. In the pre-treatment samples (3 LTS; 2 STS), the relative abundance of NK/T cells in LTS tended to be higher compared to STS (Fig. 5a), but the difference was not statistically significant (p = 0.2). However, the abundance of NK/T cells in pre-treatment samples was strongly correlated with PFS time (Fig. 6b; Spearman r = 0.97, p = 0.0048). No other cell types in the pre-treatment samples correlated significantly with PFS.

Proportions were calculated for main cell types (a) and immune cell subtypes, including T cells (b), B cells (c) and myeloid cells (d). STS short-term survivors, LTS long-term survivors.

a Correlation for all cell types displays significance for NK/T cell abundance with PFS. b Correlation between pre-treatment NK/T cell abundance and PFS. PFS, progression-free survival.
The relative abundance of B cells and myeloid cells was comparable across all tumor samples (p > 0.44). Additionally, we explored differences in the relative cell abundance of the cell-type subclusters between LTS and STS samples, including T-cell subclusters, B cells, and myeloid cells, but found no significant differences (p > 0.2) (Fig. 5b–d).
Peripheral blood immune cells
In total, 37 blood samples were collected at various time points (TP). Five patients (Pt 7, 8, 10, 11, and 14) had blood samples taken at TP1 and TP2. Six patients (Pt 1, 3, 6, 9, 15, and 16) had samples collected at TP1, TP2, TP3, and TP4. Additionally, one patient (Pt 12) had blood samples taken at TP1, TP2, and TP4. All these samples were profiled with scRNAseq to characterize the peripheral immune system. Following bioinformatics analysis, we obtained a total of 156,782 cells, including a T cell and NK cluster of 62,565 cells. In this T cell and NK cluster, multiple subclusters, comparable to those in the tumor tissue scRNA-seq data, were annotated and utilized in the downstream analysis described below.
Intra-tumoral T-cell expansion and TME-PBMC TCR sharing
Single-cell TCRseq data from three patients with paired pre- and on-treatment samples were analyzed to identify T-cell expansion upon treatment, as described by Bassez et al. (23). We detected 4308 unique TCRs (pre-treatment: 1925 TCRs; on-treatment: 2462 TCRs) from 5752 T cells. 79 TCRs from 579 T cells were shared between pre- and on-treatment samples. The number of expanded clonotypes was assessed per patient using three definitions (see Methods). Although the dataset is limited (n = 3) and does not allow for definitive conclusions, T-cell expansion seemed to be more pronounced in LTS (Pt 9 and 16), whereas almost no expansion was observed in the STS (Pt 14) (p = 0.67 for each TCR expansion criterium) (Fig. 7a). Additionally, the number of expanded TCRs showed a potential association with PFS, regardless of the definition used to define an expanded clonotype (see Methods, Fig. 7b). However, statistical analysis did not reveal a significant difference in the current dataset (Spearman r = 1, p = 0.33).

a Absolute number of expanded TCRs, derived using multiple criteria. nExp1: number of TCR clonotypes with an increase in frequency upon treatment and frequency on-treatment >2; nExp2: number of TCR clonotypes with an increase in proportion upon treatment and frequency on-treatment >2; nExp3: number of TCR clonotypes with an increase in frequency upon treatment and frequency on-treatment >5. b Spearman correlation analysis of each TCR criterion with PFS. PFS progression-free survival, TCR T-cell receptor.
To identify specific TCRs potentially linked to a strong immune response against the tumor, we analyzed T cell subclusters in pre-treatment samples (n = 5) and assessed the number of shared TCRs within TME and peripheral blood. We found 188 shared TCRs, present in 849 intra-tumoral T cells and 1170 peripheral T cells. These shared TCRs accounted for 7.7% of TCRs in the TME and 2.6% of those detected in peripheral blood. When examining survival outcomes, LTS (n = 3) seemed to have a higher proportion of shared TCR clonotypes than STS (n = 2), although the difference was not statistically significant (p = 0.2) (Fig. 8a). Mapping these shared TCRs in the TME revealed that the dominant clonotypes were primarily localized in CD8-Temra and CD8-Tem cells (Fig. 8b), with a similar distribution in the peripheral blood T cells (Fig. 8c).

a Boxplot showing the number of shared clonotypes pre-treatment stratified for response. b UMAP of pre-treatment intra-tumoral T cells with shared TCRs colored (left panel) and stratified by response (middle and right panel). c UMAP of pre-treatment T cells identified in blood with shared TCRs colored (left panel) and stratified by response (middle and right panel). The blue arrows in (a) and (b) indicate the presence of CD8-Temra cells in LTS patients. STS short-term survivors, LTS long-term survivors, UMAP uniform manifold approximation and projection.
CD8-Temra as a predictive biomarker of immunotherapy response
We integrated the current TME single-cell dataset with a separate, independent bulk transcriptome dataset of PDAC patients (n = 10) treated with immunotherapy (NCT03104439) using Scissor34. This revealed subpopulations in the single-cell dataset correlated with patient survival in the bulk data. We identified 8557 T cells in the single-cell dataset consisting of nine samples, with 5201 cells positively and 463 cells negatively associated with PFS in the bulk transcriptome dataset. By calculating relative abundances, we found that cells associated with improved survival contained significantly more CD8-Temra and CD8-Trm cells than cells associated with reduced survival (Fig. 9a). Conversely, negatively associated cells contained significantly more CD4 Treg cells compared to positively associated cells.

a Relative abundance of T-cell subtypes in pre- and on-treatment samples of long-term and short-term survivor samples associated with bulk samples. Each dot represents the percentage of cell types of either positively or negatively associated cells. Significant differences are illustrated using the following p values: p < 0.05 (*) and p < 0.01 (**). b Kaplan–Meier plots of overall survival and progression-free survival of 10 bulk gene expression PDAC samples, stratified according to CD8-Temra signature into high expression (“High”) or low expression (“Low”). TME tumor microenvironment.
To validate our CD8-Temra marker genes, we first performed a differential gene expression calculation of CD8-Temra single cells versus other T-cell subtypes and identified 10 characteristic marker genes. We then calculated CD8-Temra signature scores based on the expression of these 10 marker genes in the above-mentioned independent bulk transcriptome dataset. The pre-treatment bulk RNA samples were stratified into two groups according to median CD8-Temra signature scores (high and low) (Fig. 9b). Median PFS time was similar in both groups (p value = 0.12). Median OS time was significantly longer in patients with a high CD8−Temra signature score compared to OS time in patients with a low CD8-Temra signature score (p value = 0.02).
Discussion
This study was designed as an exploratory, early-phase II clinical trial to assess the feasibility, safety, and potential signals of efficacy of combining local ablation with immune checkpoint blockade and chemotherapy in unresectable LAPC. The generalizability of our findings is inherently limited by the small sample size, single-center setting, and selection bias due to intraoperative exclusion of patients with occult metastases. Moreover, the exploratory nature of the translational analyses, combined with the absence of a comparator arm, precludes direct extrapolation to the broader LAPC population.
We observed a median PFS time of 11.2 months (95% CI 4.0–18.4) in patients with non-metastatic unresectable LAPC who received the combination therapy of dual ICI with durvalumab and tremelimumab, LTA, and gemcitabine. According to the current literature, the median PFS time of patients with inoperable LAPC or metastatic PDAC treated with standard systemic chemotherapy is around 6 months (18). Although these results are not directly comparable due to differences in patient populations of stages of disease, the PFS time in our study seems promising. Due to slow patient recruitment, the study was terminated prematurely, and the primary endpoint was not reached. However, it is crucial to focus on the per-protocol treatment group to evaluate the effect of the entire regimen. As unexpected metastases were found in one-third of the patients at the time of laparoscopy after receiving only one dose of durvalumab and tremelimumab, these patients were not treated with LTA and were thus excluded from the study.
Despite significant advancements in cancer immunotherapy with immune checkpoint inhibitors (ICIs), outcomes for patients with PDAC remain poor. In contrast to several other cancer types, only pembrolizumab has received FDA approval for PDAC, specifically in cases involving MSI-H tumors35. To date, several randomized trials have evaluated the efficacy of ICIs, alone or combined with chemotherapy (12–15). However, none have demonstrated significant and practice-changing results. In addition, PDAC is generally considered an immunogenically “cold” tumor, with tumor-infiltrating T cells linked to better survival outcomes and immunosuppressive myeloid cells associated with poor survival36.
Although the sample size in the present study is small, the observed survival benefit may result from the synergistic effects of the combination therapy. Several studies have demonstrated positive effects of loco-regional therapies, such as LTA, in enhancing tumor immunogenicity and reducing immune suppression in PDAC, known as the abscopal effect37,38,39. Simultaneously with the destruction of the central dense desmoplastic TME of the tumor-primary, LTA may improve drug supply to the well-vascularized remaining peripheral tumor tissue. Additionally, it provides a useful antigen source for the immune system, thereby enhancing both the intra-tumoral and systemic effectiveness of immunotherapy40.
In this trial, we found a strong association between CD8-Temra cells and the survival of patients treated with ICI. Our analysis of the T cell compartment within the TME showed a higher relative abundance of NK/T cells prior to ICI therapy, which correlated significantly with long-term survival. Additionally, the number of TCRs detected in the peripheral blood of LTSs was noticeably higher than that observed in short-term survivors. This elevated presence of circulating TCRs was associated with prolonged PFS, suggesting a potential link between T cell clonal expansion and antitumor immune responses. We also found substantial overlap in TCRs and CD8-Temra cells in the TME and peripheral blood. While previous studies have reported the presence of CD8-Temra cells in PDAC41,42, this is the first study to highlight their promising role in patients treated with immunotherapy. This link between CD8-Temra cells and patient survival was also confirmed by integrating single-cell data with pre-treatment bulk transcriptome data. Stratifying the bulk dataset using CD8-Temra marker genes from the single-cell analysis revealed that LTS had higher expression of these markers prior to treatment compared to STS. However, the current study design does not allow us to determine whether these observed associations are prognostic or predictive for ICI. Without a randomized control group receiving a conventional multi-agent chemotherapy regimen or no treatment, we cannot definitively conclude whether patients with elevated CD8-Temra cell levels or increased TCR abundance directly predict benefit from ICI therapy. Additionally, the role of LTA could be uncertain, as the study was conducted after systemic therapy and was not specifically designed or timed to assess the additional benefit of LTA. Therefore, the findings from this study should be interpreted as exploratory and descriptive for now. Further studies in larger and randomized cohorts will be needed to validate these findings and clarify the potential of elevated CD8-Temra and TCR levels as predictive biomarkers or therapeutic targets in PDAC patients.
Despite significant biological differences between PDAC and other solid tumors, we sought to explore potential parallels between our findings and other cancer types in the literature. A recent scRNAseq study on advanced HCC patients treated with atezolizumab plus bevacizumab emphasized the role of cytotoxic CD8-Temra cells in responders and high TCR overlap between tumor and blood43. These parallels suggest similarities between the pre-treatment TME of PDAC and HCC that may help predict response to immunotherapy. Notably, several CD8-Temra marker genes identified in our differential gene expression analysis—including FGFBP2, PLEK, CX3CR1, and GNLY—were observed in both studies. This contrasts with other cancers, where clinical response is commonly linked to clonally expanded CD8-Tex (exhausted T-) cells. Although the study on HCC patients did not find a higher abundance of CD8-Tex cells in responders versus non-responders, they did report a significant presence of these cells in the TME of advanced HCC cancer-primaries. In our study, the abundance of CD8-Tex cells was minimal and consistent with prior PDAC studies involving chemotherapy44.
The current study has several limitations. A key limitation of our study is the relatively small number of patients, also with respect to longitudinal analysis. While eight patients were included in the overall single-cell analysis, only three patients contributed paired pre- and on-treatment tumor samples. As such, the conclusions drawn from the treatment-associated changes are based on a limited number of cases. In addition, many patients contributed multiple samples, including tumor tissue and peripheral blood, so samples from the same patients are not fully biologically independent. While we leveraged high-resolution single-cell profiling to analyze thousands of individual cells, this approach may amplify patient-specific effects and thus the results should be interpreted with caution.
The overall sample size of the MIMIPAC trial was also too small to draw final conclusions regarding survival outcomes. Although the study was conducted at the largest pancreatic cancer reference hospital in Belgium, patient recruitment was challenging as the trial started shortly before the COVID-19 pandemic, which severely hampered patient enrollment. Additionally, 4 of the initial 16 patients (25%) did not meet the inclusion criteria, and 4 of the 12 included participants (33%) had unexpected metastases at the time of laparoscopy, thus reducing the number of on-treatment tissue and blood samples available for analysis.
Another limitation was the relatively high rate of treatment-related adverse events (trAEs) in patients receiving combination therapy. One patient died due to hemorrhagic shock secondary to a post-biopsy and post-ablation pancreatic fistula, highlighting the known risks of pancreatic fistulas, a significant complication in pancreatic surgery. It remains unclear whether the fistula was caused by the direct biopsy procedure, the ablation itself, or a combination of both. Fistula formation following pancreatic biopsies may be reduced by using a transduodenal approach, although this technique is not always technically feasible to reach the tumor. More than 50% of patients experienced trAEs, a higher rate than observed in treatment with only durvalumab and tremelimumab15, likely caused by concurrent gemcitabine use. The decision not to include neoadjuvant chemotherapy in our trial was primarily driven by safety considerations. Given the novelty of combining dual immune checkpoint inhibition with MWA, we aimed to avoid excessive toxicity potentially arising from triplet therapy, which may increase the risk of peri-procedural complications such as pancreatic fistula or delayed recovery. On the other hand, this might have contributed to the modest outcomes in the overall group. The incidence of grade 3 or higher trAEs and treatment discontinuation was consistent with previous dual ICI studies, suggesting an overall safety profile similar to prior findings. These results emphasize the need for careful patient monitoring when combining systemic chemotherapy with immunotherapies to balance efficacy with safety.
In conclusion, we assessed the combination therapy of systemic durvalumab and tremelimumab, LTA of the tumor-primary, and systemic gemcitabine in patients with non-metastatic unresectable locally advanced pancreatic adenocarcinoma. Among the eight patients treated with all intended modalities, the survival outcome was promising. Despite the small sample size and the limitations in study design, the relatively promising survival might reflect the synergistic potential of this combination therapy in overcoming the immune resistance of PDAC. Patient survival was strongly linked to the high abundance of both CD8-Temra cells and TCRs. This highlights CD8-Temra cells as potential biomarkers and therapeutic targets for immunotherapy efficacy in PDAC. Local tumor ablation may enhance tumor immunogenicity and systemic anti-cancer responses via the abscopal effect, supporting their integration into future treatment strategies. Given the small, non-randomized nature of this trial, the findings should be interpreted as hypothesis-generating and warrant further evaluation in larger, controlled studies. Therefore, future prospective trials with larger patient cohorts are needed to confirm whether the abundance and activation state of CD8⁺ TEMRA cells can serve as early biomarkers of immunotherapy response. Larger longitudinal studies that combine single-cell and bulk TCR sequencing should also track in real time the dynamics of clonotypes shared between the tumour microenvironment and peripheral blood. Finally, randomized trials comparing immunotherapy alone with immunotherapy plus LTA are warranted to determine whether LTA truly enhances therapeutic efficacy.
Data availability
The source numerical data of the figures in this manuscript are provided as a Supplementary Data file. The single-cell RNA sequencing datasets used in this study are publicly available at https://lambrechtslab.sites.vib.be/en/MIMIPAC. Any additional information is available from the corresponding author upon reasonable request.
References
Rahib, L. et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921 (2014).
Rawla, P., Sunkara, T. & Gaduputi, V. Epidemiology of pancreatic cancer: global trends, etiology, and risk factors. World J. Oncol. 10, 10–27 (2019).
Mukherjee, S. et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol. 14, 317–326 (2013).
Oettle, H. et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 310, 1473–1481 (2013).
Conroy, T. et al. ESMO Guidelines Committee. Pancreatic cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 34, 987–1002 (2023).
Gill, I. S., Aron, M., Gervais, D. A. & Jewett, M. A. Clinical practice. Small renal mass. N. Engl. J. Med. 362, 624–634 (2010).
Lencioni, R., de Baere, T., Martin, R. C., Nutting, C. W. & Narayanan, G. Image-guided ablation of malignant liver tumors: recommendations for clinical validation of novel thermal and non-thermal technologies—a western perspective. Liver Cancer 4, 208–214 (2015).
Linecker, M., Pfammatter, T., Kambakamba, P. & DeOliveira, M. L. Ablation strategies for locally advanced pancreatic cancer. Dig. Surg. 33, 351–359 (2016).
Spiliotis, J. D. et al. Radiofrequency ablation combined with palliative surgery may prolong survival of patients with advanced cancer of the pancreas. Langenbecks Arch. Surg. 392, 55–60 (2007).
Holder, A. M. et al. Defining clinically useful biomarkers of immune checkpoint inhibitors in solid tumours. Nat. Rev. Cancer 24, 498–512 (2024).
Christensen, T. D. et al. Circulating immune-related proteins associated with immune checkpoint inhibitor efficacy in patients with pancreatic ductal adenocarcinoma. ESMO Open 9, 103489 (2024).
O’Reilly, E. M. et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 5, 1431–1438 (2019).
Royal, R. E. et al. Phase 2 trial of single-agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 33, 828–833 (2010).
Chen, I. M. et al. Randomized phase II study of nivolumab with or without ipilimumab combined with stereotactic body radiotherapy for refractory metastatic pancreatic cancer (CheckPAC). J. Clin. Oncol. 40, 3180–3189 (2022).
Renouf, D. J. et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat. Commun. 13, 5020 (2022).
The REDCap consortium: building an international community of software partners. J Biomed Inform. 103208. https://doi.org/10.1016/j.jbi.2019.103208 (2019).
Park, W., Chawla, A. & O’Reilly, E. M. Pancreatic cancer: a review. JAMA 326, 851–862 (2021).
Seymour, L. et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 18, e143–e152 (2017).
Dindo, D., Demartines, N. & Clavien, P. A. Classification of surgical complications: a new proposal with evaluation in a cohort of 6336 patients and results of a survey. Ann. Surg. 240, 205–213 (2004).
Bassez, A. et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27, 820–832 (2021).
Fleming, S. J. et al. Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Nat. Methods 20, 1323–1335 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–87.e29 (2021).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–37.e4 (2019).
Franken, A. et al. CD4+ T cell activation distinguishes response to anti-PD-L1+anti-CTLA4 therapy from anti-PD-L1 monotherapy. Immunity 57, 541–58.e7 (2024).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hadley W. Ggplot2. New York, NY: Springer Science+Business Media (2016).
Wei T., Simko V. R package ‘corrplot’: Visualization of a Correlation Matrix (2024).
Heaton, H. et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods 17, 615–620 (2020).
Hausser, J. & Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 20, 247–257 (2020).
Schmiedel, B. J. et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell 175, 1701–15.e16 (2018).
Monaco, G. et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 26, 1627–40.e7 (2019).
Novershtern, N. et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144, 296–309 (2011).
Parikh, A. R. et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat. Cancer 2, 1124–1135 (2021).
Sun, D. et al. Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data. Nat. Biotechnol. 40, 527–538 (2022).
André, T. et al. KEYNOTE-177 Investigators. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N. Engl. J. Med. 383, 2207–2218 (2020).
Hilmi, M. et al. The immunological landscape in pancreatic ductal adenocarcinoma and overcoming resistance to immunotherapy. Lancet Gastroenterol. Hepatol. 8, 1129–1142 (2023).
Teng, L. S., Jin, K. T., Han, N. & Cao, J. Radiofrequency ablation, heat shock protein 70 and potential anti-tumor immunity in hepatic and pancreatic cancers: a minireview. Hepatobiliary Pancreat. Dis. Int. 9, 361–365 (2010).
Granata, V. et al. Local ablation of pancreatic tumors: state of the art and future perspectives. World J. Gastroenterol. 27, 3413–3428 (2021).
Giardino, A. et al. Immunomodulation after radiofrequency ablation of locally advanced pancreatic cancer by monitoring the immune response in 10 patients. Pancreatology 17, 962–966 (2017).
Slovak, R., Ludwig, J. M., Gettinger, S. N., Herbst, R. S. & Kim, H. S. Immuno-thermal ablations - boosting the anticancer immune response. J. Immunother. Cancer 5, 78 (2017).
Mahalingam, D. evalingam et al. Combination of pembrolizumab and pelareorep promotes anti-tumour immunity in advanced pancreatic adenocarcinoma (PDAC). Br. J. Cancer 129, 782–790 (2023).
Heiduk, M. et al. TIGIT expression delineates T-cell populations with distinct functional and prognostic impact in pancreatic cancer. Clin. Cancer Res. 29, 2638–2650.44 (2023).
Cappuyns, S. et al. PD-1- CD45RA+ effector-memory CD8 T cells and CXCL10+ macrophages are associated with response to atezolizumab plus bevacizumab in advanced hepatocellular carcinoma. Nat. Commun. 14, 7825 (2023).
Werba, G. et al. Single-cell RNA sequencing reveals the effects of chemotherapy on human pancreatic adenocarcinoma and its tumor microenvironment. Nat. Commun. 14, 797 (2023).
Acknowledgements
This research was conducted with the financial support of ASTRA-ZENECA NV/SA and Stichting tegen Kanker.
Author information
Authors and Affiliations
Contributions
H.T. and B.T. conceived and designed the study. H.T., T.V., A.B., and G.C. analyzed the data. H.T., A.B., and T.V. interpreted the results and drafted the manuscript. B.T., E.V.C., S.V.D.M., and D.L. revised the manuscript critically. D.L. (translational part) and B.T. (clinical part) supervised the data analysis, interpretation, and manuscript writing. All authors approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. ASTRA-ZENECA NV/SA and Stichting tegen Kanker were not involved in any stage of the study.
Peer review
Peer review information
Communications Medicine thanks Rebekah R. White, Ulrich Mansmann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Topal, H., Venken, T., Bassez, A. et al. Progression-free survival for unresectable non-metastatic locally advanced pancreatic cancer after surgical microwave ablation plus durvalumab and tremelimumab: phase-2 non-randomized prospective clinical trial. Commun Med 5, 475 (2025). https://doi.org/10.1038/s43856-025-01186-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-01186-x
