
Abstract
Organometallic reagents are essential in organic synthesis, with organolithium compounds being most widely used. However, as lithium becomes less abundant and increasingly expensive, organosodium compounds have emerged as promising alternatives, but their use in organic synthesis is limited by their poor solubility in organic solvents, the need for pre-activated sodium sources and the necessity for highly anhydrous conditions. Here we report a mechanochemical protocol for the direct generation of organosodium compounds from cheap and shelf-stable sodium lumps and readily available organic halides under bulk, solvent-free conditions. These reactions generate an array of organosodium compounds in minutes, without special precautions against moisture or temperature control. These nucleophiles can be used directly for one-pot nucleophilic addition reactions with electrophiles and nickel-catalysed cross-coupling reactions. Furthermore, this mechanochemical approach enables the sodiation of inert C–F bonds in organic fluorides. This method is anticipated to drive progress in sodium-based synthetic chemistry.

Similar content being viewed by others
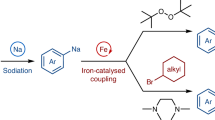

Main
Organolithium compounds have played a dominant role as carbon nucleophiles, Brønsted bases and precursors for a variety of other organometallic reagents in organic synthesis for over 100 years (Fig. 1a)1,2,3,4. Their widespread use is attributed to their relatively high stability in organic solvents, coupled with well-established methods for their preparation5,6. However, from a sustainability standpoint, there is an increasing demand for alternatives to lithium, which is becoming both less abundant and more expensive7. The growing demand for lithium-ion batteries is expected to further intensify competition for lithium resources8,9,10.

a, Comparison between organolithium compounds and organosodium compounds. b, Previous work involving mechanochemical activation of sodium metal. c, Direct mechanochemical synthesis of organosodium compounds through ball milling. E+, electrophile; SCXRD, single-crystal X-ray diffraction.
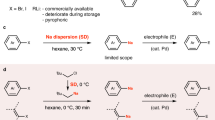
Sodium, another s-block metal, is much more earth abundant than lithium (crustal abundance 22,700 ppm versus Li abundance 18 ppm)11 and is therefore an attractive candidate for replacing lithium to explore sustainable organic synthesis (Fig. 1a)12,13,14,15,16,17,18,19,20,21,22,23. The first reports of organosodium chemistry emerged in the 1850s, pioneered by J. A. Wanklyn24, but substantial challenges exist that have prevented the widespread adoption of organosodium reagents within the field of organic synthesis25,26,27,28,29. First, the highly reactive nature of organosodium reagents makes them incompatible with many solvents, including ethers, which can react via C‒H and C‒O activation, and even aromatic solvents such as toluene and benzene29. Only alkane solvents (for example hexanes) can be considered genuinely inert towards organosodiums, but the poor solubility of organosodium reagents in hydrocarbon solvents often causes substantial problems, limiting the potential for widespread application. Second, while direct metallation of organic halides with metallic sodium is an attractive and straightforward synthetic method, it has severe drawbacks: (1) traditional solution-based methods involving direct metallation of organic halides with metallic sodium suffer from a severe side reaction, Wurtz coupling, where the newly formed organosodium (RNa) reacts with unreacted organohalide (RX) to form the homocoupling product R‒R and sodium halide (NaX)30; (2) reactions often require a large excess of sodium lumps or unstable, expensive pre-activated sodium sources such as sodium dispersion13,17. To avoid Wurtz coupling, the mainstream method to prepare organosodiums is Li–Na exchange, by reacting organolithium (RLi) with sodium tert-butoxide (NaOtBu) to generate RNa and LiOtBu in a hydrocarbon solvent (usually hexanes), driven by the poor solubility of RNa in these solvents (see above)31. This Li–Na exchange still requires Li, which eliminates the sustainability merit of organosodiums. The need for rigorously dry and deoxygenated organic solvents and relatively complex setups involving inert-gas atmospheres presents additional practical drawbacks.
Mechanochemical organic synthesis using ball-milling techniques has emerged as a new methodology to carry out organic synthesis under solvent-free conditions32,33,34,35,36,37,38,39,40,41,42,43. The advantages of this protocol include reduced solvent waste, fast reaction kinetics and operational simplicity under ambient conditions. Furthermore, recent studies have shown that vigorous mechanical agitation can activate zero-valent metals by removing the unreactive surface oxide layer and increasing the reactive surface area44,45,46,47,48,49,50,51,52,53. These mechanochemically activated, zero-valent metals readily undergo metal–surface reactions with organic halides to produce the corresponding organometallic species efficiently51. Surprisingly, by utilizing this strategy, organometallic compounds previously thought to be synthesizable only in solution have been successfully prepared under mechanochemical conditions, including Grignard reagents54,55,56,57, organozinc58,59,60, organocalcium61,62,63, organomanganese64,65, organobarium66 and organolithium reagents67. However, the direct mechanochemical synthesis of organosodium compounds has yet to be explored. Recently, mechanochemical activation of sodium lumps has been utilized for highly efficient ammonia-free Birch reduction and the preparation of a sodium anion complex (Fig. 1b)68,69. Building on these successful results, we envisioned that a mechanochemical protocol could offer a practical and efficient solution to address the challenges associated with conventional solution-based methods for the preparation and application of organosodium compounds in synthetic chemistry.
In this Article, we report a mechanochemical generation of organosodium compounds from various organic halides and commercially available, cheap and stable sodium lumps and their application to organic synthesis (Fig. 1c). This protocol can be performed under ambient conditions without the need for pre-activated sodium metal, large amounts of anhydrous organic solvents or complex synthetic procedures that require precautions against moisture and strict temperature control. Furthermore, these rapid reactions complete within minutes. This is likely to be due to the mechanical activation of sodium metal in situ. The resulting organosodium species readily react with a wide range of electrophiles in a one-pot mechanochemical process. Nickel-catalysed direct cross-coupling of organosodium compounds with aryl halides also proceeded under mechanochemical conditions. Additionally, this mechanochemical strategy was applicable to the direct sodiation of poorly soluble aromatic halides as well as organic fluorides, which are unreactive towards sodiation under conventional solution-based conditions. Single-crystal X-ray diffraction (SCXRD) analysis and NMR spectroscopy studies were successfully performed to confirm the identity of the key mechanochemically generated organosodium intermediates. Overall, the present study provides a synthetic platform centred on organosodium compounds, which has the potential to replace conventional organolithium-based synthesis with a more sustainable, cost-effective and environmentally friendly approach.
Results
Mechanochemical generation of aryl sodium compounds and subsequent reactions with electrophiles
We initially attempted the generation of organosodium species through the reaction of bromobenzene (1a) with sodium lumps using a Retsch MM400 mixer mill (5 ml stainless-steel milling jar with one stainless-steel ball; ball diameter: 10 mm). The mineral oil on the sodium lumps was removed by wiping them with a paper towel, before the lumps were cut into pieces approximately 4–5 mm in width and depth. Subsequently, the sodium-metal pieces were weighed and introduced into the jar, followed by addition of 1a under atmospheric conditions. After optimization (see Supplementary Table 1 for details), we established the following mechanochemical protocol for generating organosodium compounds (Fig. 2): ball milling of 1a (2.0 equiv.) and sodium lumps (4.4 equiv.) in the presence of n-hexane (4.4 equiv.) as a liquid additive for efficient grinding was conducted for 5 min. The jar was opened in air and N-benzylideneaniline 2a (1.0 mmol) was added to the reaction mixture. The jar was then quickly closed (without any inert gas purging) and a second phase of ball milling was conducted for 5 min, yielding the desired amine 3a in excellent yield (87%). The scope of the aromatic halides and electrophiles using this one-pot, two-step mechanochemical protocol is shown in Fig. 2. Phenylsodium could also be generated from chlorobenzene, and subsequent reaction with 2a produced 3a in 85% yield. Organosodium species were prepared from various alkylated aryl halides (1b–1f) and reacted with 2a smoothly to produce the corresponding amines (3b–3f) in moderate to excellent yields (50–91%). Biphenyl bromides (1g and 1h) were also readily metallated, and subsequent reaction with 2a afforded 3g and 3h in 53 and 61% yields, respectively. Aryl halides bearing trimethylsilyl, dimethylamino and methoxy groups were compatible with this reaction, and the corresponding amines (3i–3k) were obtained in moderate to high yields (34–73%). We tested the generation of organosodium compounds from sterically hindered aryl bromides such as mesityl bromide (1l) and 2-bromo-1,3,5-triisopropylbenzene (1m). Mesityl sodium was efficiently generated, and subsequent nucleophilic addition to 2a proceeded to give 3l in 90% yield. A more sterically hindered aryl halide 1m also underwent direct sodiation under mechanochemical conditions to afford the desired product 3m in 45% yield. N-(4-Methoxybenzylidene)aniline (2b) also reacted with the mechanochemically generated aryl sodium compound, 4-tolyl sodium, to give 3n in 66% yield.

Conditions: 1 (2.0 mmol), Na (4.4 mmol), n-hexane (4.4 mmol) and electrophile (1.0 mmol) in a stainless-steel ball-milling jar (5 ml) with a stainless-steel ball (10 mm). Ball milling (30 Hz) was carried out. Isolated yields are reported as percentages. a15 min for first step. b15 min for first step and 30 min for second step. c1 (3.0 mmol), Na (6.0 mmol) and n-hexane (6.0 mmol). dFirst step: 1 (1.0 mmol), Na (2.2 mmol) and n-hexane (2.2 mmol) for 5 min. Second step: dry ice (excess) for 30 min. eFirst step: 1 (2.5 mmol), Na (5.0 mmol) and n-hexane (5.0 mmol) for 5 min. Second step: Ar–Cl (1.0 mmol), NiCl2(dppe) (0.10 mmol). iPr, isopropyl; tBu, tert-butyl; TMS, trimethylsilyl; pin, pinacol.
In addition to imines as trapping reagents, aromatic and aliphatic aldehydes also smoothly reacted with mechanochemically generated organosodium compounds to afford the corresponding alcohols (3o–3s) in moderate to excellent yields (56–81%). Methyl benzoate could also be used as an electrophile and the desired tertiary alcohol 3t was obtained in 97% yield. Nucleophilic additions to aromatic ketones also proceeded rapidly, providing the corresponding alcohols (3u–3x) in moderate to excellent yields (42–80%). Additionally, various aromatic ketones (3y–3aa) were successfully prepared by reactions between organosodium compounds and Weinreb amides in good to excellent yields (65–85%). Morpholine amides were also reactive with the organosodium compounds, and the desired ketone 3ab and aldehyde 3ac were obtained in 53 and 67% yields, respectively. Reactions of dimethylphenyl silane (HSiMe2Ph) with the organosodium compounds bearing biphenyl and fluorene moieties provided the corresponding silylated products 3ad and 3ae in 92 and 89% yields, respectively. Mechanochemically generated phenylsodium could be trapped by dry ice, and benzoic acid 3af was obtained in 83% yield. The feasibility of mechanochemical nickel-catalysed cross-coupling reaction with organosodium compounds was also investigated70. Following catalyst optimization (see Supplementary Table 2 for details), we found that NiCl2(dppe) (dppe, 1,2-bis(diphenylphosphino)ethane) was an ideal catalyst for this reaction, and the cross-coupling between phenylsodium and 2-chlorolonapthalene gave the corresponding coupled product 3ag in 43% yield. In our substrate scope studies, side products such as homocoupled products of organic halides were not detected.
One-pot, one-step reactions for various organosodium compounds
Unfortunately, unlike aryl halides, alkyl halides proved difficult to use in one-pot, two-step reactions, and the desired products were not obtained. This was probably due to rapid decomposition of the reactive alkyl sodium intermediates formed during the metallation step. To address this limitation, we explored one-pot, one-step transformations in which the electrophile was introduced at the start of the reaction, enabling trapping of the reactive organosodium intermediate as soon as it was formed (Fig. 3). Pleasingly, we found that employing this Barbier-type approach, a reaction between 1-chlorohexane, sodium lumps and imine 2a proceeded smoothly to provide the corresponding amine 3ah in 95% yield. Shorter and branched primary alkyl chlorides could also be used for this reaction, and the desired products 3ai and 3aj were obtained in 87 and 90% yields, respectively. Reactions using secondary and tertiary alkyl chlorides produced the corresponding amines 3ak and 3al in 63 and 18% yields, respectively. Fortunately, morpholine amides were also compatible under these strongly reducing conditions, enabling nucleophilic acyl substitution with n-butyl sodium to deliver the corresponding ketones (3am–3ap) in 42–66% yield. Nucleophilic addition to cyclopropyl phenyl ketone also readily proceeded and alcohol 3aq was obtained in 70% yield. Aldehyde electrophiles were also compatible, enabling access to secondary alcohol products (3ar–3at) in moderate to good yields (55–60%). Additionally, mechanochemical borylation of in situ-generated primary alkyl sodium species provided the corresponding boronic esters 3au and 3av in yields of 75 and 62%, respectively (in the latter case, 5% of Wurtz-type homocoupled product was detected as a minor side product).

Conditions: 1 (2.0 mmol), Na (4.4 mmol), n-hexane (4.4 mmol) and electrophile (1.0 mmol) in a stainless-steel ball-milling jar (5 ml) with a stainless-steel ball (10 mm). Ball milling (30 Hz) was carried out. Isolated yields are reported as percentages. aAddition of n-hexane (4.4 mmol). b1 (1.0 mmol), Na (2.2–4.0 equiv.) and iPrO–B(pin) (2.0 mmol). iPr, isopropyl; tBu, tert-butyl; pin, pinacol.
We also applied this operationally simple one-pot, one-step protocol to reactions using aryl sodium compounds (Fig. 3). Under our optimized conditions, both 4-chlorotoluene and 4-tert-butylbromobenzene reacted with morpholine amides to furnish the corresponding ketones 3ab and 3aw in 76 and 74% yields, respectively. Nucleophilic additions of aryl sodium compounds to aldimine 2a and 4-tert-butylbenzaldehyde were also investigated, and the desired products 3b and 3o were successfully obtained in 77 and 40% yields, respectively. One-pot, one-step borylations were applied to 4-bromotoluene and 4-bromoanisole to deliver boronic esters 3ax and 3ay in 64 and 56% yields, respectively. These results are comparable to the corresponding one-pot, two-step reactions (Fig. 2).
Gram-scale reactions and applications to prepare sodium 2,2,6,6-tetramethylpiperidide
To underline the practical utility of our developed mechanochemical protocol, we investigated its use in preparative-scale reactions (Fig. 4). 4-Tolyl sodium was successfully prepared on a 12-mmol scale under mechanochemical conditions, and its nucleophilic addition to aldimine 2a gave 3b without any decrease in efficiency (96%, 1.582 g) compared to small-scale synthesis. Likewise, a one-pot, one-step reaction between 1-chlorobutane and 4-benzoylmorpholine was also carried out on a gram scale to furnish 3am in 69% yield. These results underscore the practical utility of this protocol.

Isolated yields are reported as percentages. See Supplementary Section 4 for full details.
To further expand the range of synthetically accessible organosodium compounds, we explored C–H sodiation of heteroaromatic arenes using a mechanochemically generated sodium amide (Fig. 5)71. We were delighted to find that sodium 2,2,6,6-tetramethylpiperidide (NaTMP) could be generated by the reaction between phenylsodium and 2,2,6,6-tetramethylpiperidine (TMPH), and employed to deprotonate benzothiophene. Subsequent addition to aldimine 2a proceeded smoothly to afford 3az in 97% yield. Benzofuran and 1-methylindole were also deprotonated by mechanochemically generated NaTMP and the resulting organosodium compounds reacted with aldimine 2a to produce 3ba and 3bb in good yields. Aldehyde and Weinreb amide electrophiles could also be employed as electrophiles, delivering the corresponding alcohol (3bc) and ketone (3bd) products in yields of 93 and 85%, respectively.

Isolated yields are reported as percentages. Conditions: 1a (1.0 mmol), Na (2.2 mmol), n-hexane (2.2 mmol), TMPH (1.0 mmol), 1 (1.0 mmol) and electrophile (0.5 mmol) in a stainless-steel ball-milling jar (5 ml) with a stainless-steel ball (10 mm). Ball milling (30 Hz) was carried out. See Supplementary Section 3F for full details.
To highlight the synthetic utility of our developed mechanochemical protocol, we applied it to synthesize the drug molecule orphenadrine, an anticholinergic agent used to treat painful muscle spasms (Fig. 6). After minor optimization, the generation of phenylsodium, followed by nucleophilic addition to 2-methylbenzaldehyde under mechanochemical conditions, afforded the intermediate 3q in 86% yield. Subsequent alkylation and reduction with LiAlH4 yielded orphenadrine in 85% yield over two steps.

Isolated yields are reported as percentages. TMPH, 2,2,6,6-tetramethylpiperidine; THF, tetrahydrofuran; r.t., room temperature. See Supplementary Section 6 for full details.
Preparation of organosodium compounds from poorly soluble aromatic halides and organic fluorides via C–F bond cleavage
Due to the high basicity of organosodium species, the scope of available solvents is limited to simple hydrocarbon solvents. Therefore, aryl halides containing a large π-conjugated system, which are often sparingly soluble in such solvents, are difficult to directly sodiate under conventional solution-based conditions. For example, 1-bromo-3,5-diphenylbenzene (1be) is poorly soluble in n-hexane, and an attempted solution-state sodiation followed by addition to cyclopropyl phenyl ketone did not afford the desired product 3be (Fig. 7a). By contrast, we were delighted to find that our mechanochemical protocol was successfully able to deliver alcohol 3be in excellent yield (89%), highlighting the potential of this approach to access organosodium compounds that cannot be accessed via conventional solution-based reactions.

a, Generation of organosodium compounds from a poorly soluble halide and nucleophilic addition to ketone. b, Generation of organosodium compounds from an organofluoride and nucleophilic addition to 2a. c, Various Na-meditated transformations of C–F bonds. Isolated yields are reported as percentages. Integrated 1H NMR spectroscopy yields are shown in parentheses. TMS, trimethylsilyl. See Supplementary Section 5 for details.
In a similar vein, we also explored the generation of organosodium species from organic fluorides via inert C–F bond cleavage (Fig. 7b)72. Direct metallation of organofluorine compounds is challenging because it involves breaking a strong C–F bond, and reports on the direct sodiation of fluoroarenes with unactivated sodium lumps are lacking73. Indeed, an attempted solution-phase (n-hexane) sodiation of fluorobenzene followed by addition of aldimine (2a) resulted in no product formation. On the other hand, we discovered that phenylsodium was rapidly generated from fluorobenzene in 5 min under mechanochemical conditions, and subsequent nucleophilic addition to aldimine 2a furnished 3a in 96% yield. We found that other sodium-meditated transformations via C–F bond activation were feasible under mechanochemical conditions (Fig. 7c). For example, sodiation of 4-fluorobiphenyl also proceeded smoothly, and nucleophilic addition to phenyl ethyl ketone produced 3u in 59% yield. Likewise, fluoroarenes bearing trimethylsilyl (TMS) and methyl groups could also be successfully sodiated, and nucleophilic substitutions with Weinreb amides afforded the corresponding ketones 3bf and 3bg in 76 and 53% yields, respectively. A Barbier-type reaction between [1,1’-biphenyl]-4-yl(morpholino)methanone and 1-fluoropentane afforded 3bh in 35% yield. 4-Tolyl sodium could be generated from 4-fluorotoluene and reacted with HSiMe2Ph to give the silylation product 3bi in 70% yield.
Structural evidence for the generation of organosodium species through isolation of a mechanochemically generated organosodium compound and structural studies
Finally, to offer unambiguous evidence for the mechanochemical generation of the Na‒C bond during our reactions, we attempted to isolate and characterize a mechanochemically generated organosodium compound. For these structural studies, we selected a model aromatic organosodium bearing a lipophilic tert-butyl substituent, which we hoped would ensure sufficient hydrocarbon solubility to enable crystallization. Given that organosodium compounds are well known to form poorly soluble aggregates74 that could hamper their study by SCXRD, we also introduced a neutral amine ligand, namely N,N,Nʹ,Nʹ,Nʹ-pentamethyldiethylenetriamine (PMDTA), to break up these aggregates and form a soluble compound, allowing for crystallization.
Accordingly, under an argon atmosphere, 3-chloro-tert-butylbenzene and 2.2 equiv. of Na metal were ball milled with 2.2 equiv. of n-hexane according to our standard optimized conditions (Fig. 8). The crude product was treated with 0.6 equiv. of PMDTA solution in n-hexane followed by filtration to remove NaCl. Concentration followed by crystallization from n-hexane solution at −35 °C afforded a dimeric PMDTA-coordinated organosodium compound [Na(μC-3-tBu-C6H4)(κ3-N,Nʹ,Nʹ-PMDTA)]2 (4) (28% yield based on 1-(tert-butyl)-3-chlorobenzene). The single-crystal X-ray diffraction structure of 4 is shown in Fig. 8b, featuring two bridging phenyl groups, with Na‒C bond lengths within the range 2.61–2.65 Å. Solution state characterization of 4 was also performed by 1H, 13C and 23Na NMR spectroscopy in d12-cyclohexane. The 23Na NMR spectrum of 4 features a broad signal at –14.90 ppm (line width at half height LW1/2 approximately 10 ppm), which matches reported Na+ cation signals69. The 1H NMR spectrum of 4 clearly exhibits a characteristic pattern of four protons consistent with a 1,3-disubstituted phenyl group, and the PMDTA remains coordinated (Fig. 8c). The 13C NMR spectrum (d12-cyclohexane) of 4 features a Na‒Csp2 quaternary carbon signal at 195.1 ppm similar to other PMDTA-ligated aryl sodium complexes described in the literature75. The NMR spectra indicate that the SCXRD structure of 4 persists in solution in cyclohexane—this is particularly important in organo-alkali metal chemistry, where fast equilibria are often prevalent, such as ligand coordination–dissociation and deprotonation–reprotonation. The isolation and characterization of 4 unambiguously supports the formation of a Na‒C bond during the mechanochemical sodiation reactions in our methodology.

a, Mechanochemical synthesis of [3-(tert-butyl)phenyl]sodium followed by ligation with PMDTA and crystallization to afford [Na(μC-3-tBu-C6H4)(κ3-N,Nʹ,Nʹ-PMDTA]2 (4). b, Single-crystal X-ray structure of 4. For clarity only the disordered components with the highest occupancy are depicted and hydrogen atoms are omitted. Key bond lengths (Å): Na1‒C3 2.616(2), Na1‒C3A 2.643(2), Na1‒N1 2.663(9), Na1‒N2 2.565(5), Na1‒N3 2.547(9). c, 1H NMR spectrum of 4 (d12-cyclohexane, 298 K). The colours in the 1H NMR spectrum indicate which signals correspond to the protons of compound 4. PMDTA, N,N,Nʹ,Nʹ,Nʹ-pentamethyldiethylenetriamine.
Discussion
Organosodium compounds have garnered substantial attention as promising, sustainable alternatives to organolithium reagents. However, their practical and widespread application in organic synthesis has been hindered by the lack of efficient generation methods using easy-to-handle sodium sources and their limited solubility in organic solvents. This study demonstrates that a mechanochemical protocol can overcome these challenges, enabling rapid synthesis of a wide range of organosodium compounds, including aryl, primary, secondary alkyl and benzylic examples from inexpensive, abundant sodium lumps and organic halides within minutes, without the need for large volumes of solvents or complex inert-gas techniques. The resulting mechanochemically generated organosodium compounds react smoothly with various electrophiles in a one-pot manner (either stepwise or Barbier-type conditions), negating any requirement to isolate the organosodium intermediates. The operational simplicity of the procedure, in particular the ability to proceed without exclusion of air is especially noteworthy, substantially lowering the barrier for synthetic chemists to make and apply these reagents. Notably, this method facilitated the direct sodiation of poorly soluble halides, which are typically unreactive to sodiation under solution-based conditions. Furthermore, the study revealed that the sodiation of organofluorides, which are generally inert in solution, could be achieved via C–F bond cleavage under mechanochemical conditions. These findings highlight the effectiveness of this mechanochemical approach in expanding the accessible scope of organosodium chemistry. Finally, the isolation and characterization of mechanochemically generated organosodium compounds by single-crystal X-ray analysis and NMR spectroscopy was performed, providing unambiguous support for the key organosodium species proposed in this methodology. Given the increasing demand for organosodium compounds, this efficient and straightforward mechanical strategy is expected to advance the development of sodium-based synthetic chemistry, contributing to more sustainable syntheses of value-added molecules.
Methods
Representative procedure for the virtually solvent-less synthesis of sodium-based carbon nucleophiles and their reactions with electrophiles
Sodium lumps (4.4 equiv.) were cut into small pieces (approximately 4–5 mm in width and depth) and weighed in air after wiping off the mineral oil on them with paper. These were then added into a milling jar (5 ml) with a ball (10 mm diameter). An organic halide (2.0 equiv.) and n-hexane (4.4 equiv.) were added to the jar. After the jar was closed without purging with inert gas, it was placed in the ball mill (Retsch MM400, 30 Hz). After grinding for 5 min, the jar was opened in air and charged with an electrophile (1.0 mmol, 1.0 equiv.) as quickly as possible. The jar was then closed without purging with inert gas, and was placed in the ball mill (Retsch MM400, 30 Hz). After grinding for 5 min, the reaction mixture was quenched with a saturated aqueous solution of NH4Cl and extracted with EtOAc (30 ml × 3). The solution was dried over MgSO4, filtered and evaporated to dryness under reduced pressure. The crude material was purified by flash chromatography (SiO2, typically EtOAc/n-hexane, typically 0–10:90) to give the corresponding product.
Data availability
The data that support the findings of this study are available in the Article and its Supplementary Information. For full characterization data, including NMR spectra of the new compounds and experimental details, see the Supplementary Information. The X-ray crystallographic coordinates for the structure reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number CCDC 2423000 (4). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
References
Rappoport, Z. & Marek, I. (eds) The Chemistry of Organolithium Compounds (Wiley, 2004).
Clayden, J. Organolithiums: Selectivity for Synthesis (Elsevier, 2002).
Hodgson, D. M. (ed) Organolithiums in Enantioselective Synthesis (Springer, 2003).
Wu, G. & Huang, M. Organolithium reagents in pharmaceutical asymmetric processes. Chem. Rev. 106, 2596–2616 (2006).
Screttas, C. G., Steele, B. R., Micha-Screttas, M. & Heropoulos, G. A. Aryllithiums with increasing steric crowding and lipophilicity prepared from chlorides in diethyl ether. The first directly prepared room-temperature-stable dilithioarenes. Org. Lett. 14, 5680–5683 (2012).
Crockett, M. P., Aguirre, L. S., Jimenez, L. B., Hsu, H.-H. & Thomas, A. A. Preparation of highly reactive lithium metal dendrites for the synthesis of organolithium reagents. J. Am. Chem. Soc. 144, 16631–16637 (2022).
Tarascon, J.-M. Is lithium the new gold? Nat. Chem. 2, 510 (2010).
Kim, T., Song, W., Son, D.-Y., Ono, L. K. & Qi, Y. Lithium-ion batteries: outlook on present, future, and hybridized technologies. J. Mater. Chem. A 7, 2942–2964 (2019).
Weiss, M. et al. Fast charging of lithium-ion batteries: a review of materials aspects. Adv. Energy Matter. 11, 2101126 (2021).
Wulandari, T., Fawcett, D., Majumder, S. B. & Poinern, G. E. J. Lithium-based batteries, history, current status, challenges, and future perspectives. Battery Energy 2, 20230030 (2023).
Greenwood, N. N. & Earnshaw, A. (eds) Chemistry of the Elements 2nd edn (Butterworth-Heinemann, 1997).
Weidmann, N., Ketels, M. & Knochel, P. Sodiation of arenes and heteroarenes in continuous flow. Angew. Chem. Int. Ed. 57, 10748–10751 (2018).
Asako, S., Nakajima, H. & Takai, K. Organosodium compounds for catalytic cross-coupling. Nat. Catal. 2, 297–303 (2019).
Woltornist, R. A. et al. Structure, reactivity, and synthetic applications of sodium diisopropylamide. Synthesis 52, 1478–1488 (2020).
Fukazawa, M., Takahashi, F., Nogi, K., Sasamori, T. & Yorimitsu, H. Reductive difunctionalization of aryl alkenes with sodium metal and reduction-resistant alkoxy-substituted electrophiles. Org. Lett. 22, 2303–2307 (2020).
Asako, S., Takahashi, I., Nakajima, H., Ilies, L. & Takai, K. Halogen-sodium exchange enables efficient access to organosodium compounds. Commun. Chem. 4, 76 (2021).
Harenberg, J. H. et al. (2-Ethylhexyl)sodium: A hexane-soluble reagent for Br/Na-exchanges and directed metalations in continuous flow. Angew. Chem. Int. Ed. 60, 14296–14301 (2021).
De, P. B., Asako, S. & Ilies, L. Recent advances in the use of sodium dispersion for organic synthesis. Synthesis 53, 3180–3192 (2021).
Bole, L. J., Tortajada, A. & Hevia, E. Enhancing metalating efficiency of the sodium amide NaTMP in arene borylation applications. Angew. Chem. Int. Ed. 61, e202204262 (2022).
Tortajada, A. & Hevia, E. Perdeuteration of arenes via hydrogen isotope exchange catalyzed by the superbasic sodium amide donor species NaTMP‧PMDETA. J. Am. Chem. Soc. 144, 20237–20242 (2022).
Anderson, D. E., Tortajada, A. & Hevia, E. Highly reactive hydrocarbon soluble alkylsodium reagents for benzylic aroylation of toluenes using Weinreb amides. Angew. Chem. Int. Ed. 62, e202218498 (2023).
Davison, N. et al. Divergent reaction patterns between organolithium and organosodium complexes and ligand-catalyzed ketone/aldehyde methylenation. J. Am. Chem. Soc. 145, 6562–6576 (2023).
Dilauro, G. et al. Introducing water and deep eutectic solvents in organosodium chemistry: Chemoselective nucleophilic functionalizations in air. Angew. Chem. Int. Ed. 62, e202304720 (2023).
Wanklyn, J. A. XXII. On some new ethyl-compounds containing the alkalimetals. Proc. R. Soc. London 9, 341–345 (1858).
Benkeser, R. A., Foster, D. J., Sauve, D. M. & Nobis, J. F. Metalations with organosodium compounds. Chem. Rev. 57, 867–894 (1957).
Schlosser, M. Organosodium and organopotassium compounds part Ⅰ: properties and reactions. Angew. Chem. Int. Ed. 3, 287–306 (1964).
Schlosser, M. Organosodium and organopotassium compounds part Ⅱ: Preparation and synthetic applications. Angew. Chem. Int. Ed. 3, 362–373 (1964).
Gentner, T. X. & Mulvey, R. E. Alkali-metal mediation: diversity of applications in main-group organometallic chemistry. Angew. Chem. Int. Ed. 60, 9247–9262 (2021).
Anderson, D. E., Tortajada, A. & Hevia, E. New frontiers in organosodium chemistry as sustainable alternatives to organolithium reagents. Angew. Chem. Int. Ed. 63, e202313556 (2024).
Wurtz, A. Sur une nouvelle classe de radicaux organiques. Ann. Chim. Phys. 44, 275–312 (1855).
Tortajada, A., Anderson, D. & Hevia, E. Gram-scale synthesis, isolation and characterisation of sodium organometallics: nBuNa and NaTMP. Helv. Chim. Acta 105, e202200060 (2022).
James, S. L. et al. Mechanochemistry: opportunities for new and cleaner synthesis. Chem. Soc. Rev. 41, 413–447 (2012).
Do, J.-L. & Friščić, T. Mechanochemistry: a force of synthesis. ACS Cent. Sci. 3, 13–19 (2017).
Hernández, J. G. & Bolm, C. Altering product selectivity by mechanochemistry. J. Org. Chem. 82, 4007–4019 (2017).
Kubota, K. & Ito, H. Mechanochemical cross-coupling reactions. Trends Chem. 2, 1066–1081 (2020).
Tan, D. & García, F. Main group mechanochemistry: from curiosity to established protocols. Chem. Soc. Rev. 48, 2274–2292 (2019).
Bolm, C. & Hernández, J. G. Mechanochemistry of gaseous reactants. Angew. Chem. Int. Ed. 58, 3285–3299 (2019).
Porcheddu, A., Colacino, E., De Luca, L. & Delog, F. Metal-mediated and metal-catalyzed reactions under mechanochemical conditions. ACS Catal. 10, 8344–8394 (2020).
Ardila-Fierro, K. J. & Hernández, J. G. Sustainability assessment of mechanochemistry by using the twelve principles of green chemistry. ChemSusChem 14, 2145–2162 (2021).
Bolt, R. R. A., Leitch, J. A., Jones, A. C., Nicholson, W. I. & Browne, D. L. Continuous flow mechanochemistry: reactive extrusion as an enabling technology in organic synthesis. Chem. Soc. Rev. 51, 4243–4260 (2022).
Cuccu, F. et al. Mechanochemistry: New tools to navigate the uncharted territory of ‘impossible’ reactions. ChemSusChem 15, e202200362 (2022).
Martinez, V., Stolar, T., Karadeniz, B., Brekalo, I. & Užarević, K. Advancing mechanochemical synthesis by combining milling with different energy sources. Nat. Rev. Chem. 7, 51–65 (2023).
Alić, J., Schlegel, M.-C., Emmerling, F. & Stolar, T. Meeting the UN sustainable development goals with mechanochemistry. Angew. Chem. Int. Ed. 63, e202414745 (2024).
Jones, A. C., Leitch, J. A., Raby-Buck, S. E. & Browne, D. L. Mechanochemical techniques for the activation and use of zero-valent metals in synthesis. Nat. Synth. 1, 763–775 (2022).
Davison, N. et al. A room-temperature-stable electride and its reactivity: Reductive benzene/pyridine couplings and solvent-free Birch reductions. Chem 9, 576–591 (2023).
Gao, Y., Kubota, K. & Ito, H. Mechanochemical approach for air-tolerant and extremely fast lithium-based Birch reductions in minutes. Angew. Chem. Int. Ed. 62, e202217723 (2023).
Fujishiro, K. et al. Lithium-mediated mechanochemical cyclodehydrogenation. J. Am. Chem. Soc. 145, 8163–8175 (2023).
Davison, N., Waddell, P. G. & Lu, E. Reduction of K+ or Li+ in the heterobimetallic electride K+[LiN(SiMe3)2]e−. J. Am. Chem. Soc. 145, 17007–17012 (2023).
Nallaparaju, J. V. et al. Mechanochemical Birch reduction with low alkaline earth metals. Angew. Chem. Int. Ed. 63, e202319449 (2024).
Kubota, K., Fukuzawa, Y., Kondo, K., Gao, Y. & Ito, H. Highly efficient and air-tolerant calcium-based Birch reduction using mechanochemistry. Chem. Lett. 53, upae060 (2024).
Bhattacharjee, D., Jana, S. K. & Maji, B. Solid-state mechanochemical Clemmensen reduction. Synthesis 57, 84–90 (2025).
Kubota, K., Nagao, A. & Ito, H. Solvent-free zinc meditated Béchamp reduction using mechanochemistry. RSC Mechanochem. 2, 389–393 (2025).
Wada, S., Hayashi, N. & Suzuki, H. Noticeable facilitation of the bismuth-mediated Barbier-type allylation of aromatic carbonyl compounds under solvent-free conditions. Org. Biomol. Chem. 1, 2160–2163 (2003).
Harrowfield, J. M., Hart, R. J. & Whitaker, C. R. Magnesium and aromatics: mechanically-induced Grignard and McMurry reactions. Aust. J. Chem. 54, 423–425 (2001).
Takahashi, R. et al. Mechanochemical synthesis of magnesium-based carbon nucleophiles in air and their use in organic synthesis. Nat. Commun. 12, 6691 (2021).
Pfennig, V. S., Villella, R. C., Nikodemus, J. & Bolm, C. Mechanochemical Grignard reactions with gaseous CO2 and sodium methyl carbonate. Angew. Chem. Int. Ed. 61, e202116514 (2022).
Nallaparaju, J. V. et al. Mechanochemistry-amended Barbier reaction as an expedient alternative to Grignard synthesis. Angew. Chem. Int. Ed. 62, e202305775 (2023).
Cao, Q., Howard, J. L., Wheatley, E. & Browne, D. L. Mechanochemical activation of zinc and application to Negishi cross-coupling. Angew. Chem. Int. Ed. 57, 11339–11343 (2018).
Cao, Q., Stark, R. T., Fallis, I. A. & Browne, D. L. A ball-milling-enabled Reformatsky reaction. ChemSusChem 12, 2554–2557 (2019).
Yin, J., Stark, R. T., Faills, I. A. & Browne, D. L. A mechanochemical zinc-meditated Barbier-type allylation reaction under ball-milling conditions. J. Org. Chem. 85, 2347–2354 (2020).
Gao, P., Jiang, J., Maeda, S., Kubota, K. & Ito, H. Mechanochemically generated calcium-based heavy Grignard reagents and their application to carbon–carbon bond-forming reactions. Angew. Chem. Int. Ed. 61, e2022207118 (2022).
Gao, P. et al. Direct arylation of alkyl fluorides using in situ mechanochemically generated calcium-based heavy Grignard reagents. RSC Mechanochem. 1, 486–491 (2024).
Wang, X. et al. Direct arylation of gem-difluorostyrenes using in situ mechanochemically generated calcium-based heavy Grignard reagents. RSC Mechanochem. 2, 256–262 (2025).
Nicholson, W. I. et al. Ball-milling-enabled reactivity of manganese metal. Angew. Chem. Int. Ed. 60, 23128–23133 (2021).
Takahashi, R., Gao, P., Kubota, K. & Ito, H. Mechanochemical protocol facilitates the generation of arylmanganese nucleophiles from unactivated manganese metal. Chem. Sci. 14, 499–505 (2023).
Kubota, K., Kawamura, S., Jiang, J., Maeda, S. & Ito, H. Mechanochemical generation of aryl barium nucleophiles from unactivated barium metal. Chem. Sci. 15, 17453–17459 (2024).
Kondo, K., Kubota, K. & Ito, H. Mechanochemical activation of metallic lithium for the generation and application of organolithium compounds in air. Nat. Synth. https://doi.org/10.1038/s44160-025-00753-3 (2025).
Kondo, K., Kubota, K. & Ito, H. Mechanochemistry enabling highly efficient Birch reduction using sodium lumps and D-(+)-glucose. Chem. Sci. 15, 4452–4457 (2024).
Davison, N. et al. Mechanochemical synthesis of a sodium anion complex [Na+(2,2,2-cryptand)Na−] and studies of its reactivity: Two-electron and one-electron reductions. Inorg. Chem. 63, 15247–15258 (2024).
Inoue, T. et al. Cross-coupling polymerization of organosodium for polythiophene synthesis. Organometallics 40, 3506–3510 (2021).
Asako, S., Kodera, M., Nakajima, H. & Takai, K. Lithium-free synthesis of sodium 2,2,6,6,-tetramethylpiperidine and its synthetic applications. Adv. Synth. Catal. 361, 3120–3123 (2019).
Ito, S., Takahashi, F. & Yorimitsu, H. Defluorinative diborasodiation of benzotrifluorides with bis(pinacolato)diboron and sodium. Asian J. Org. Chem. 10, 1440–1443 (2021).
Lei, P. et al. A practical and chemoselective ammonia-free Birch reduction. Org. Lett. 20, 3439–3442 (2018).
Davison, N. & Lu, E. The quest for organo-alkali metal monomers: unscrambling the structure–reactivity relationship. Dalton Trans. 52, 8172–8192 (2023).
Garden, J. A. et al. Donor-activated lithiation and sodiation of trifluoromethylbenzene: structural, spectroscopic, and theoretical insights. Organometallics 32, 5481–5490 (2013).
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) via KAKENHI grants 22H00318 (H.I.), 24H00453 (K.K.), 24H01050 (K.K.), 24H01832 (K.K.) and 22K18333 (H.I.), by the JST via CREST grant JPMJCR19R1 (H.I.) and FOREST grant JPMJFR201I (K.K.) and by the Institute for Chemical Reaction Design and Discovery (ICReDD), which was established by the World Premier International Research Initiative (WPI), MEXT, Japan. E.L. and R.J.A. thank the Leverhulme Trust for generous financial support via research grants RPG-2022-231 (E.L. and N.D.) and RPG-2023-159 (E.L., R.J.A. and M.L.).
Author information
Authors and Affiliations
Contributions
R.J.A., E.L., K.K. and H.I. conceived and designed the study. All authors co-wrote the paper. K.K. and M.L. performed chemical experiments and analysed the data. N.D. performed preliminary piloting experiments at an early stage of this project. P.G.W. collected and refined the single crystal structure of 4. All authors discussed the results and the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Andrea Porcheddu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Peter Seavill, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Sections 1–15, Figs. 1–10, Tables 1–3 and Experimental details.
Supplementary Data 1
X-ray crystallographic data for compound 4, CCDC 2423000.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kondo, K., Lowe, M., Davison, N. et al. Mechanochemical synthesis of organosodium compounds through direct sodiation of organic halides. Nat. Synth (2025). https://doi.org/10.1038/s44160-025-00949-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44160-025-00949-7
