
Abstract
Depression is a risk factor for the later development of Alzheimer’s disease (AD), but evidence for the genetic relationship is mixed. Assessing depression symptom-specific genetic associations may better clarify this relationship. To address this, we conducted genome-wide meta-analysis (a genome-wide association study, GWAS) of the nine depression symptom items, plus their sum score, on the Patient Health Questionnaire (PHQ-9) (GWAS-equivalent N: 224,535–308,421) using data from UK Biobank, the GLAD study and PROTECT, identifying 37 genomic risk loci. Using six AD GWASs with varying proportions of clinical and proxy (family history) case ascertainment, we identified 20 significant genetic correlations with depression/depression symptoms. However, only one of these was identified with a clinical AD GWAS. Local genetic correlations were detected in 14 regions. No statistical colocalization was identified in these regions. However, the region of the transmembrane protein 106B gene (TMEM106B) showed colocalization between multiple depression phenotypes and both clinical-only and clinical + proxy AD. Mendelian randomization and polygenic risk score analyses did not yield significant results after multiple testing correction in either direction. Our findings do not demonstrate a causal role of depression/depression symptoms on AD and suggest that previous evidence of genetic overlap between depression and AD may be driven by the inclusion of family history-based proxy cases/controls. However, colocalization at TMEM106B warrants further investigation.
Similar content being viewed by others

Main
Epidemiological studies suggest that a diagnosis of depression is a risk factor for the later development of dementia1,2,3,4, of which Alzheimer’s disease (AD) is the most common form, accounting for ~80% of the over 40 million global cases5. Establishing the underlying mechanisms by which depression confers increased risk for AD offers a pathway by which new interventions might be implemented and the global dementia burden reduced6.
As twin studies have demonstrated, both depression and AD are substantially heritable—approximately 40% and 80%, respectively7,8. Furthermore, large-scale genome-wide association studies (GWASs) have demonstrated high polygenicity, identifying over 70 genomic risk loci for AD and nearly 200 for depression9,10,11,12,13,14,15. It is therefore possible that their phenotypic association is partially due to a shared genetic architecture. However, results from previous investigations into the genetic overlap between the two disorders have been mixed. For example, some findings indicate non-significant genetic overlap16,17, others a significant—if modest—genetic correlation of ~16–17% and a risk-increasing causal effect of depression on AD18,19,20.
According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-5), diagnosis of a major depressive episode requires the presence of at least five of a possible nine symptoms for ≥2 weeks, including one of the two cardinal symptoms—depressed mood or anhedonia21. Potentially hundreds of symptom combinations are possible to meet these diagnosis criteria22. As such, heterogeneity poses challenges to researchers seeking to better understand differences in the genetic contribution to depression and its subtypes23. However, the decomposition of depression into individual symptoms has provided insight into unique patterns of genome-wide significant loci and cross-trait genetic associations, as demonstrated in a recent GWAS of depression symptoms on the Patient Health Questionnaire (PHQ-9) by Thorp and colleagues24.
A number of studies suggest that anhedonia may be a better predictor of dementia than depressed mood25,26. Furthermore, several depression symptoms, including appetite changes, psychomotor dysfunction and sleep disruption, are commonly observed in non-depressed patients with dementia27,28,29. Taking this into account alongside the mixed nature of previous findings examining the genetic overlap between depression and AD, it is possible that leveraging depression symptom-level genetic information may offer greater insight into the disorders’ shared genetic architecture.
However, any association between depression and AD must also consider the potential influence of differences in case/control ascertainment in AD GWASs. A review by Escott-Price and colleagues30 notes that recent large-scale AD GWASs contain a relatively small proportion of clinically ascertained cases/controls, with a large percentage of cases ascertained by proxy, that is, cases and controls are defined as individuals with and without a self-reported parental history of AD/dementia, respectively. The combination of clinical and proxy samples in AD GWAS meta-analyses has proved an effective way of boosting sample size and variant discovery13,14,15,31. However, evidence suggests that this has come at the expense of specificity in regard to genomic risk loci and an apparent stagnation in the percentage of variance explained by common variants30. Most importantly for cross-trait analysis, recent studies indicate that the direction of Mendelian randomization (MR) causal estimates for AD risk factors on AD can be in the opposite direction depending on whether the AD outcome GWAS contains both clinical and proxy cases/controls or is more strictly clinically ascertained32,33.
To address these points, here we report a large genome-wide meta-analysis of PHQ-9 depression symptom items using data from the Genetic Links to Anxiety and Depression (GLAD) Study34, the PROTECT Study35 and two questionnaires from UK Biobank (UKB)36. We obtained summary statistics from previous large-scale GWAS for clinical9 and broad10 depression, and six AD GWASs (three with clinical + proxy case/control ascertainment13,14,15, one with proxy-only31 and two with clinical-only12,37). We used these GWASs to assess the presence, strength and differences in genetic overlap between depression, depression symptoms and AD, with the additional aim of better understanding the influence of different AD case ascertainment strategies on associations.
Results
For a flowchart of this study, see Fig. 1. For details on depression and AD GWAS summary statistics obtained for this study, see Methods.

Flowchart describing the analyses undertaken following genetic and phenotypic quality control (see Methods) for each of the depression symptom items of the PHQ-9 in each of the four samples—UKB (the Mental Health Questionnaire and Experience of Pain Questionnaire), the GLAD Study and the PROTECT Study. MTAG, multi-trait analysis of GWAS; FUMA, functional mapping and annotation; LAVA, local analysis of [co]variant association; ADNI, Alzheimer’s disease neuroimaging initiative; PC, genetic principal components.
PHQ-9 genome-wide meta-analyses
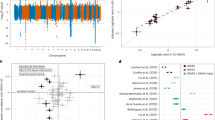
The final genome-wide meta-analysis—conducted using multi-trait analysis of GWAS (MTAG)38—identified a total of 40 genomic risk loci between the 10 PHQ-9 phenotypes (GWAS-equivalent N range: 224,535–308,421). Only one depression symptom—suicidal thoughts—identified no genome-wide significant variants. Three lead single nucleotide polymorphisms (SNPs) were shared with more than one PHQ-9 phenotype, leaving a total of 37 unique genomic risk loci (Table 1). The significance of each of the lead variants in each of the samples contributing to the meta-analysis is provided in Supplementary Table 1. Expression quantitative trait loci (eQTL) mapping in functional mapping and annotation (FUMA) mapped lead variants at genomic risk loci to 76 genes (Supplementary Table 2). The SNP heritability (h2SNP) value for the MTAG-PHQ-9 GWAS ranged from 1.12% for suicidal thoughts to 6.78% for the PHQ-9 sum score. h2SNP z-scores were all >4 (range 6.59–18.50) (Supplementary Table 3), indicating sufficient heritability to obtain reliable genetic correlation estimates in downstream analyses39. The genomic inflation factors (λGC) ranged from 1.0638 to 1.2156, with linkage disequilibrium score regression (LDSC) intercepts ranging from 0.9997 to 1.0007, indicating that inflation was due to the polygenic signal as opposed to confounding due to population stratification40. Manhattan and quantile–quantile (QQ) plots are presented in Fig. 2.

The red lines on the Manhattan plots indicate genome-wide significance (P ≤ 5 × 10−8) and the blue lines suggestive significance (P ≤ 1 × 10−5). No further study-wide multiple testing correction was applied. The P-value estimates are derived from a linear regression model with a two-sided test. The FUMA-identified nearest gene to the top variant at each locus is labeled.
Genetic correlations
Of the 72 bivariate genetic correlations (rg) calculated between the 12 depression phenotypes and the six AD GWASs, 24 were nominally significant and 20 remained significant after false discovery rate (FDR) correction (PFDR ≤ 0.05) (rg range −0.25–0.35; P-value range 1.25 × 10−2–4.01 × 10−5; PFDR range 4.5 × 10−2–1.9 × 10−3). Of these, 19 were identified when the AD GWAS in the pair contained either clinical + proxy cases and controls, or proxy-only cases and controls (Fig. 3 and Supplementary Table 4). Only one PFDR significant association was found when using a clinical AD GWAS—between suicidal thoughts and Wightman et al. (excl. UKB) (rg = −0.25, P = 6.78 × 10−3, PFDR = 3.48 × 10−2). All depression phenotypes were significantly genetically correlated with each other (rg range, 0.57–0.98; P ≤ 3.71 × 10−23) (Supplementary Table 5 and Supplementary Material 1). Only one PHQ-9 symptom pair—concentration problems and psychomotor changes—showed a genetic correlation that was not statistically different from one (95% confidence interval (CI) included one), indicating genetic heterogeneity across depression symptoms.

The color of each square indicates the strength of the correlation on a scale of −1 to 1. Genetic correlations significant at PFDR ≤ 0.05 are circled in black.
Local genetic correlations
After univariate testing, a total of 4,271 bivariate local genetic correlation tests were conducted in local analysis of [co]variant association (LAVA)41 across 324 genomic loci. Of these, 716 were nominally significant and 15 remained significant at PFDR ≤ 0.05 across 14 unique genomic loci (local rg range −0.81–0.82; P-value range 1.48 × 10−4–4.2 × 10−6; PFDR range 4.22 × 10−2–1.38 × 10−2) (Supplementary Table 6). Of the 15 statistically significant tests, ten were identified when using clinical + proxy/proxy-only AD GWASs. No depression phenotype showed a statistically significant association at the same genomic locus with more than one AD GWAS. However, for 10 of the 15 statistically significant tests, nominally significant local genetic correlation was observed between the depression phenotype and at least one additional AD GWAS at the same locus (Supplementary Table 7). Only locus 1790 (chr12: 51769420–53039987) showed a significant PFDR local genetic correlation with more than one depression phenotype—concentration and sleep problems—both with the clinical-only Wightman et al. GWAS. The numbers of positively and negatively correlated loci identified between each phenotype pair are presented in Supplementary Table 8.
Colocalization
Following LAVA, 14 PFDR-significant regions of local genetic correlation were passed to the COLOC-reporter pipeline42 across 15 depression–AD phenotype pairs. A further 14 colocalization tests were conducted where a nominally significant local genetic correlation was observed at a PFDR-significant locus between the same depression phenotype and a different AD GWAS. As such, a total of 29 statistical colocalization tests were conducted to follow up the LAVA results. No 95% credible sets were identified by sum of single effects (SuSiE) for any phenotype pairs in these regions. All analyses were therefore conducted under the single causal variant assumption of coloc.abf. No colocalization was identified at any of these loci (mean posterior probability for hypothesis 4 (PP.H4 ) = 0.59%) (Supplementary Table 7). All but two of these tests indicated no causal variant present in either phenotype (PP.H0 > 0.8). The two tests in locus 319 (chr2: 126754028–127895644) indicated a strong probability of a causal variant for the Kunkle et al.12 and Wightman et al.37 clinical AD GWASs (PP.H2 > 0.9). This locus contains BIN1, a known risk gene for AD that is involved in tau regulation43,44.
An additional 762 colocalization tests were conducted with the six AD GWASs using regions ±250 kb (r2 > 0.1) of lead variants from the MTAG-PHQ-9, broad and clinical depression GWAS. SuSiE identified evidence of colocalization in regions ±250 kb of lead variants at genomic risk loci 14 (depressed mood), 15 (appetite change) and 16 (PHQ-9 sum score) and for broad depression at chr7: 12000402–12500402 (PP.H4 range 0.79–0.85), all with the same three AD GWASs—Bellenguez et al.15, Wightman et al.14 and Wightman et al. (excluding the UKB)37 (Supplementary Table 9). These colocalizations were all in the region of the transmembrane protein 106B gene (TMEM106B), which is visualized using LocusZoom45 in Fig. 4. Colocalization was also identified for the same phenotype pairs using coloc.abf (Supplementary Table 10). The same depression phenotypes and loci were suggestive of colocalization with Jansen et al.13 (PP.H4 > 0.6).

a–g, LocusZoom plots for broad depression (a), appetite changes (b), depressed mood (c), the PHQ-9 sum score (d), Bellenguez et al. (e), Wightman et al. (f) and Wightman et al. excluding UKB (g). The most significant variant for each phenotype is labeled in the respective plots.
In a follow-up analysis, we assessed statistical colocalization for ±250 kb TMEM106B (chr7: 12000920–12532993) between these four AD GWASs and all remaining depression phenotypes. Additional colocalization was identified at TMEM106B between both fatigue and psychomotor changes with the Bellenguez et al.15, Wightman et al.14 and Wightman et al. (excluding the UKB)37 AD GWASs (Supplementary Table 11), and was suggestive for fatigue with Jansen et al.13 (Supplementary Table 21).
SMR analysis using gene expression in the TMEM106B region
We further followed-up colocalizing regions using summary-based Mendelian randomization (SMR)46 to integrate eQTLs. In total, 50 tests were conducted for prefrontal cortex and peripheral blood eQTL probes within chr7: 12000920–12532993 (genes TMEM1106B and VWDE) and the ten AD/depression phenotypes implicated in cross-trait colocalization. Of these, 11 associations remained significant after Bonferroni correction (P ≤ 0.001), all with expression levels of TMEM106B (Supplementary Table 13). Peripheral blood TMEM106B expression was positively associated with broad depression (bSMR [s.e.] = 0.029 [0.004], P = 2.30 × 10−7) and showed evidence of colocalization (PHEIDI = 0.108). Prefrontal cortex TMEM106B expression was significantly associated with all ten of the AD/depression phenotypes. Significant associations with AD were consistently positive (bSMR range 0.029–0.15; P-value range 1.042 × 10−5–3.395 × 10−4). Conversely, significant associations with depression phenotypes were consistently negative (bSMR range −0.097 to −0.033; P-value range 2.302 × 10−7–4.08 × 10−5). All brain-based associations showed evidence of colocalization (PHEIDI ≥ 0.05).
Mendelian randomization
We conducted 144 MR tests to assess bidirectional causal effects between the depression phenotypes and AD (72 in each direction). In our primary MR method, CAUSE47, no significant causal effects were identified between any of the depression items and AD in either direction, even at nominal significance (Supplementary Table 14).
F-statistics indicated that instrument strength was sufficient (FMean range 22.43–63.36; FMin range 20.84–31.56; FMax range 26.37–402.86). Measurement error, as indicated by the IGX2 statistics, was low, indicating instrument suitability for MR-Egger (IGX2 range 0.91–0.98). PFDR ≤ 0.05 was applied in each of the other MR methods to correct for the 144 tests conducted, after which no statistically significant associations were observed for any method (Supplementary Table 15).
No evidence of colocalization was observed within the APOE region between any depression phenotype and any AD GWAS, with a maximum PP.H4 of 16.58% observed in the region (Supplementary Table 16).
Polygenic risk scores
No statistically significant associations were detected between any depression phenotype polygenic risk score (PRS) and AD case/control status in any of the three AD target samples (PFDR ≤ 0.05, corrected within each target sample). Exclusion of the APOE region had no effect on results. (Supplementary Table 17 and Supplementary Material 2).
Similarly, no significant associations were observed between any AD-PRS and PHQ-9 depression items within the GLAD (Supplementary Table 18) or PROTECT (Supplementary Table 19) samples after FDR correction, with or without the APOE region.
Discussion
This study presents a genome-wide meta-analysis of PHQ-9 depression symptom items (GWAS-equivalent N range: 224,535–308,421), identifying 37 genomic risk loci. Subsequent genetic correlation analysis identified 20 significant global correlations and 15 significant local correlations at 14 loci with AD, across six AD GWASs with varying proportions of clinical case/control ascertainment. Significant global genetic correlations were primarily found with AD GWASs containing proxy cases and controls. Although no colocalization was identified at any of the regions of local genetic correlation, strong evidence of colocalization was observed between several depression phenotypes and AD in the region of TMEM106B. MR and PRS analyses did not yield significant results, and no evidence of colocalization was observed between depression phenotypes and any AD GWAS in the region of APOE.
The increased power of our PHQ-9 GWAS allowed for the identification of 28 more genomic risk loci than the previous PHQ-9 GWAS24. Several loci identified in this study have shown previous associations with related phenotypes. For example, SHISA4—identified in association with fatigue symptoms—was implicated as playing a role in disrupted sleep48 and daytime napping49. The top variant for sleep problems at genomic risk loci 6 (MEIS1)—rs113851554 (chr2: 66750564)—was also the top variant in a GWAS of insomnia and restless leg syndrome50. Additionally, the obesity gene FTO51 was identified as a genomic risk locus for appetite changes. Although the role of FTO in depression is inconclusive52, it has been linked to anxiety and depression symptoms in individuals with anorexia nervosa (AN)53. Its identification in association with appetite change symptoms—a phenotype relevant to eating behaviors—suggests that symptom-based genetic analysis can help identify the phenotype-relevant biology of individual depression symptoms.
Our findings also highlight cross-symptom genetic similarities. For example, TMEM106B—a gene identified in previous depression GWASs9,10—was the nearest gene to lead variants for three PHQ-9 items—appetite changes (rs13234970), depressed mood (rs3807866) and the PHQ-9 sum score (rs12699338). TMEM106B was strongly suggested as a causal gene in a recent multi-ancestry depression GWAS54. Furthermore, dysregulation of TMEM106B expression has been implicated in association with major depressive disorder (MDD)55 as well as with the anxious and weight gain MDD subtypes, both of which are associated with treatment resistance56. TMEM106B has also been implicated in self-reported diagnosis of anxiety disorder57, neuroticism58 and in a latent factor GWAS of depressive, manic and psychotic symptoms/disorders59, suggesting a link to psychiatric risk more generally.
The observed colocalization at TMEM106B between multiple depression phenotypes and both proxy + clinical and clinical-only AD is therefore of particular interest. Two previous studies have identified TMEM106B as playing a role in both depression and AD18,19. TMEM106B is involved in lysosomal function—particularly in motor neurons60—and is classically considered a frontotemporal dementia risk gene61. As well as being identified in recent AD GWASs14,15, it is also associated with brain aging, cognitive decline and neurodegeneration across other brain disorders, including amyotrophic lateral sclerosis, multiple sclerosis and Parkinson’s disease62,63,64,65. TMEM106B is also linked to higher levels of cerebrospinal fluid (CSF) neurofilament light (NfL) chain66—itself predictive of cognitive decline, brain atrophy and cortical amyloid burden in individuals with AD and mild cognitive impairment67. Higher levels of plasma NfL are also observed in individuals with depression68. Accordingly, colocalization between depression phenotypes and AD at TMEM106B indicates that depression may be genetically linked to overall brain health and the resulting general dementia risk. Our study suggests that this overlap may be driven by the genetic architecture of specific depression symptoms, highlighting the benefits of symptom-level genetic analysis. However, SMR analysis indicates that levels of TMEM106B expression as measured in brain have directionally opposite effects for depression phenotypes and AD. As such, further work is required to better understand the role of TMEM106B in brain disorders.
Depression/depression symptom PRSs were not predictive of AD case/control status in three clinical samples, and we did not find evidence of any MR causal associations. Although in contradiction to the study by Harerimana et al.20, these MR findings are consistent with previous studies17,69. The overall lack of evidence in our analyses versus the relationship observed in previous epidemiological studies suggests the relationship is subject to unidentified confounding. The investigation of this is an important step for future research.
Previous studies have shown changes in the direction of MR effects depending on whether the outcome AD GWAS contains proxy or clinical cases/controls32, but this study differs in that it demonstrates a similar effect with genetic correlations. Of the significant genetic correlations we identified, 95% were identified in proxy + clinical or proxy-only AD GWASs. Where two previous studies18,20 identified a genetic correlation between depression and AD, it is noticeable that they used the Jansen et al. proxy + clinical AD GWAS as their primary outcome.
Exactly why depression/depression symptoms show differences in genetic correlation between proxy and clinical AD is a matter of interest, particularly as no genetic correlations were identified with the Bellenguez et al.15 GWAS, despite this also containing proxy + clinical phenotyping. As mentioned, the Bellenguez et al.15 GWAS defines proxy cases/controls as a binary phenotype, whereas Wightman et al.14 and Jansen et al.13 define proxy cases/controls as a continuous phenotype. These phenotyping differences probably partially explain the differences in the genetic correlation results, given that all three GWASs use the same proxy cases/controls from UKB. However, genetic correlations were also observed with the proxy-only Marioni et al.31 GWAS—a meta-analysis of maternal and paternal AD status where parental age-at-diagnosis/age-of-death is controlled for in the GWAS model, instead of being used for weighting the proxy phenotype before analysis. Considering that depression is itself associated with all-cause mortality70, it is plausible that including age-at-diagnosis/age-of-death in proxy AD phenotyping induces a form of bias in later cross-trait analyses when the other trait is itself associated with longevity. Further investigation of this issue is required.
Nonetheless, conflicting results such as these pose a problem to researchers seeking to identify genetic relationships between AD and its risk factors. Large differences in the presence or direction of effects depending on which AD GWAS is used to assess associations increases the difficulty in discerning true associations. As such, a sensible approach for future cross-trait genetic studies of AD would be to conduct primary analyses using a clinically ascertained AD phenotype, with different proxy/clinical ascertainment GWASs used to examine consistency. Although this approach would limit researchers to AD GWASs with smaller sample sizes for primary analyses, it would also ensure that the results are driven not by AD proxy phenotyping alone.
This study has several limitations. Despite it being such a large meta-analysis of PHQ-9 items, the ability to detect genome-wide significant variants was probably limited by small sample sizes relative to other psychiatric conditions. Additionally, our analyses were restricted to individuals of European ancestry. The GWAS results may therefore have poor transferability to other ancestry groups. Furthermore, our study uses data from UKB, which is known to be affected by healthy volunteer bias and as a consequence is not fully representative of the wider population71. We also note that recent work by Huang and colleagues72 has suggested that the PHQ-9 capture does not capture the symptom-level genetic heterogeneity underlying depression as accurately as the Composite International Diagnostic Interview Short-Form (CIDI-SF). Although the present study is better powered due to a larger overall sample size, we suggest that future GWAS meta-analyses of individual depression symptoms would benefit from utilizing multiple rating scales.
This study focused on depression as a risk factor for AD. However, there is evidence that some late-life depression represents a prodromal phase of dementia onset6,73, possibly related to dementia biomarker levels74. Therefore, dementia-related depression may be biologically distinct from depression as a mental health disorder. Future genomic studies of dementia-related depression—as undertaken with psychosis in AD75—could prove illuminating.
In conclusion, this study describes a genome-wide meta-analysis of PHQ-9 depression symptom items (GWAS-equivalent N range: 224,535–308,421), identifying 37 unique genomic risk loci. Genetic correlations between depression/depression symptoms and AD were primarily observed when the AD GWAS contained clinical + proxy or proxy-only AD case/control ascertainment. Despite null results in MR and PRS, colocalization in the TMEM106B region between four depression phenotypes and AD across both proxy and clinical AD GWASs suggests that future research is warranted into the shared biological mechanisms underlying the role of this locus in depression and AD.
Methods
GWAS in the UK Biobank, GLAD and PROTECT
Patient Health Questionnaire-9 phenotypes
The PHQ-9 is a well-validated clinical screening questionnaire used to assess depression symptom severity on nine individual symptoms in the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV)76. The severity of each symptom is measured by the self-reported persistence of that symptom over the preceding two weeks, on a scale of 0 to 3. Scores of 3 indicate an individual experienced that symptom nearly every day, 2 indicates an individual experienced that symptom on more than half the days, 1 indicates an individual experienced that symptom for several days, and 0 indicates no experience of that symptom at all. The sum of an individual’s scores over all nine items (sum score) ranges from 0 to 27. For an overview of the PHQ-9 items and response distribution for each sample, see Supplementary Table 20. Supplementary Table 21 provides sum-score distributions.
Study population
In each GWAS sample, individuals were only retained if they had reported European ancestry and provided a valid response to all PHQ-9 items. Individuals were excluded if they had reported a previous professional diagnosis of schizophrenia, psychosis, mania, hypomania, bipolar or manic depression (UKB field ID 20544) or a previous prescription of medication for a psychotic experience (UKB field ID 20466).
GWAS software
GWAS analyses were conducted using REGENIE v3.1.377. In step one of REGENIE, ridge regression is applied to a subset of quality-controlled variants to fit, combine and decompose a set of leave-one-chromosome-out (LOCO) predictions. Here, quality control for step one was undertaken using PLINK v1.978. In step two, imputed variants are tested for association with the phenotype. LOCO predictions from step one are included as covariates to control for proximal contamination. For all GWASs, genotyping batch, sex, age and age-squared were included as covariates, as were the maximum available genetic principal components (PCs) for GLAD (10 PCs) and PROTECT (20 PCs) to control for population stratification. For the UKB analyses, 16 PCs were included, as recommended by Privé and colleagues79. Assessment center was also included as a covariate for UKB analyses.
A total of 40 GWASs were conducted for the meta-analyses—one for each of the nine PHQ-9 depression symptom phenotypes as well as the sum score across all nine items in each of the four samples. To maximize the statistical power, PHQ-9 phenotypes were treated as continuous (range of 0–3 for individual items and 0–27 for the sum score) and analyzed using linear regression. Analyses were restricted to the autosomes.
GWAS with UK Biobank
UKB is a large-scale biomedical database and research resource consisting of ~500,000 individuals with data across a broad range of phenotypes, including mental health outcomes36. Individuals in UKB have been genotyped on the custom UK Biobank Axiom or UKBiLEVE arrays, with imputed data available for ~90 million variants imputed with IMPUTE2 using the Haplotype Reference Consortium (HRC)80 and combined UK10K + 1000 Genomes Phase 3 reference panels81.
UKB participants completed the PHQ-9 in two online surveys. In total, 157,345 individuals provided responses as part of the Mental Health Questionnaire (UKB-MHQ) (category 136) between 2016 and 2017, and 167,199 individuals provided responses as part of the Experience of Pain Questionnaire (UKB-EoP) (category 154) between 2019 and 2020.
After filtering for self-reported European ancestry, valid PHQ-9 responses and previous diagnosis/prescription exclusions, 144,630 (UKB-MHQ) and 155,027 (UKB-EoP) individuals remained before genetic quality control for REGENIE. In step one, SNPs with a call rate of >98%, minor allele frequency (MAF) > 1% and Hardy–Weinberg equilibrium test P > 1 × 10−8 were retained, as were individuals with variant missingness < 2%, no unusual levels of heterozygosity and not mismatched on sex. Individuals were retained if they were determined to be of European ancestry based on 4-means clustering on the first PCs.
For the final GWAS analyses, 143,171 (mean age [s.d.] = 63.70 [7.68]; % female = 56.38%) and 152,932 (mean age [s.d.] = 65.95 [7.63]; % female = 56.57%) individuals proceeded from the MHQ and EoP questionnaires, respectively. Of these, 108,601 individuals had provided responses on both questionnaires. In step two, a total of 9,746,698 imputed variants were retained with MAF ≥ 0.01 and imputation quality (INFO) score ≥ 0.7.
GWAS with the Genetic Links to Anxiety and Depression study
The GLAD study has the specific goal of recruiting a large cohort of recontactable individuals with anxiety or depression into the National Institute for Health and Care Research (NIHR) Mental Health BioResource, with genetic, environmental and phenotypic data collected34. Genotyping for GLAD was conducted using the UKB v2 Axiom array and imputed using the TopMed imputation pipeline82.
After filtering for self-reported European ancestry, valid PHQ-9 responses and previous diagnosis/prescription exclusions, 15,472 individuals remained before genetic quality control for REGENIE step one. Genotype data were provided by the study team and had been filtered to retain SNPs with a genotype call rate > 95%, MAF > 1%, Hardy–Weinberg equilibrium test P > 1 × 10−10, and individuals with genotype missingness < 5%. Individuals were also excluded if they had unusual levels of heterozygosity, were mismatched on sex and were of non-European ancestry based on 4-means clustering. A total of 15,171 individuals (mean age [s.d.] = 39.27 [14.61]; % female = 78.30%) were retained for the final analysis. In step two, a total of 13,979,187 imputed variants with MAF ≥ 0.001 and INFO ≥ 0.7 were analyzed.
GWAS with the PROTECT study
PROTECT is an online registry of ~25,000 UK-based individuals that aims to track cognitive health in older adults. Individuals were only considered eligible for inclusion in PROTECT if they were older than 50 years, had no previous dementia diagnosis and had internet access. Genetic data are available alongside phenotypic data for ~10,000 of the participants. These individuals were genotyped on the Illumina Infinium Global Screening Array and imputed on the 1000 Genomes reference panel83 using the Michigan imputation server and genotype phasing using Eagle.
After filtering for self-reported European ancestry, valid PHQ-9 responses and previous diagnosis/prescription exclusions, 7,589 individuals remained for genetic quality control for step one of REGENIE. Genetic data in PROTECT had been quality-controlled previously before imputation to only retain individuals and variants with a call rate of >98%, Hardy–Weinberg equilibrium test P > 0.00001 and excluding unusual heterozygosity35. Variants used in step one were down-sampled from the imputed data using a snplist from the Illumina Infinium Global Screening Array provided by the PROTECT investigators. Variants were retained if they had MAF > 1%. After mismatched sex and 4-means clustering ancestry exclusions, a total of 7,589 individuals (mean age [s.d.] = 61.96 [7.07]; % female = 75.13%) proceeded to step two. In step two, 9,388,534 imputed variants with MAF ≥ 0.001 and imputation INFO score ≥ 0.7 were analyzed.
GWAS summary statistics
An overview of additional summary statistics obtained for this study is provided in Table 2.
Clinical and broad depression
To examine potential differences in genetic overlap with AD between depression as a disorder compared to individual depression symptoms, summary statistics for two previously conducted GWASs of clinical and broad depression were obtained from the Psychiatric Genomics Consortium (PGC; https://pgc.unc.edu/for-researchers/download-results/). For clinical depression, we used a subsample of the MDD GWAS by Wray et al.9 that excluded samples from the UKB and 23andMe, and contained only individuals for whom case ascertainment was defined through structured diagnostic interview or electronic health records. For the broad definition depression GWAS, we used a subsample of the depression GWAS by Howard et al.10, which also excluded samples from 23andMe. In addition to clinical cases and controls used by Wray et al.9, this broad depression GWAS included individuals in the UKB for whom case–control ascertainment was based on self-reported responses to the questions ‘Have you ever seen a general practitioner for nerves, anxiety tension or depression?’ and ‘Have you ever seen a psychiatrist for nerves anxiety, tension or depression?’
Alzheimer’s disease
Summary statistics were obtained from six previously conducted AD GWASs: three with proxy + clinical, one with proxy-only and two with clinical-only case ascertainment. All three of the proxy + clinical AD GWASs (Bellenguez et al.15, Wightman et al.14 and Jansen et al.13) and the proxy-only AD GWAS (Marioni et al.31) used data from the UKB for proxy AD samples.
There are some key differences in the way these AD GWASs define proxy cases and controls. Bellenguez et al.15 define proxy cases/controls as a binary phenotype, whereby individuals reporting a parent with AD or dementia are considered cases and those reporting no parental history are considered controls. Wightman et al.14 and Jansen et al.13 instead define proxy cases/control as a continuous phenotype, summing the number of parents an individual has reported with dementia and down-weighting unaffected parents by their age (or age of death).
For the proxy-only Marioni et al.31 GWAS, summary statistics were obtained from a meta-analysis of paternal and maternal AD. Here, proxy phenotyping was based on the self-report of either maternal or paternal AD, including the parent’s age at the time of reporting/age of death as a covariate.
Summary statistics from a clinical-only subsample of the GWAS by Wightman et al. that excluded proxy cases/controls from the UKB37 were obtained from the authors. Summary statistics for a final clinical-only AD GWAS were obtained from stage 1 of the GWAS by Kunkle and colleagues12.
Summary statistic standardization
Summary statistics from all 40 depression symptom GWASs, the two depression GWASs and the six AD GWASs were standardized using MungeSumstats84 in R version 4.2.1. Using dbSNP 141 and the BSgenome.Hsapiens.1000genomes.hs37d5 reference genome, missing rsIDs were corrected, duplicates and multi-allelic variants removed, effect alleles and the direction of their effects aligned to the reference genome, and variants filtered at an INFO score of ≥0.7 and MAF ≥ 0.01. The GLAD Study and Bellenguez et al.15 summary statistics were lifted over from GRCh38 to GRCh37.
SNP heritability
SNP heritability (h2SNP) estimates were calculated using LDSC39. Briefly, LDSC calculates h2SNP by regressing the effect sizes from GWAS summary statistics on their LD score as computed in a reference panel—in this case HapMap3 variants contained within the European sample of 1000 Genomes Phase 3. Liability scale h2SNP was calculated naïvely from the standardized depression GWAS and AD GWAS using a 15% and 5% population prevalence, respectively9,14. Heritability z-scores were calculated for all phenotypes by dividing the h2SNP estimates by their standard error.
GWAS meta-analysis of depression symptoms
To leverage the maximum genetic information available controlling for the sample overlap between the UKB-MHQ and UKB-EoP samples, the REGENIE output for each PHQ-9 phenotype from the UKB-EoP, GLAD Study and PROTECT were first subject to inverse variance weighted (IVW) meta-analysis using METAL85. All available variants were included, for a total of 8,425,618 (N = 175,692). Multi-trait analysis of GWAS (MTAG)38 v1.0.8 was then used to meta-analyze the METAL output with the UKB-MHQ sample. Although MTAG is commonly used for the joint genetic analysis of multiple traits or multiple measurements of the same trait, by assuming the heritability of included phenotypes are equal (--equal-h2) and their genetic correlation is one (--perfect-gencov), MTAG performs an IVW meta-analysis of the same measures of the same trait, accounting for sample overlap using the cross-trait intercept from LDSC39,40. Heritability estimates for all samples, plus the METAL meta-analysis, are shown in Supplementary Table 22. Genetic correlations between the UKB-MHQ and METAL GWAS are provided in Supplementary Table 23. For greater detail on the IVW function of MTAG, see the online methods of the original MTAG paper38. A total of 8,196,874 SNPs with MAF > 0.01 were available for MTAG analysis.
This MTAG function provides one set of summary statistics and two GWAS-equivalent sample sizes—one for each original sample included. A single, weighted GWAS-equivalent N was obtained for each PHQ-9-MTAG GWAS, using the following formula:
where N1 and N2 represents the UKB-MHQ and METAL GWASs, respectively, pre is the mean sample size prior to inclusion in MTAG, and post is the GWAS-equivalent sample estimated by MTAG following analysis.
Genomic risk loci and gene annotation
The GWAS meta-analysis results were annotated using FUMA GWAS86 v3.1.6a. Genome-wide significance was set at P ≤ 5 × 10−8. Lead variants at genomic risk loci were defined by clumping all variants correlated at r2 > 0.1, 250 kb either side, performed using the European sample of the 1000 Genomes Phase 3 reference panel. Lead variants were mapped to genes within 10 kb using positional mapping and eQTLs from four brain (BrainSeq87, PsychENCODE88, CommonMind89 and BRAINEAC90) and five blood (BloodeQTL91, BIOS92, eQTLGen cis and trans93, Twins UK94 and xQTLServer95) eQTL datasets, alongside all 54 tissue-type eQTLs from GTEx v8 (https://gtexportal.org/home/tissueSummaryPage).
Genetic correlations
Genetic correlation can be understood as the genome-wide correlation of genetic effects between two phenotypes, and as such can be viewed as an estimate of pleiotropy39.
Genetic correlations were calculated between each depression phenotype and the six AD GWASs using High Definition Likelihood v1.4.1 (HDL)96. HDL extends the LDSC framework by leveraging LD information from across the entire LD reference panel through eigen decomposition, thus shrinking standard errors and improving precision. A pre-computed eigenvector/value LD reference panel calculated from 335,265 individuals of European ancestry in the UKB was obtained via the HDL GitHub entry (https://github.com/zhenin/HDL/wiki/Reference-panels). This reference panel was calculated using 1,029,876 imputed, autosomal HapMap3 SNPs, with bi-allelic SNPs outside the major histocompatibility (MHC) region, MAF > 5%, call rate > 95% and INFO > 0.9 retained.
Local genetic correlations
Local genetic correlation assesses the correlation of genetic effects between two phenotypes in a specific region of the genome. It provides a more refined examination of genetic overlap, allowing for the identification of key regions driving a shared genetic architecture.
LAVA41 was used to assess regions of local genetic correlation between each depression phenotype and the six AD GWASs across 2,495 semi-independent, predefined LD blocks of at least 2,500 base pairs (https://github.com/josefin-werme/LAVA). Loci-specific heritability estimates were calculated for each phenotype for each block. If both a depression phenotype and an AD GWAS showed significant local heritability at a specific locus (Bonferroni-corrected P ≤ 2 × 10−5 (0.05/2,495)), bivariate local genetic correlation was tested. Bivariate results were considered significant at PFDR ≤ 0.05, correcting for the total number of bivariate tests. Sample overlap was accounted for using an LDSC intercept matrix. Analysis was restricted to the 5,531,969 non-strand-ambiguous variants shared across all GWAS summary statistics and on the European ancestry 1000 Genomes Phase 3 reference panel.
Colocalization
Colocalization using COLOC is a Bayesian statistical method to assess the probability of two phenotypes sharing a causal variant in a predefined genomic region. Within the colocalization framework, posterior probability is assessed for five hypotheses:
H0: there is no causal variant in the region for either phenotype
H1: there is a causal variant in the region for the first phenotype
H2: there is a causal variant in the region for the second phenotype
H3: there are distinct causal variants for each trait in the region
H4: both traits share a causal variant.
Colocalization analysis was conducted using the COLOC-reporter pipeline42 (https://github.com/ThomasPSpargo/COLOC-reporter). COLOC-reporter extracts variants in user-defined genomic regions and calculates the LD matrix for this region from a user-defined reference panel. In this study, this panel is the European ancestry sample of 1000 Genomes Phase 3 (N = 503). It then harmonizes the summary statistics to match the allele order of the reference panel, flipping effect directions accordingly. Observed versus expected z-scores are assessed using the diagnostic tools provided in the susieR R package97. The z-score outliers are omitted. Following this quality control, SuSiE fine-mapping97 is conducted to identify 95% credible sets in these regions for each phenotype. Identification of credible sets in both phenotypes allows for relaxation of the single causal variant assumption. All possible credible sets are then assessed pairwise for a shared signal between phenotypes, improving the resolution for colocalization inference in regions containing multiple signals98. Should no 95% credible set be identified or only identified for one phenotype, colocalization under the single causal variant assumption is performed using coloc.abf99. A posterior probability of ≥80% for H4 was considered evidence of colocalization between two phenotypes (PP.H4 ≥ 0.8). The SuSiE model assumed at most ten causal variants (L = 10) per credible set. We used default priors (P1 = 1 × 10−4, P2 = 1 × 10−4, P12 = 5 × 10−6).
Trait pairs with LAVA correlations significant at PFDR ≤ 0.05 were passed to COLOC-reporter. Where the same depression phenotype showed nominally significant local genetic correlation at the same locus but with different AD GWASs, these phenotype pairs were included as a sensitivity analysis. Regions ±250 kb (r2 ≥ 0.1) from lead variants at genome-wide significant loci from the MTAG-PHQ-9, broad and clinical depression GWASs were also examined for evidence of colocalization with the six AD GWASs.
Follow-up SMR analysis at TMEM106B
Identified cross-trait colocalization between multiple depression phenotypes and AD (both clinical only and clinical + proxy) at 12,000,920–12,532,993 was followed up using SMR46 software v1.3.1. SMR integrates eQTL and GWAS summary data within an MR framework to identify genes with expression levels linked to a phenotype of interest through pleiotropy. Resulting associations can be interpreted as assessing whether the effect of a variant on a phenotype is mediated through gene expression. In our analysis, only probes for genes in colocalizing region chr7: 12000920–12532993 (TMEM106B and VWDE) were assessed. We used eQTL summary data for gene expression in peripheral blood from ref. 100 (N = 2,765) and prefrontal cortex from PsychENCODE101 (N = 1,387) (https://yanglab.westlake.edu.cn/software/smr/#eQTLsummarydata). We included only probes for which there was ≥1 cis-eQTL significant at P ≤ 5 × 10−8. We used the heterogeneity in dependent instruments (HEIDI) test to identify association driven by LD as opposed to pleiotropy (PHEIDI ≥ 0.05 genuine pleiotropy). The HEIDI test is in essence a test of colocalization.
Mendelian randomization
MR is a statistical method that uses genetic variants associated with an exposure as instrumental variables to assess the causal effect of that exposure on an outcome of interest102. The two-sample MR framework estimates the causal relationships between an exposure and outcome using GWAS summary statistics. Valid MR instruments are defined by three key assumptions: (1) relevance—instrumental variables (IVs) are strongly associated with the exposure of interest; (2) independence—there are no confounders in the association between IVs and the outcome of interest; and (3) exclusion restriction—instruments are not associated to the outcome other than via exposure, for example, through horizontal pleiotropy102.
Sample overlap is a known source of bias in TwoSampleMR103. Given the likelihood of sample overlap between the depression phenotypes and the AD GWASs containing proxy cases/controls due to participants from the UKB, MR analysis was primarily conducted using ‘causal analysis using summary effect’ estimates (CAUSE)47 v1.2.0. CAUSE is a Bayesian MR method robust to sample overlap and correlated and uncorrelated pleiotropy. As such, it is robust to exclusion restriction assumption violations104. CAUSE uses a larger set of instruments than traditional MR methods. As such, clumping was performed with the default setting at r2 ≥ 0.01 and P ≤ 0.001 within a 10,000-kb window.
Causal estimates were also calculated using the traditional IVW method. IVW estimates are biased by the presence of horizontal pleiotropy105. Sensitivity analyses were therefore conducted using MR-Egger, weighted-median and weighted-mode MR. These methods allow for varying degrees of pleiotropy while providing unbiased causal estimates106,107,108. MR-PRESSO109 was also implemented. MR-PRESSO identifies and excludes outlying instruments based on their contribution to heterogeneity and provides a corrected causal estimate. For these tests, instruments were clumped at r2 ≥ 0.001 and P ≤ 5 × 10−8 within 10,000 kb. Where no or fewer than five instruments were available at P ≤ 5 × 10−8, a P ≤ 5 × 10−6 threshold was used. These analyses were conducted using the TwoSampleMR package v0.5.6
Pleiotropy was assessed using the MR-Egger intercept test110 (significant pleiotropy, P ≤ 0.05). Heterogeneity tests were also conducted for IVW and MR-Egger estimates using Cochran’s Q tests111 (significant heterogeneity, P ≤ 0.05). Instrument strength was calculated via the F-statistic (recommended F-statistic ≥ 10)112 (β2/s.e.2). IGX2 was calculated to ensure that the measurement error was sufficiently low so as to ensure the validity of results from MR-Egger (recommended IGX2 ≥ 0.90)113. For all analyses, instruments were clumped using data from individuals of European ancestry in 1000 Genomes Phase 3.
The APOE gene is known to be associated with non-AD phenotypes such as type 2 diabetes114, which has been linked to depression in previous MR analyses115. As such, the inclusion of APOE violates MR’s independence assumption. All MR analyses were therefore conducted excluding variants in the APOE region (chr19: 45020859–45844508 (GRCh37)) as per ref. 116. To investigate the involvement of the APOE region in both depression/depression symptoms and AD, we perform a separate cross-trait colocalization analysis in the same manner detailed above across all depression and AD phenotype trait-pairs at chr19: 45020859–45844508.
Polygenic risk scores
PRSs describe the sum of an individual’s risk alleles, weighted by their effect size117. The ten PHQ-9-MTAG, clinical depression and broad definition depression GWASs were processed for PRSs using the BayesR-SS function of MegaPRS, implemented in LDAK v5.2.1118. BayesR-SS assumes the BLD-LDAK heritability model, incorporating 65 genome annotations such as whether the variants are in coding regions or highly conserved118. Annotation files were obtained from the LDAK website (http://dougspeed.com/bldldak/). PRS calculation was restricted to the 1,217,311 HapMap3 SNPs, with strand-ambiguous SNPs excluded.
The predictive utility of PRSs for AD case/control status was assessed using logistic regression in three clinically ascertained AD cohorts: AddNeuroMed and Dementia Case Register Studies (ANM)119 (Ncases = 564; Ncontrols = 345) (mean age [s.e.] = 78.8 [6.92], % female = 58.65%), the Alzheimer’s Disease Neuroimaging Initiative (ADNI; https://adni.loni.usc.edu) (Ncases = 356; Ncontrols = 360) (mean age [s.e.] = 78.6 [6.55], % female = 44.55%) and the Genetic and Environmental Risk in Alzheimer’s Disease (GERAD1) Consortium (https://portal.dementiasplatform.uk/CohortDirectory/Item?fingerPrintID=GERAD) (Ncases = 2,661; Ncontrols = 1,124) (mean age [s.e.] = 76.6 [6.77], % female = 65.01%) (Supplementary Table 24). All analyses controlled for age, sex and ten PCs, and were restricted to individuals aged ≥65 years to provide clean controls. As a sensitivity analysis, PRSs were also calculated in these cohorts excluding the APOE region. Genetic quality control and imputation steps for these cohorts can be viewed in detail in the study by Lord and colleagues120.
We also assessed the predictive utility of AD PRSs on all ten PHQ-9 outcomes using linear regression, controlling for the same covariates. We used all six AD summary statistics to calculate scores including and excluding the APOE region. To avoid sample overlap between base and target datasets, we use the GLAD and PROTECT samples as the target dataset. We further filtered GLAD to exclude genetically related individuals up to third degree using a pi_hat cutoff value of 0.1875. The final GLAD PRS target sample contained 14,900 individuals (mean age [s.d.] = 39.20 [14.56], % female = 78.22). No such further exclusions were required for PROTECT.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this Article.
Data availability
All GWAS summary statistics generated in the process of conducting this study have been deposited on Zenodo at https://doi.org/10.5281/zenodo.13828101 (ref. 121). Individual-level data from UK Biobank, GLAD and PROTECT are subject to restrictions. Data are available on reasonable request from UK Biobank (https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/contact-us) through application to the NIHR BioResource for GLAD (https://bioresource.nihr.ac.uk/using-our-bioresource/academic-and-clinical-researchers/apply-for-bioresource-data/) and through the PROTECT data team (https://medicine.exeter.ac.uk/clinical-biomedical/research/protect/). The Alzheimer’s disease GWAS summary statistics used in this study are publically available through the GWAS catalog (https://www.ebi.ac.uk/gwas/efotraits/MONDO_0004975). GWAS summary statistics for the Wightman et al. GWAS excluding the UK Biobank are available at https://vu.data.surfsara.nl/index.php/s/LGjeIk6phQ6zw8I. For clinical and broad depression, summary statistics are available through the Psychiatric Genomic Consortium (https://pgc.unc.edu). eQTL summary datasets used in SMR analysis from Lloyd-Jones et al.100 and PsychENCODE101 can be obtained from the website of the Yang laboratory (https://yanglab.westlake.edu.cn/software/smr/#eQTLsummarydata). This study has been pre-registered on the Open Science Framework (https://osf.io/94q35/?view_only=e77f72d4100d47eea7f3ef07dfa9c059).
Code availability
Code for performing these analyses has been deposited on GitHub (https://github.com/lpgilchrist/PHQ-9_AD_genetic_overlap_project). This study made use of the following publicly available analysis software: CAUSE (https://jean997.github.io/cause/index.html); coloc (https://chr1swallace.github.io/coloc/); COLOC-reporter (https://github.com/ThomasPSpargo/COLOC-reporter); FUMA GWAS (https://fuma.ctglab.nl); HDL (https://github.com/zhenin/HDL); LAVA (https://github.com/josefin-werme/LAVA); LDSC (https://github.com/bulik/ldsc); MegaPRS (https://dougspeed.com/megaprs/); METAL (https://genome.sph.umich.edu/wiki/METAL_Documentation); MTAG (https://github.com/JonJala/mtag); MungeSumstats (https://github.com/Al-Murphy/MungeSumstats); REGENIE (https://rgcgithub.github.io/regenie/); SMR (https://yanglab.westlake.edu.cn/software/smr/); susieR (https://stephenslab.github.io/susieR/index.html); TwoSampleMR (https://mrcieu.github.io/TwoSampleMR/).
References
Livingston, G. et al. Dementia prevention, intervention and care: 2020 report of the Lancet Commission. Lancet 396, 413–446 (2020).
Yang, W. et al. Association of life-course depression with the risk of dementia in late life: a nationwide twin study. Alzheimers Dement. 17, 1383–1390 (2021).
Richmond-Rakerd, L. S., D’Souza, S., Milne, B. J., Caspi, A. & Moffitt, T. E. Longitudinal associations of mental disorders with dementia: 30-year analysis of 1.7 million New Zealand citizens. JAMA Psychiatry 79, 333–340 (2022).
Yang, L. et al. Depression, depression treatments and risk of incident dementia: a prospective cohort study of 354,313 participants. Biol. Psychiatry https://doi.org/10.1016/j.biopsych.2022.08.026 (2022).
GBD 2016 Dementia Collaborators Global, regional and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 88–106 (2019).
Dafsari, F. S. & Jessen, F. Depression—an underrecognized target for prevention of dementia in Alzheimer’s disease. Transl. Psychiatry 10, 160 (2020).
Corfield, E. C., Yang, Y., Martin, N. G. & Nyholt, D. R. A continuum of genetic liability for minor and major depression. Transl. Psychiatry 7, e1131 (2017).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Levey, D. F. et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat. Neurosci. 24, 954–963 (2021).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Gibson, J. et al. Assessing the presence of shared genetic architecture between Alzheimer’s disease and major depressive disorder using genome-wide association data. Transl. Psychiatry 7, e1094 (2017).
Andrews, S. J. et al. Causal associations between modifiable risk factors and the Alzheimer’s phenome. Ann. Neurol. 89, 54–65 (2021).
Santos, F. C. D., Mendes-Silva, A. P., Nikolova, Y. C., Sibille, E. & Diniz, B. S. Genetic overlap between major depression, bipolar disorder and Alzheimer’s disease. Preprint at medRxiv https://doi.org/10.1101/2021.05.01.21256220 (2021).
Monereo-Sánchez, J. et al. Genetic overlap between Alzheimer’s disease and depression mapped onto the brain. Front. Neurosci. 15, 653130 (2021).
Harerimana, N. V. et al. Genetic evidence supporting a causal role of depression in Alzheimer’s disease. Biol. Psychiatry 92, 25–33 (2022).
Diagnostic and Statistical Manual of Mental Disorders (American Psychiatric Association, 2013); https://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596
Fried, E. I. & Nesse, R. M. Depression is not a consistent syndrome: an investigation of unique symptom patterns in the STAR*D study. J. Affect. Disord. 172, 96–102 (2015).
Cai, N., Choi, K. W. & Fried, E. I. Reviewing the genetics of heterogeneity in depression: operationalizations, manifestations and etiologies. Hum. Mol. Genet. 29, R10–R18 (2020).
Thorp, J. G. et al. Genetic heterogeneity in self-reported depressive symptoms identified through genetic analyses of the PHQ-9. Psychol. Med. 50, 2385–2396 (2020).
Lee, J. R. et al. Anhedonia and dysphoria are differentially associated with the risk of dementia in the cognitively normal elderly individuals: a prospective cohort study. Psychiatry Investig. 16, 575–580 (2019).
Vaquero-Puyuelo, D. et al. Anhedonia as a potential risk factor of Alzheimer’s disease in a community-dwelling elderly sample: results from the ZARADEMP Project. Int. J. Environ. Res. Public Health 18, 1370 (2021).
Ikeda, M., Brown, J., Holland, A. J., Fukuhara, R. & Hodges, J. R. Changes in appetite, food preference and eating habits in frontotemporal dementia and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 73, 371–376 (2002).
Bailon, O., Roussel, M., Boucart, M., Krystkowiak, P. & Godefroy, O. Psychomotor slowing in mild cognitive impairment, Alzheimer’s disease and Lewy body dementia: mechanisms and diagnostic value. Dement. Geriatr. Cogn. Disord. 29, 388–396 (2010).
Koren, T., Fisher, E., Webster, L., Livingston, G. & Rapaport, P. Prevalence of sleep disturbances in people with dementia living in the community: a systematic review and meta-analysis. Ageing Res. Rev. 83, 101782 (2023).
Escott-Price, V. & Hardy, J. Genome-wide association studies for Alzheimer’s disease: bigger is not always better. Brain Commun. 4, fcac125 (2022).
Marioni, R. E. et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 99 (2018).
Liu, H. et al. Mendelian randomization highlights significant difference and genetic heterogeneity in clinically diagnosed Alzheimer’s disease GWAS and self-report proxy phenotype GWAX. Alzheimers Res. Ther. 14, 17 (2022).
European Alzheimer’s & Dementia Biobank Mendelian Randomization (EADB-MR) Collaboration Genetic associations between modifiable risk factors and Alzheimer disease. JAMA Netw. Open 6, e2313734 (2023).
Davies, M. R. et al. The Genetic Links to Anxiety and Depression (GLAD) Study: online recruitment into the largest recontactable study of depression and anxiety. Behav. Res. Ther. 123, 103503 (2019).
Creese, B. et al. Genetic risk for Alzheimer’s disease, cognition, and mild behavioral impairment in healthy older adults. Alzheimers Dement. (Amst.) 13, e12164 (2021).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Wightman, D. P. et al. The genetic overlap between Alzheimer’s disease, amyotrophic lateral sclerosis, Lewy body dementia and Parkinson’s disease. Neurobiol. Aging 127, 99–112 (2023).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Bulik-Sullivan, B. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Werme, J., van der Sluis, S., Posthuma, D. & de Leeuw, C. A. An integrated framework for local genetic correlation analysis. Nat. Genet. 54, 274–282 (2022).
Spargo, T. P. et al. Statistical examination of shared loci in neuropsychiatric diseases using genome-wide association study summary statistics. eLife 12, RP88768 (2023).
Voskobiynyk, Y. et al. Alzheimer’s disease risk gene BIN1 induces tau-dependent network hyperexcitability. eLife 9, e57354 (2020).
Lambert, E. et al. The Alzheimer susceptibility gene BIN1 induces isoform-dependent neurotoxicity through early endosome defects. Acta Neuropathol. Commun. 10, 4 (2022).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Morrison, J., Knoblauch, N., Marcus, J. H., Stephens, M. & He, X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat. Genet. 52, 740–747 (2020).
Dashti, H. S. et al. Genetic determinants of daytime napping and effects on cardiometabolic health. Nat. Commun. 12, 900 (2021).
Jansen, P. R. et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet. 51, 394–403 (2019).
Hammerschlag, A. R. et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat. Genet. 49, 1584–1592 (2017).
Loos, R. J. F. & Yeo, G. S. H. The genetics of obesity: from discovery to biology. Nat. Rev. Genet. 23, 120–133 (2022).
Zarza-Rebollo, J. A., Molina, E. & Rivera, M. The role of the FTO gene in the relationship between depression and obesity. A systematic review. Neurosci. Biobehav. Rev. 127, 630–637 (2021).
González, L. M. et al. Variants in the obesity-linked FTO gene locus modulates psychopathological features of patients with anorexia nervosa. Gene 783, 145572 (2021).
Li, Y. et al. Cross-ancestry genom e-wide association study and systems-level integrative analyses implicate new risk genes and therapeutic targets for depression. Preprint at medRxiv https://doi.org/10.1101/2023.02.24.23286411 (2023).
Dall’Aglio, L., Lewis, C. M. & Pain, O. Delineating the genetic component of gene expression in major depression. Biol. Psychiatry 89, 627–636 (2021).
Fabbri, C., Pain, O., Hagenaars, S. P., Lewis, C. M. & Serretti, A. Transcriptome-wide association study of treatment-resistant depression and depression subtypes for drug repurposing. Neuropsychopharmacology 46, 1821–1829 (2021).
Koskinen, M.-K. & Hovatta, I. Genetic insights into the neurobiology of anxiety. Trends Neurosci. 46, 318–331 (2023).
Nagel, M. et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 50, 920–927 (2018).
Mallard, T. T. et al. Multivariate GWAS of psychiatric disorders and their cardinal symptoms reveal two dimensions of cross-cutting genetic liabilities. Cell Genomics 2, 100140 (2022).
Lüningschrör, P. et al. The FTLD risk factor TMEM106B regulates the transport of lysosomes at the axon initial segment of motoneurons. Cell Rep. 30, 3506–3519.e6 (2020).
Van Deerlin, V. M. et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239 (2010).
Feng, T., Lacrampe, A. & Hu, F. Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol. 141, 327–339 (2021).
Perneel, J. & Rademakers, R. Identification of TMEM106B amyloid fibrils provides an updated view of TMEM106B biology in health and disease. Acta Neuropathol. 144, 807–819 (2022).
Vass, R. et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 121, 373–380 (2011).
Shafit-Zagardo, B. et al. TMEM106B puncta is increased in multiple sclerosis plaques, and reduced protein in mice results in delayed lipid clearance following CNS injury. Cells 12, 1734 (2023).
Hong, S. et al. TMEM106B and CPOX are genetic determinants of cerebrospinal fluid Alzheimer’s disease biomarker levels. Alzheimers Dement. 17, 1628–1640 (2021).
Dhiman, K. et al. Cerebrospinal fluid neurofilament light concentration predicts brain atrophy and cognition in Alzheimer’s disease. Alzheimers Dement. (Amst.) 12, e12005 (2020).
Chen, M.-H. et al. Neurofilament light chain is a novel biomarker for major depression and related executive dysfunction. Int. J. Neuropsychopharmacol. 25, 99–105 (2022).
Desai, R. et al. Examining the Lancet Commission risk factors for dementia using Mendelian randomisation. BMJ Ment. Health 26, e300555 (2023).
Chesney, E., Goodwin, G. M. & Fazel, S. Risks of all-cause and suicide mortality in mental disorders: a meta-review. World Psychiatry 13, 153–160 (2014).
Fry, A. et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am. J. Epidemiol. 186, 1026–1034 (2017).
Huang, L. et al. Polygenic analyses show important differences between major depressive disorder symptoms measured using various instruments. Biol. Psychiatry 95, 1110–1121 (2024).
Brommelhoff, J. A. et al. Depression as a risk factor or prodromal feature for dementia? Findings in a population-based sample of Swedish twins. Psychol. Aging 24, 373–384 (2009).
Creese, B. & Ismail, Z. Mild behavioral impairment: measurement and clinical correlates of a novel marker of preclinical Alzheimer’s disease. Alzheimers Res. Ther. 14, 2 (2022).
DeMichele-Sweet, M. A. A. et al. Genome-wide association identifies the first risk loci for psychosis in Alzheimer disease. Mol. Psychiatry 26, 5797–5811 (2021).
Kroenke, K., Spitzer, R. L. & Williams, J. B. The PHQ-9: validity of a brief depression severity measure. J. Gen. Intern. Med. 16, 606–613 (2001).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Privé, F., Luu, K., Blum, M. G. B., McGrath, J. J. & Vilhjálmsson, B. J. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics 36, 4449–4457 (2020).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Chou, W.-C. et al. A combined reference panel from the 1,000 Genomes and UK10K projects improved rare variant imputation in European and Chinese samples. Sci. Rep. 6, 39313 (2016).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Murphy, A. E., Schilder, B. M. & Skene, N. G. MungeSumstats: a bioconductor package for the standardisation and quality control of many GWAS summary statistics. Bioinformatics https://doi.org/10.1093/bioinformatics/btab665 (2021).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Jaffe, A. E. et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci. 21, 1117–1125 (2018).
Wang, D. et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464 (2018).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Westra, H.-J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Zhernakova, D. V. et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 49, 139–145 (2017).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Buil, A. et al. Gene–gene and gene–environment interactions detected by transcriptome sequence analysis in twins. Nat. Genet. 47, 88–91 (2015).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017).
Ning, Z., Pawitan, Y. & Shen, X. High-definition likelihood inference of genetic correlations across human complex traits. Nat. Genet. 52, 859–864 (2020).
Zou, Y., Carbonetto, P., Wang, G. & Stephens, M. Fine-mapping from summary data with the ‘Sum of Single Effects’ model. PLoS Genet. 18, e1010299 (2022).
Wallace, C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 17, e1009440 (2021).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Lloyd-Jones, L. R. et al. The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet. 100, 228–237 (2017).
Gandal, M. J. et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia and bipolar disorder. Science 362, eaat8127 (2018).
Sanderson, E. et al. Mendelian randomization. Nat. Rev. Methods Primers 2, 6 (2022).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 40, 597–608 (2016).
Hemani, G., Bowden, J. & Davey Smith, G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 27, R195–R208 (2018).
Slob, E. A. W. & Burgess, S. A comparison of robust Mendelian randomization methods using summary data. Genet. Epidemiol. 44, 313–329 (2020).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Hartwig, F. P., Davey Smith, G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998 (2017).
Verbanck, M., Chen, C.-Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389 (2017).
Burgess, S., Bowden, J., Fall, T., Ingelsson, E. & Thompson, S. G. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology 28, 30–42 (2017).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. C., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Bowden, J. et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int. J. Epidemiol. 45, 1961–1974 (2016).
Liu, S., Liu, J., Weng, R., Gu, X. & Zhong, Z. Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc. Disord. 19, 213 (2019).
Xuan, L. et al. Type 2 diabetes is causally associated with depression: a Mendelian randomization analysis. Front. Med. 12, 678–687 (2018).
Lord, J. et al. Mendelian randomization identifies blood metabolites previously linked to midlife cognition as causal candidates in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 118, e2009808118 (2021).
Choi, S. W., Mak, T. S.-H. & O’Reilly, P. F. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 15, 2759–2772 (2020).
Zhang, Q., Privé, F., Vilhjálmsson, B. & Speed, D. Improved genetic prediction of complex traits from individual-level data or summary statistics. Nat. Commun. 12, 4192 (2021).
Lovestone, S. et al. AddNeuroMed—the European collaboration for the discovery of novel biomarkers for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1180, 36–46 (2009).
Lord, J. et al. Disentangling independent and mediated causal relationships between blood metabolites, cognitive factors and Alzheimer’s disease. Biol. Psychiatry Global Open Sci. 2, 167–179 (2022).
Gilchrist, L. Summary statistics for PHQ-9 items from ‘Depression symptom-specific genetic associations in clinically diagnosed and proxy case Alzheimer’s disease’. Zenodo https://doi.org/10.5281/zenodo.13828101 (2023).
Acknowledgements
L.G. is funded by the King’s College London DRIVE-Health Centre for Doctoral Training and the Perron Institute for Neurological and Translational Science. P.P. is funded by Alzheimer’s Research UK. S.K. is funded by MSWA and the Perron Institute. H.L.D. acknowledges funding from the Economic and Social Research Council (ESRC). D.M.H. is supported by a Sir Henry Wellcome Postdoctoral Fellowship (ref. 213674/Z/18/Z). B.N.A. acknowledges funding from an NIHR pre-doctoral fellowship (NIHR301067). A.I. is funded by the Motor Neurone Disease Association (MNDA), MND Scotland, Darby Rimmer MND Foundation, Rosetrees Trust, Alzheimer’s Research UK, Spastic Paraplegia Foundation, LifeArc and The NIHR Maudsley Biomedical Research Centre. T.P.S. acknowledges funding from the MNDA. This Article represents independent research that was part funded by the NIHR Maudsley Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. This research was conducted using the UK Biobank Resource under application no. 18177. We thank the UK Biobank Team for collecting the data and making it available. We also thank the UK Biobank participants. We thank the GLAD Study volunteers for their participation, and gratefully acknowledge the NIHR BioResource centers, NHS Trusts and staff for their contribution. We thank the National Institute for Health Research, NHS Blood and Transplant, and Health Data Research UK as part of the Digital Innovation Hub Programme. This study presents independent research funded by the NIHR Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. Further information can be found at https://www.maudsleybrc.nihr.ac.uk/facilities/bioresource/. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, the HSC R&D Division, King’s College London or the Department of Health and Social Care. The PROTECT study was funded/supported by the National Institute of Health and Care Research Exeter Biomedical Research Centre. PROTECT genetic data were funded in part by the University of Exeter through the MRC Proximity to Discovery: Industry Engagement Fund (External Collaboration, Innovation and Entrepreneurism: Translational Medicine in Exeter 2 (EXCITEME2) ref. MC_PC_17189). Genotyping was performed at deCODE Genetics.
As data used in this study was obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database, the ADNI investigators contributed to the conception of the sample and acquisition of the data, but were not participants in other parts of this study, such as conceptualization, data analysis or writing. A full acknowledgment list of ADNI investigators is available at https://adni.loni.usc.edu/wp-content/uploads/2024/07/ADNI-Acknowledgement-List_July2024.pdf. Details on ADNI data access can be found at https://adni.loni.usc.edu/data-samples/adni-data/#AccessData. Data collection and sharing for this project was funded by the ADNI (National Institutes of Health grant U01 AG024904) and DOD ADNI (Department of Defense award no. W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai, Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research are providing funds to support ADNI clinical sites in Canada. Private-sector contributions are facilitated by the Foundation for the National Institutes of Health (https://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Similarly, data were obtained from the GERAD1 Consortium, and GERAD1 investigators contributed to the conception of the cohort and acquisition of the data, but did not participate in the conceptualization, analysis or writing of this study. A full list of GERAD1 collaborators can be found in Supplementary Section 3. For GERAD1 data, Cardiff University was supported by the Wellcome Trust, MRC, ARUK and the Welsh Assembly Government. Cambridge University and King’s College London acknowledge support from the MRC. ARUK supported sample collections at the South West Dementia Bank and the Universities of Nottingham, Manchester and Belfast. The Belfast group acknowledges support from the Alzheimer’s Society, Ulster Garden Villages, Northern Ireland R&D Office and the Royal College of Physicians/Dunhill Medical Trust. The MRC and Mercer’s Institute for Research on Ageing supported the Trinity College group. The South West Dementia Brain Bank acknowledges support from Bristol Research into Alzheimer’s and Care of the Elderly. The Charles Wolfson Charitable Trust supported the OPTIMA group. Washington University was funded by National Institutes of Health (NIH) grants, the Barnes Jewish Foundation and the Charles and Joanne Knight Alzheimer’s Research Initiative. Patient recruitment for the MRC Prion Unit/University College London(UCL) Department of Neurodegenerative Disease collection was supported by the UCL Hospitals/UCL Biomedical Centre and NIHR Queen Square Dementia Biomedical Research Unit. LASER-AD was funded by Lundbeck SA. The Bonn group was supported by the German Federal Ministry of Education and Research, Competence Network Dementia and Competence Network Degenerative Dementia, and Alfried Krupp von Bohlen und Halbach-Stiftung. The Genetic and Environmental Risk for Alzheimer’s Disease (GERAD1) Consortium also used samples ascertained by the National Institute of Mental Health Alzheimer’s Disease Genetics Initiative. The i-Select chip was funded by the French National Foundation on Alzheimer’s disease and related disorders. The European Alzheimer’s Disease Initiative was supported by a LABEX (Laboratory of Excellence Program Investment for the Future) DISTALZ grant, the Institut National de la Santé et de la Recherche Médicale, Institut Pasteur de Lille, Université de Lille 2 and the Lille University Hospital. The GERAD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium was supported by the MRC (grant no. 503480), ARUK (grant no. 503176), the Wellcome Trust (grant no. 082604/2/07/Z) and the German Federal Ministry of Education and Research (Competence Network Dementia grant nos. 01GI0102, 01GI0711 and 01GI0420). The Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium was partly supported by NIH/NIA grant no. R01 AG033193; NIA grant no. AG081220; AGES contract no. N01-AG-12100; National Heart, Lung and Blood Institute grant no. R01 HL105756; the Icelandic Heart Association; and the Erasmus Medical Center and Erasmus University. The Alzheimer’s Disease Genetics Consortium was supported by NIH/NIA grant nos. U01 AG032984, U24 AG021886 and U01 AG016976; and Alzheimer’s Association grant no. ADGC-10-196728.
ANM data are accessible via Synapse (https://www.synapse.org/Synapse:syn22252881). The AddNeuroMed study was supported by InnoMed (Innovative Medicines in Europe)—an Integrated Project funded by the European Union of the Sixth Framework program priority FP6-2004-LIFESCIHEALTH-5, Life Sciences, Genomics and Biotechnology for Health.
Compensation was not provided for participants in any of the above studies.
We also thank and acknowledge the contribution and use of the CREATE high-performance computing cluster at King’s College London (King’s Computational Research, Engineering and Technology Environment (CREATE); retrieved 23 May 2023 from https://doi.org/10.18742/rnvf-m076).
The analysis flowchart in Fig. 1 was created and licensed in BioRender (https://www.BioRender.com/z76j516).
The ethics committee/IRB of King’s College London gave ethical approval for this work. Ethical approval for the UK Biobank study was granted by the National Information Governance Board for Health and Social Care and the NHS North West Multicentre Research Ethics Committee (11/NW/0382). Data access permission was granted under UK Biobank application 18177. Written informed consent was obtained from all participants by UK Biobank. The GLAD Study was approved by the London–Fulham Research Ethics Committee on 21 August 2018 (REC ref. 18/LO/1218) following a full review by the committee. The NIHR BioResource has been approved as a Research Tissue Bank by the East of England–Cambridge Central Committee (REC ref. 17/EE/0025). The PROTECT study received ethical approval from the UK London Bridge National Research Ethics Committee (ref. 13/LO/1578). Data were obtained from ADNI, AddNeuroMed and GERAD1 following formal request to each consortia. Permission was granted for the use of data.
For the purposes of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any accepted author manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
Formal analysis and data visualization were conducted by L.G., with the study conceptualized under the supervision of S.K., C.M.L. and P.P. T.P.S., R.E.G., J.R.I.C., D.M.H., J.G.T. and A.I. provided conceptual and methodological guidance and computing resources. J.L. performed data curation and provided resources for ADNI, ANM and GERAD. R.A., A.C., C.B., H.B., B.C., D.A. and A.H. collected and curated data for PROTECT and provided resource access for this study. L.V. collected and curated data for ANM. B.N.A., H.L.D., J.M., A.R.t.K., M.R.D., C.H., S.B., S.H.L., H.R., C.C., S.K., C.M.M. and G.K. performed data collection and curation for GLAD under the supervision of T.C.E. and G.B., who provided data resource access for this study. The original draft of this Article was written by L.G. with initial editing from S.K., C.M.L. and P.P. All co-authors reviewed the manuscript and provided further guidance on editing.
Corresponding authors
Ethics declarations
Competing interests
C.M.L. is on the Scientific Advisory Board of Myriad Neuroscience and has received honoraria for consultancy from UCB. The other authors declare no competing interests.
Peer review
Peer review information
Nature Mental Health thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Materials 1–3.
Supplementary Tables 1–24 (download XLSX )
This file contains all supplementary tables for the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gilchrist, L., Spargo, T.P., Green, R.E. et al. Depression symptom-specific genetic associations in clinically diagnosed and proxy case Alzheimer’s disease. Nat. Mental Health 3, 212–228 (2025). https://doi.org/10.1038/s44220-024-00369-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44220-024-00369-0
