
Abstract
Expansion microscopy is a groundbreaking technique that enables nanoscale imaging of biological specimens using standard optical microscopes. By embedding specimens in swellable hydrogels, it achieves sub-diffraction resolution. Compatible with various tissue types, it offers 3D, multi-colour visualisation of cellular and sub-cellular structures. While challenges remain, like sample isotropy and preservation of molecular integrity, expansion microscopy is a transformative tool for cell and neurobiology. Here, we discuss its potential for virology.
Similar content being viewed by others

Principles of expansion microscopy
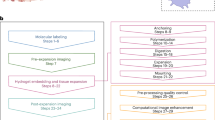
The standard expansion microscopy protocol involves several key steps (Fig. 1): (1) fixation of the specimen, (2) labelling of biomolecules of interest, (3) anchoring chemical linkers to biomolecules, (4) embedding in a hydrogel, (5) homogenisation through enzymatic digestion or denaturation of the tissue, and (6) subsequent expansion by immersion in water or an expansion buffer. These steps result in the isotropic enlargement of the sample, preserving the native tissue architecture while enabling visualisation at a significantly higher resolution - typically on the order of 70 nm and less. The exact resolution attained depends on the specific expansion protocol, as different methods yield varying expansion factors (commonly 4x, but protocols achieving 10x and more have been developed) and may also involve iterative rounds of expansion.

Schematic overview of the expansion microscopy workflow. The workflow consists of several consecutive steps that need to be adjusted and optimised with regard to the nature of the sample and the target of interest.
Several practical considerations must be made regarding each individual step. Based on the specific structure of interest, the most suitable reagent should be chosen for fixation. These are usually the same as commonly used for immunocytochemistry, such as methanol and aldehydes (e.g., paraformaldehyde, glutaraldehyde, glyoxal). Cryo-fixation provides the most accurate preservation of biological samples in their native state1; however, it requires more time and skilled handling than conventional chemical fixation methods.
The selection of fluorescent labels is crucial: some free-radical-induced dye degradation is inevitable during the polymerisation step of forming the hydrogel backbone, but some dyes are more photo- and chemically stable than others. For example, rhodamine-based dyes (e.g., Atto647N) show much better stability than cyanine dyes (e.g., Alexa Fluor 647)2 which should best be avoided for expansion microscopy. Autofluorescent and self-labelling proteins can stay intact if digestion is not too prolonged, so the digestion time must be adjusted for each sample type like single cells or tissue sections3,4,5,6. Although enzymatic homogenisation generally improves expansion, excessive digestion can degrade these proteins, especially autofluorescent proteins and SNAP-tags. There are further strategies to preserve and enhance fluorescence signals in expanded samples. Label retention might be improved by using small, inert trifunctional probes that bind the target, anchor to the hydrogel and carry a fluorophore5. Signal amplification can be achieved using fluorescent nanobodies or tyramide signal amplification7 to boost fluorescence intensity. Post-expansion labelling can improve labelling efficiency due to better epitope accessibility8, often using treatment with sodium dodecyl sulfate (SDS) for long durations and/or high temperature treatment9,10 instead of enzymatic digestion.
The anchoring step requires thorough incubation of the sample with the linker solution to ensure uniform distribution within the sample. Slow polymerisation at lower temperatures during embedding promotes uniform reactions and homogeneity. For expansion microscopy, labelling and anchoring strategies must be compatible with the functional groups present on the target molecules. Proteins are typically anchored to the hydrogel matrix using N-hydroxysuccinimidyl (NHS) ester-functionalised linkers like 6-((acryloyl)amino)hexanoic acid (acryloyl-X)4 or methacrylic acid3. The NHS ester moiety reacts with the primary amines in the lysine side chains whereas the polymerizable handle of the linker covalently binds to the hydrogel during polymerisation. Protein lLabels - such as antibodies, streptavidin, or genetically encoded tags like self-labelling or fluorescent proteins - are covalently anchored to the matrix along with their targets. Non-specific lysine staining with NHS-activated fluorescent dyes enables visualisation of protein densities (ultrastructure) similar to electron microscopy11. RNA and DNA can be visualised using fluorescence in situ hybridisation (FISH), which employs short fluorescent oligonucleotides that hybridise to specific RNA or DNA sequences. The guanine-reactive Label-IT reagent can anchor nucleic acid targets as well as their probes and through functionalisation with a polymerizable handle covalently attach them to the hydrogel12. DNA can be labelled non-specifically with high-affinity intercalating dyes (e.g., DAPI, SYTO dyes) for high-resolution imaging of chromatin and sub-nuclear structures. Lipid labelling and anchoring is more challenging due to membrane disruption by detergents which are needed to permeabilise samples for monomer infiltration. However, it can be achieved using mild detergents combined with amine-13,14, azide-15,16 or alkyne-17 (for click-chemistry) as well as biotin16 -functionalised lipid probes that allow coupling to the gel matrix. Universal linkers such as methacrolein9 or glycidyl methacrylate18 are designed to work with a broad range of targets (proteins, RNA, lipids) with minimal or no need for target-specific chemistry. More specialised multifunctional linkers integrate targeting, anchoring, and detection in one molecule to improve multiplexing capacities14,19. Another strategy uses various clickable probes in combination with biotin click-labelling and fluorescent streptavidin staining to target a range of biomolecules (lipids, glycans, proteins, DNA, RNA) using conventional protein retention hydrogels17.
Assuming sufficient labelling density, the resolution improvement in expansion microscopy depends mainly on the expansion factor, which is defined by the gel recipe and describes the size ratio between the expanded and original sample. Since its original description20, many variations of expansion microscopy protocols have been developed to further increase the expansion factor and hence the effective image resolution21. Two major strategies have been developed to extend the original protocol. The first approach focuses on modifying the polymer network to enable greater expansion. By reducing cross-linker concentration in bis-acrylamide-cross-linked hydrogels typically used in the standard 4-5x expansion protocols, the expansion factor can be increased to around 10x9,13. Achieving such high expansion factors necessitates enhancing structural integrity. Therefore, mechanically robust and elastic hydrogels composed of N,N-dimethylacrylamide and sodium acrylate have been developed. By removing oxygen from the gelation solution, performing gelation in an oxygen-depleted environment, and optimising both gelation time and initiator concentration, expansion factors between 10x and 20x were achieved with this monomer chemistry22,23,24,25. The second approach increases the expansion factor by using multiple rounds of expansion, a method called iterative expansion microscopy26. The first gel(s) are coupled to a final gel, which is expanded after disrupting the first gel(s). Several iterative approaches have demonstrated electron microscopy-like resolution using measured expansion factors ranging from 15 to 20x11,26,27.
While expansion microscopy can achieve substantial magnification, practical limitations might arise due to sample distortion. Local inhomogeneities and deformations in the polyacrylate or other hydrogel matrix might lead to non-isotropic expansion, especially beyond expansion factors of 4-5x. Heat treatment24 or alternative enzymes for digestion7 might improve homogenisation. Isotropic expansion can be verified using biological rulers such as microtubules, nuclear pore complexes (NPCs) and centrioles, which are also commonly measured to determine the expansion factor. Isotropy of expansion might e.g. be measured as the ratio between centriole length and diameter28 or as a localisation error from comparing the same structure before and after expansion, most often microtubules. By adapting open-source software29, post-expansion images can first be aligned to pre-expansion ones using translation, rotation, and scaling corrections, followed by non-rigid registration to account for deformations like stretching and bending. The deformation field generated can then be used to calculate localisation errors3,4,30. Artificial molecular rulers such as a gel-embedded reference grid have also been developed for quality control and to intrinsically calibrate the polymer matrix31. This approach allows distortions to be corrected without pre-expansion reference images through software tools using landmark-based deformable image alignment like BigWarp32 and simultaneously embeds a coordinate system that facilitates sample navigation.
Swollen gels can be stored short-term (up to about a week) in pure water at 4 °C in the dark, which maintains full expansion but leaves them prone to drying and degradation. For longer-term storage, gels are usually shrunk in buffers with sodium azide at 4 °C; HEPES buffer is preferred to preserve fluorescence. Before imaging, gels are re-expanded in water. Less commonly, gels can be stored in protective agents and frozen (e.g., 50% glycerol for a sample of algae33), but freeze–thaw cycles and residual glycerol may hinder re-expansion and increase background.
A wide range of expansion microscopy protocols exists, differing in their polymer chemistry, anchoring strategies, digestion conditions, and achievable expansion factors. This diversity can make the methodological landscape difficult to navigate, especially for new users. For this reason, it is useful to begin with the original, basic expansion microscopy workflow20,34,35 to become familiar with the fundamental steps - anchoring, gelation, digestion, and expansion - before moving on to specialised variants. To help readers orient themselves and choose the most appropriate protocol for their application, we recommend consulting recent comprehensive reviews on expansion microscopy21,36. These reviews provide clear overviews of available variants (e.g., Magnify9, X1022, TREx13, BOOST37), discuss their strengths and limitations, summarise advances and offer a solid foundation for selecting and optimising an expansion microscopy protocol for specific experimental needs.
Imaging strategies for expanded samples
A central parameter in expansion microscopy is the expansion factor. In first approximation, the optical resolution improves linearly with the inverse of the expansion factor. With standard fourfold expansion microscopy protocols in combination with conventional optical microscopes such as a confocal microscope (diffraction limit ~300 nm), a resolution improvement of down to ~75 nm is achievable. More advanced protocols with expansion factors of 10-20 should attain a resolution improvement in the range of 15-30 nm in theory, achieving resolution capabilities closer to those offered by electron microscopy (Fig. 2).

Theoretical resolution limit calculated for a confocal microscope setup equipped with a 1.2 NA water objective lens, with NA = numerical aperture. The Rayleigh resolution limit is defined as 0.61*λ/NA, with λ = emission wavelength. The resolution is dependent on the emission wavelength and, in first approximation, on the expansion factor.
While this theoretical calculation provides a simple and intuitive relationship, in practice, several factors can significantly affect the actual resolution using expansion microscopy. To achieve optimal imaging performance and reach the theoretical optical resolution limit when imaging expanded samples, several factors must be carefully considered.
First, the theoretical gain in resolution assumes that the fluorescent label is small relative to the structure of interest. However, commonly used labels (e.g., primary and secondary antibodies) are ~10-20 nm in size, which becomes a significant source of localisation uncertainty when attempting to resolve structures at tens of nanometres. Here, post-expansion instead of pre-expansion labelling leads to an improvement in accuracy. For post-expansion labelling, the linkage error (i.e. the physical separation between the target and the fluorophore) is reduced proportional to the expansion factor since the affinity reagent (e.g., antibodies) does not undergo expansion so that the fluorophore is closer to the target compared to pre-expansion labelling8,9.
Furthermore, sufficient labelling density is required to satisfy the Nyquist sampling criterion (i.e. the minimum sampling that preserves all available resolution). Sparse or uneven labelling may result in incomplete or misleading reconstructions, regardless of the expansion factor. Post-labelling can also help in this case as it improves labelling efficiency.
Optimal resolution and image quality in light microscopy also depend on minimising refractive index mismatch between the sample and the immersion medium. Expansion microscopy reduces local refractive index heterogeneity and light scattering through its inherent sample clearing38 and decrowding39 properties. This also improves sample transparency and hence imaging depth, especially important for big samples. Due to the water-based environment of expanded samples, it is usually recommended to use water-immersion objectives. High numerical aperture (NA > 1.0) objectives increase the resolution but also improve fluorescence light gathering due to their large collection angles. Efficient light detection is essential, as the expansion process lowers fluorophore density by decrowding the samples molecules, thereby decreasing the overall brightness. A beneficial side effect though can be an improvement of the signal-to-noise ratio due to reduced crowding and background fluorescence39. The limited working distance of typical high-NA objectives however constrains the maximum imaging depth - an important consideration for expanded samples - and can introduce mechanical interference or compromise focus precision.
Deciding which microscope is most appropriate for different samples involves not only assessing resolution and imaging depth, but also imaging speed, optical sectioning capability, sample compatibility, user-friendliness, photobleaching and field of view (Fig. 3a). Standard microscopy techniques, such as widefield and confocal microscopy, are commonly used for imaging expanded samples due to their accessibility (Fig. 3b and c). Typical inverted configurations however limit imaging depth, restricting observations mainly to expanded single cell and tissue layers.

Trade-off among different microscope techniques for imaging physically expanded, biological samples and schematic illustration of their beam path geometries. Different imaging techniques offer distinct advantages but also have some limitations (a). Shown are the optical configurations of (b) widefield microscopy, c confocal laser scanning microscopy, d light sheet fluorescence microscopy, and e oblique plane microscopy. For each technique, the excitation (blue) and detection paths (yellow) are indicated, highlighting differences in excitation geometry, optical sectioning and image generation.
Widefield microscopy offers rapid, multi-channel imaging with minimal excitation power requirements. However, its lack of optical sectioning leads to the detection of out-of-focus fluorescence, resulting in low contrast and signal-to-noise ratio. Deconvolution algorithms are often applied post-acquisition to enhance contrast and resolution40. These algorithms require user-informed application based on the relevant image acquisition parameters. Deconvolution accuracy depends critically on how well the point spread function (PSF) of the imaging system is known or modelled. If the PSF is mismatched, the algorithm can produce distortions or artifacts. Over-iteration or improper settings can also generate artificial features and mislead quantitative interpretation, while incorrect use may amplify noise and obscure true structures41.
Confocal microscopy, on the other hand, achieves optical sectioning through the use of spatial pinholes that significantly reduce out-of-focus light42. In conventional point-scanning systems, the inherently low acquisition speed can limit throughput. Slow imaging is particularly problematic for expanded samples, as their increased size together with multi-colour imaging requirements and potential need for volumetric acquisition can substantially prolong total acquisition time. Spinning disk confocal microscopy offers a higher-speed alternative, but it typically exhibits less efficient out-of-focus light rejection, increased background noise, and lower sensitivity. These factors might be limiting when imaging the inherently dim and diluted fluorescent signals characteristic of expanded samples.
In comparison to these techniques, light sheet and oblique plane microscopy offer some distinct advantages (Fig. 3d and e). They combine the speed of widefield microscopy with the optical sectioning capability of confocal microscopes. Optical sections are generated by illuminating the sample with a thin sheet of light. Low photo-bleaching, highly light-efficient detection and fast volumetric imaging make them in principle ideally suited for imaging expanded gels43,44,45. Light sheet microscopy has typically been employed for volumetric imaging of large samples like organoids or organisms such as zebrafish46. The features of interest in these samples lie in the micrometer range and therefore require only micrometer-level resolution, which conventional light-sheet setups typically provide. However, the axial resolution of light-sheet microscopes can be tuned through sheet thickness and beam profile to reach values as low as ~250 nm47. This makes light sheet microscopy a versatile technique capable of spanning imaging scales from the mesoscopic to the nanoscale.
For light sheet microscopy, illumination and detection light path are separated geometrically (Fig. 3d), usually by mounting two objectives perpendicular to each other around the sample in different geometries dependent on the type of specimen48. Integration with expansion microscopy has proven particularly powerful for mapping of neuron architecture in organoids49 as well as in animal brains43,50,51,52,53,54. A challenge of the optical pathway geometry apart from sample mounting is that either stage or objectives must be repositioned during imaging to maintain focus throughout the entire depth of the sample.
Oblique plane microscopy (OPM) is a variant of light-sheet fluorescence microscopy designed to image adherent cells and samples on flat substrates such as coverslips55,56. It has been demonstrated suitable for expanded samples by imaging a ~ 4x expanded brain organoid57. The light sheet is delivered at an angle through the same high-NA objective used for detection, creating a tilted illumination plane near the coverslip (Fig. 3e). OPM works with conventionally mounted specimens on inverted microscopes and allows high-speed acquisition without moving parts. Although technically challenging, its adaptability to various sample types and sizes, along with advanced variants offering improved resolution, field of view, and modalities, makes it a promising tool to enhance expansion microscopy.
Sample expansion may furthermore be integrated with super-resolution microscopes58,59 to further extend the achievable resolution in biological imaging60,61 (Fig. 4), though the intrinsic restrictions of each method must be taken into account. Through technically advanced setups, often paired with complex data processing, super-resolution microscopes modulate fluorescence emission in space or time to surpass the diffraction limit of optical microscopes. It is possible to reveal the molecular architecture of sub-cellular ultrastructure with near electron microscopic precision (ca. 10-20 nm) by coupling standard expansion microscopy (4-5x) with e.g. stimulated emission depletion (STED)28 or direct stochastic optical reconstruction microscopy (dSTORM)8 (Fig. 4). Recently, it was shown, that protein shapes could be revealed improving the resolution below 10 nm39 by coupling 10x expansion with super-resolution radial fluctuation (SRFF) analysis of the fluorescence signal62,63. Limitations depending on the technique include the need for compatible fluorophores and specialised buffers rather than water - which is required for maximal expansion - as well as motion artifacts and photobleaching caused by long imaging times and the often-high laser powers required. Despite these challenges, the advantages are significant: by combining different methods, it becomes attainable to resolve the structure of viruses whose diameters lie well below the traditional diffraction limit of light microscopes (Fig. 4).

Combination of light microscopy techniques and expansion strategies can improve the resolution below 10 nm. In comparison, physical sizes of selected viruses are indicated. Viruses are positioned on the resolution scale according to their approximate diameter.
Single virus expansion microscopy
Directly visualising the structure of viral particles is essential for advancing our understanding of the viral lifecycle. It informs about how viruses attach to and penetrate host cells, copy and process their genomes and assemble proteins into virions. Identifying structural features of viral particles can help to reveal neutralizing epitopes, guide the design of antigen-based tests and structure-based drug design.
Electron microscopy was and still is the benchmark technique for imaging and analysing virus particles. Its principal strength lies in its exceptionally high resolution at near-atomic level, which surpasses that of any other currently available imaging method. The extremely good resolution, however, comes at a cost: it has low contrast, sample preparation is time-consuming and requires a high level of skill, and there is little, or no, molecular identification of specific proteins. Furthermore, labelling and detection of specific proteins inside large assemblies and cells is challenging with electron microscopy. Fluorescence optical microscopy provides several significant advantages for the study of virus structure and morphology, including high-contrast images, multi-colour acquisition and high specificity through the use of specific fluorescent labels for viral components (capsid and envelope proteins as well as nucleic acids).
Virus particles vary in size across different virus species (Fig. 4). The size of most virions and their sub-structures is below the resolution limit of conventional light microscopes. However, super-resolution microscopes with resolution capabilities of up to 20 nm, using techniques such as stimulated emission depletion (STED) and single molecule localisation microscopy (SMLM), can provide nanoscale insights into virus structure64. The direct imaging of virions with these advanced light microscope methodologies has expanded our scientific knowledge into various human pathogens65,66,67,68,69.
The potential of expansion microscopy for imaging single virions has only just started to be explored. Expansion microscopy has already been employed to examine single virions of herpes simplex virus-1 (HSV-1), Epstein-Barr virus (EBV) and human immunodeficiency virus-1 (HIV). It is possible to visualise the capsid layer around the double-stranded DNA of HSV-1 particles by use of a standard 4x expansion protocol in combination with AiryScan confocal microscopy (an advanced form of confocal microscopy that utilises a specialised detector to improve spatial resolution beyond the diffraction limit) as shown in Fig. 5. Measurement of the tip-to-tip distance of isotropically expanded, antibody-labelled HSV-1 capsids within infected cells yielded an average capsid size of 125.4 nm70, consistent with electron microscopy data71. Using 12x expansion microscopy, the viral architecture of EBV could be resolved and the inner and outer diameter of EBV virions was determined (162 and 76 nm respectively)72, in line with previous electron microscopy data73. Applying the same expansion protocol, sites of HIV-1 genome integration could be visualised by distinguishing monomers and dimers of unspliced viral RNA and co-localisation with the viral Gag structural protein72.

HFF-hTERT cells were infected with HSV-1 (multiplicity of infection (MOI) 2) for 16 h. Pre-expansion images (a–c) show VP26–YFP–labelled viral capsids, with (b) and (c) providing magnified views of regions in (a) and (b), respectively. Post-expansion images (d–f; expansion factor 4.2, protocol according to Chen et al.20) were acquired under the same conditions, with (e) and (f) providing corresponding magnified views of (d) and (e). In the expanded sample, both the capsid structure and the enclosed viral DNA are resolved (f), features that are indistinguishable in the unexpanded state (c).
These examples show that expansion microscopy is ready to be used for detailed examination of virion layers and architecture. The method has a huge advantage since virion architecture can be readily observed within the context of infected cells which is not easy to achieve with electron microscopy. It might be applied to reveal key information about all stages in the viral life cycle, including cell entry, assembly, release, maturation and cell-to-cell transport.
Of course, high enough resolution and localisation accuracy are essential for single virion imaging. For rigid macromolecular assemblies with highly ordered and symmetric architectures - such as viral capsids - it is especially critical that all expansion steps are efficient. This ensures isotropic swelling of the sample and a sufficiently high labelling density to accurately retain structural features. By using an optimised polymer matrix and iterative ~10x expansion, the spherical shape of HSV-1 virions was better preserved and the median spatial error for localising the viral envelope layer was improved from 14.3 to 9.2 nm compared to standard protocols74.
Testing the limits of expansion microscopy, the combination of 20x one-step expansion microscopy and computational super-resolution using videos of temporal fluorophore fluctuations has recently proven that resolution of protein shapes is possible39. Achieving single-protein resolution with standard confocal microscopy would require an expansion greater than 20-fold, ideally in the range of at least 50-fold. By iterating protocols with high expansion factors, it is feasible that expansion factors of 50 or more can be achieved in the near future.
Nanoscale insights into viral infection through expansion microscopy
The rapidly advancing field of viral optical imaging - which encompasses investigations into virus-host interactions, viral transmission, replication, assembly, and egress, as well as the characterisation of viral vaccines and antiviral strategies - continues to evolve and expand (see reviews75,76,77,78). Within this field, expansion microscopy is emerging as a powerful tool to study complex viral processes such as viral replication and host immune evasion. Offering greater accessibility than super-resolution microscopes and fewer limitations related to sample preparation and imaging conditions, expansion microscopy holds significant promise for broad adoption in laboratories.
Expansion microscopy enables high-precision sub-cellular imaging, allowing for detailed quantitative analysis of viral processes. It facilitates for example the measurement of viral particle numbers and co-localisation with host proteins. This enables quantitative measurements with substantially higher confidence compared to conventional light microscopy. Due to its compatibility with multi-spectral imaging, expansion microscopy also allows for an increase in the number of distinguishable labels - from the typical three or four to up to eight and more. This could allow a more detailed investigation into complex biomolecular interaction patterns in the future. Furthermore, the spatial distribution of viral proteins within the host cell and its compartments can be mapped in great detail, which aids e.g. in deciphering mechanisms of viral trafficking and genome delivery. Such capabilities are crucial for advancing our understanding of virus-host interactions at the molecular level.
For several of the most virulent and extensively studied human pathogens, including high-priority viruses of global health concern, expansion microscopy has already been successfully applied to elucidate key aspects of their replication and host interactions.
Hepatitis virus
Expansion microscopy was used to study the exact role of the multifunctional hepatitis C virus (HCV) phosphoprotein NS5A which is essential for HCV RNA replication and virion assembly79. The mechanism by which its phosphorylation controls these functions has been poorly understood. The study uncovered that phosphorylation at serine 225 orchestrates the nanoscale structure and distribution of NS5A assemblies. Applying ~4-5x sample expansion ( ~ 70 nm spatial resolution), puncta size, shape, and cell surface localisation of serine 225-phosphorylated NS5A was visualised and quantified (Fig. 6a). The spatial control of NS5A is essential for forming effective HCV replication complexes and sustaining viral replication - highlighting the value of expansion microscopy in revealing spatial regulation of viral proteins within infected cells.

Representative examples of expansion microscopy in the study of virus biology. a Localisation of the hepatitis C virus (HCV) pS225-NS5A protein (phosphorylated proteoform of the viral NS5A protein) with respect to the total amount of NS5A in Huh7 cells79. Post-expansion confocal images show NS5A and pS225-NS5A (left, scale bar in magnified views: 1 µm) as well as three-dimensional reconstructions of NS5A puncta (right). NS5A is a multifunctional phosphoprotein essential for HCV genome replication and assembly, and expansion microscopy enables nanoscale analysis of its spatial organisation. b Expansion microscopy of HIV-1 assembly sites in HeLa cells84. Maximum intensity projections before (pre-expansion, left) and after expansion (post-expansion, right) reveal nanoscale co-clustering of the viral structural protein Gag with host transmembrane proteins such as PSGL-1, regulated by local enrichment of the acidic phospholipid PIP₂. c Confocal imaging of SARS-CoV-2–infected A549-ACE2 cells at 24 h post-infection. Pre-expansion images (left) show viral nucleocapsid protein (N), spike protein (S), and host stress granule marker G3BP1, while post-expansion images (right) resolve nanoscale co-localisation of N with G3BP1 and S, highlighting the interplay between viral components and host cell structures during infection91. In all post-expansion images, the scale bars correspond to pre-expansion scales. Images are taken from the cited articles under CC BY 4.0 international licenses.
Herpesvirus
Herpesviruses are particularly adept at remodelling their host cell environment to support their replication mechanisms. Studying these processes is fundamental for understanding herpesvirus assembly and egress. During infection with the prototypic herpesvirus herpes simplex virus 1 (HSV-1), especially the nucleus as the site of viral DNA replication, gene expression and capsid assembly undergoes extensive structural reorganisation. Using expansion microscopy, it was precisely mapped how HSV-1 forces chromatin rearrangement to create egress routes for viral capsids80. In a stepwise, time-dependent mechanism, chromatin is first compacted before channels are formed. These low-density interchromatin channels emerge at late infection stage within compacted peripheral chromatin, likely providing pathways for capsid movement. HSV-1 not only remodels chromatin architecture, but also organisation of nucleoli81. High-resolution 3D mapping by expansion microscopy showed furthermore how chromatin redistributes around displaced nucleoli and that key nucleolar proteins are delocalised during infection before relocating together with marginalised chromatin. These rearrangements coincide with impaired rRNA synthesis and processing, suggesting that viral infection actively hijacks nucleolar structure and function. But structural rearrangements also occur in the cytoplasm during HSV-1 infection. Mitochondria, which normally show a reticular network, were observed via expansion microscopy to undergo progressive condensation and collapse toward the perinuclear region during late infection82. This redistribution may support energy demands or help remodel membranes during virus assembly.
Human immunodeficiency virus (HIV)
Expansion microscopy has enabled refined visualisation of HIV-1 replication. A recent study uncovered distinct morphologies of HIV-1 capsid assemblies inside nuclei through ~4x isotropic expansion in multiple cell types: HeLa-derived lines, primary CD4⁺ T-cells, and monocyte-derived macrophages (key reservoirs for HIV-1)83. These structural features were previously obscured by standard immunofluorescence. The findings suggest a wider diversity in nuclear capsid structures than previously observed. This provides fresh insight into post-entry processes such as uncoating and nuclear import - key to deciphering HIV-1 replication strategy. A new mechanism involving lipid composition in HIV-1 assembly was uncovered by showing that the incorporation of host proteins depends on phosphatidylinositol 4,5-bisphosphate (PIP2)-dependent co-clustering with HIV-1 Gag at the plasma membrane84. The acidic phospholipid PIP2 is crucial for recruiting the host transmembrane proteins CD43, PSGL-1, and CD44 into budding virions, likely through electrostatic clustering. When PIP2 is depleted, these proteins are no longer incorporated into HIV particles - even though Gag is still expressed - disrupting virion composition and potentially reducing infectivity. Critically, only through ~4-5x sample expansion could these nanoscale co-clustering events in the range of ~10-100 nm be directly visualised and quantified (Fig. 6b). Expansion microscopy was also employed to develop tools for investigating HIV-1 latency85. By engineering a hybrid sensor, the PromA sequence within the U3 region of the HIV-1 long terminal repeat (LTR) was targeted and validated as a specific and conserved biomarker of infected cells. PromA transcripts are short non-coding RNAs and are transcribed even when HIV is transcriptionally silent. By ~4.5x isotropic expansion, 3D spatial mapping of PromA captured the sub-cellular distribution of HIV-1 RNA. This information may aid in identifying cells that harbour latent HIV, which typically evade immune detection and antiretroviral therapy. Latent reservoirs can be distinguished from actively replicating virus based on their distinct localisation patterns - punctate nuclear clusters versus diffuse signals, respectively.
Influenza virus
Expansion microscopy has enabled high-resolution mapping of host structures involved in influenza virus replication relative to viral components. Detailed imaging using super-resolution microscopes and expansion microscopy revealed thin, F-actin–rich nanotubes (50-200 nm in diameter) connecting influenza A virus (IAV)-infected lung epithelial cells with naïve cells86. IAV does not only use theses nanotubes to transfer viral proteins (e.g., NP, HA) but also viral genomes (vRNPs) to recipient cells, resulting in productive viral replication. These observations indicate that influenza viruses use these intercellular networks to bypass immune defences. By applying ∼4x expansion, the fine-scale localisation of viral proteins to cytoskeletal structures was monitored44. This provided insights into how viral components may associate with microtubules or other cellular networks - information not resolved by conventional confocal microscopy. To understand how IAV enters the host cytoplasm after endocytosis, expansion microscopy was used to explore virus-endosome interactions with enhanced resolution by mapping the spatial separation between virus and membrane87. This quantitative approach demonstrated that in the absence of the host proteins light chain 3 proteins (LC3s) or pericentrin, virus particles remained further from the endosome membrane - signalling a failure to uncoat and enter the cytoplasm. As a response of IAV infection, the formation of PANoptosomes is triggered. PANoptosomes are macromolecular complexes that regulate a unique innate immune inflammatory cell death pathway. By applying expansion microscopy, it was demonstrated how IAV infection promotes the assembly and co-localisation of PANoptosome components, including inflammasome adaptor protein ASC, apoptotic caspase-8 (CASP8), and necroptotic RIPK388. The results support PANoptosome role in inflammatory responses and offering insight into the cellular mechanisms underlying viral-induced immune activation.
SARS-CoV-2
Replication organelles and viral assembly sites represent dynamic and densely packed hubs of viral and host protein activity during SARS-CoV-2 infection. These compartments support viral RNA synthesis and coordinate the recruitment of structural proteins for virion assembly. However, the close apposition of membranes, proteins, and RNA within these confined regions presents a major challenge for ultrastructural characterisation by electron microscopy alone, particularly when attempting to resolve the spatial organisation of specific viral proteins. Recent work employing expansion microscopy has begun to overcome these limitations.
Tenfold expansion microscopy allowed the quantification of spatial relationships among viral structural proteins, organelles and epithelial surface protrusions in an intact three-dimensional context89. Imaging of SARS-CoV-2–infected human airway cells revealed Golgi fragmentation, virus-laden multivesicular bodies and apical membrane as well as ciliary remodelling. By physically enlarging fixed SARS-CoV-2-infected cells 4x and applying light-sheet imaging, it was shown that the nucleocapsid protein accumulates in layered, vesicle-like compartments closely associated with replication organelles90. Viral RNA replication foci (dsRNA) were found embedded within the outer layers of the N-positive compartments, confirming these as active sites of viral RNA synthesis. Stress granule formation is an effect of SARS-CoV-2 infection. However, the stress granule proteins G3BP1 and G3BP2 are not passive bystanders but are actively recruited into SARS-CoV-2 particles during viral assembly. These host proteins localise within toroidal ERGIC-derived vesicles, which serve as key sites of virion formation. Expansion microscopy provided the spatial resolution necessary to detect and characterise these highly packed, multi-component organelles91 (Fig. 6c). Depletion of G3BPs disrupted assembly site formation, underscoring their proviral role and highlighting them as potential antiviral targets.
Detection of pathogenic features in viral infections with expansion microscopy
Pathology plays a critical role in virology by providing the structural and functional context in which viral infections manifest within host tissues. Through histological, molecular, and imaging techniques, pathology enables the identification of virus-induced cellular damage, inflammation, and tissue remodelling. This helps to link specific viral mechanisms - such as immune evasion, latency, or cytopathic effects - with clinical outcomes. Conversely, virology informs pathology by investigating the life cycles, tropisms, and host interactions of viruses, guiding the interpretation of disease patterns and aiding in the development of diagnostic markers and targeted therapies.
Expansion microscopy can enhance diagnostic precision due to a better resolution of pathological features. Specialised protocols have been developed to accommodate various sample types, including the reuse of formalin-fixed, paraffin-embedded (FFPE) tissue slices commonly archived in clinical settings92,93. Also, recent protocols have improved both the speed and expansion factor of the technique, including the development of an optimised, broadly compatible biomolecular anchor37,94,95. In addition to the expansion factor itself, effective optical clearing in expanded samples plays a key role in reducing light scattering. Thus, besides the resolution improvement, expansion microscopy can also enhance imaging depth in dense or heterogeneous tissues. A further advantage is the improved labelling efficiency achieved when staining in the expanded state, as epitopes become more accessible. In some applications, resolution may be less critical, allowing samples to be shrunk back afterward for faster imaging - an important consideration for diagnostic workflows. This approach can enhance the visibility of disease or infection markers, supporting more reliable assessments.
A pathological case study demonstrated the value of expansion microscopy by revealing uncontrolled mpox virus (MPXV) infection in the gastrocnemius muscle of an immunocompromised mpox patient96. The patient, who had severe HIV and a low CD4⁺ count, showed a lack of effective immune response in the infected tissue. Threefold expansion of the formalin-fixed tissue enabled high-resolution immunofluorescence imaging, even within the densely packed, necrotic regions. This approach revealed viral structures and infected cells that would have been difficult to detect using conventional microscopy, allowing precise localisation of MPXV antigens and exposing the lack of effective immune cell engagement with infected cells.
Outlook
Expansion microscopy has already demonstrated significant promise for studying virus biology as well as virion morphology by enabling high-resolution imaging using standard diffraction-limited microscopes. It offers a more accessible and user-friendly alternative to optical super-resolution microscopes, without compromising spatial detail. Its low equipment demands and compatibility with common optical setups like confocal microscopes, make it straightforward to implement. In most current studies, the straightforward fourfold expansion is employed, which already allows detailed analysis of viral structures in complex tissue environments. However, newly developed protocols that further increase the expansion factor and shorten preparation time - along with more robust and universally compatible biomolecular anchors - could unlock the full potential of expansion microscopy for virology. Beyond the increase in resolution, additional advantages of expansion protocols - such as tissue clearing and enhanced labelling efficiency - can be especially valuable for analysing pathological samples. If these advances prove consistent and reliable in sample handling and dimensional control, they could provide unprecedented access to nanoscale features of virus biology, including capsid structure, host-virus interactions, and viral replication sites, across a broad range of sample types.
Data availability
No datasets were generated or analysed during the current study.
References
Laporte, M. H., Klena, N., Hamel, V. & Guichard, P. Visualizing the native cellular organization by coupling cryofixation with expansion microscopy (Cryo-ExM). Nat. Methods 19, 216–222 (2022).
Wen, G., Leen, V., Jia, Y., Rohand, T. & Hofkens, J. Improved dye survival in expansion microscopy through stabilizer-conjugated linkers. Chem. A Eur. J. 28, e202202404 (2022).
Chozinski, T. J. et al. Expansion microscopy with conventional antibodies and fluorescent proteins. Nat. Methods 13, 485–488 (2016).
Tillberg, P. W. et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol. 34, 987–992 (2016).
Shi, X. et al. Label-retention expansion microscopy. J. Cell Biol. 220, e202105067 (2021).
Campbell, L. A., Pannoni, K. E., Savory, N. A., Lal, D. & Farris, S. Protein-retention expansion microscopy for visualizing subcellular organelles in fixed brain tissue. J. Neurosci. Methods 361, 109285 (2021).
Wang, U.-T. T. et al. Protein and lipid expansion microscopy with trypsin and tyramide signal amplification for 3D imaging. Sci. Rep. 13, 21922 (2023).
Zwettler, F. U. et al. Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM). Nat. Commun. 11, 3388 (2020).
Klimas, A. et al. Magnify is a universal molecular anchoring strategy for expansion microscopy. Nat. Biotechnol. 41, 858–869 (2023).
Valdes, P. A. et al. Improved immunostaining of nanostructures and cells in human brain specimens through expansion-mediated protein decrowding. Sci. Transl. Med. 16, eabo0049 (2024).
M’Saad, O. & Bewersdorf, J. Light microscopy of proteins in their ultrastructural context. Nat. Commun. 11, 3850 (2020).
Chen, F. et al. Nanoscale imaging of RNA with expansion microscopy. Nat. Methods 13, 679–684 (2016).
Damstra, H. G. et al. Visualizing cellular and tissue ultrastructure using Ten-fold Robust Expansion Microscopy (TREx). eLife 11, e73775 (2022).
Wen, G. et al. Evaluation of Direct Grafting Strategies via Trivalent Anchoring for Enabling Lipid Membrane and Cytoskeleton Staining in Expansion Microscopy. ACS Nano 14, 7860–7867 (2020).
Götz, R. et al. Nanoscale imaging of bacterial infections by sphingolipid expansion microscopy. Nat. Commun. 11, 6173 (2020).
Shin, T. W. et al. Dense, continuous membrane labeling and expansion microscopy visualization of ultrastructure in tissues. Nat. Commun. 16, 1579 (2025).
Sun, D. et al. Click-ExM enables expansion microscopy for all biomolecules. Nat. Methods 18, 107–113 (2021).
Cui, Y. et al. Expansion microscopy using a single anchor molecule for high-yield multiplexed imaging of proteins and RNAs. PLoS ONE 18, e0291506 (2023).
Wen, G. et al. A Universal Labeling Strategy for Nucleic Acids in Expansion Microscopy. J. Am. Chem. Soc. 143, 13782–13789 (2021).
Chen, F., Tillberg, P. W. & Boyden, E. S. Expansion microscopy. Science 347, 543–548 (2015).
Hümpfer, N., Thielhorn, R. & Ewers, H. Expanding boundaries – a cell biologist’s guide to expansion microscopy. J. Cell Sci. 137, jcs260765 (2024).
Truckenbrodt, S. et al. X10 expansion microscopy enables 25-nm resolution on conventional microscopes. EMBO Rep. 19, e45836 (2018).
Truckenbrodt, S., Sommer, C., Rizzoli, S. O. & Danzl, J. G. A practical guide to optimization in X10 expansion microscopy. Nat. Protoc. 14, 832–863 (2019).
Saal, K. A. et al. Heat denaturation enables multicolor X10-STED microscopy. Sci. Rep. 13, 5366 (2023).
Wang, S. et al. Single-shot 20-fold expansion microscopy. Nat. Methods 21, 2128–2134 (2024).
Chang, J.-B. et al. Iterative expansion microscopy. Nat. Methods 14, 593–599 (2017).
Louvel, V. et al. iU-ExM: nanoscopy of organelles and tissues with iterative ultrastructure expansion microscopy. Nat. Commun. 14, 7893 (2023).
Gambarotto, D. et al. Imaging cellular ultrastructures using expansion microscopy (U-ExM). Nat. Methods 16, 71–74 (2019).
Klein, S., Staring, M., Murphy, K., Viergever, M. A. & Pluim, J. elastix: A Toolbox for Intensity-Based Medical Image Registration. IEEE Trans. Med. Imaging 29, 196–205 (2010).
Gao, M. et al. Expansion Stimulated Emission Depletion Microscopy (ExSTED). ACS Nano 12, 4178–4185 (2018).
Damstra, H. G. J. et al. GelMap: intrinsic calibration and deformation mapping for expansion microscopy. Nat. Methods 20, 1573–1580 (2023).
Bogovic, J. A., Hanslovsky, P., Wong, A. & Saalfeld, S. Robust registration of calcium images by learned contrast synthesis. In 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI) 1123–1126 (IEEE, Prague, Czech Republic, https://doi.org/10.1109/ISBI.2016.7493463. 2016).
Klena, N. et al. An In-depth Guide to the Ultrastructural Expansion Microscopy (U-ExM) of Chlamydomonas reinhardtii. BIO-PROTOCOL 13, (2023).
Gao, R., Asano, S. M. & Boyden, E. S. Q.&A. : Expansion microscopy. BMC Biol. 15, 50 (2017).
Wassie, A. T., Zhao, Y. & Boyden, E. S. Expansion microscopy: principles and uses in biological research. Nat. Methods 16, 33–41 (2019).
Jia, D. et al. Derivative Technologies of Expansion Microscopy and Applications in Biomedicine. ChemBioChem 26, e202400795 (2025).
Guo, J. et al. BOOST: a robust ten-fold expansion method on hour-scale. Nat Commun 16, (2025).
Chuang, Y. et al. Super-Resolution Imaging in Collagen-Abundant Thick Tissues. Small Struct. 5, 2400231 (2024).
Shaib, A. H. et al. One-step nanoscale expansion microscopy reveals individual protein shapes. Nat Biotechnol https://doi.org/10.1038/s41587-024-02431-9 (2024).
Quantitative Fluorescence Microscopy and Image Deconvolution. In Methods in Cell Biology 407–426 (Elsevier, https://doi.org/10.1016/b978-0-12-407761-4.00017-8. 2013).
Wallace, W., Schaefer, L. H. & Swedlow, J. R. A Workingperson’s Guide to Deconvolution in Light Microscopy. BioTechniques 31, 1076–1097 (2001).
Elliott, A. D. Confocal Microscopy: Principles and Modern Practices. Curr. Protoc. Cytom. 92, e68 (2020).
Bürgers, J. et al. Light-sheet fluorescence expansion microscopy: fast mapping of neural circuits at super resolution. Neurophoton 6, 1 (2019).
Mascheroni, L. et al. Combining sample expansion and light sheet microscopy for the volumetric imaging of virus-infected cells with super-resolution. Biomed. Opt. Express 11, 5032 (2020).
Pesce, L., Ricci, P., Sportelli, G., Belcari, N. & Sancataldo, G. Expansion and Light-Sheet Microscopy for Nanoscale 3D Imaging. Small Methods 2301715 https://doi.org/10.1002/smtd.202301715 (2024).
Keller, P. J., Schmidt, A. D., Wittbrodt, J. & Stelzer, E. H. K. Reconstruction of Zebrafish Early Embryonic Development by Scanned Light Sheet Microscopy. Science 322, 1065–1069 (2008).
Chen, B.-C. et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346, 1257998 (2014).
Stelzer, E. H. K. et al. Light sheet fluorescence microscopy. Nat Rev Methods Primers 1, (2021).
Rodriguez-Gatica, J. E. et al. Imaging three-dimensional brain organoid architecture from meso- to nanoscale across development. Development 149, dev200439 (2022).
Düring, D. N., Rocha, M. D., Dittrich, F., Gahr, M. & Hahnloser, R. H. R. Expansion Light Sheet Microscopy Resolves Subcellular Structures in Large Portions of the Songbird Brain. Front. Neuroanat. 13, 2 (2019).
Tian, X. et al. Rapid lightsheet fluorescence imaging of whole Drosophila brains at nanoscale resolution by potassium acrylate-based expansion microscopy. Nat. Commun. 15, 10911 (2024).
Scardigli, M. et al. Comparison of Different Tissue Clearing Methods for Three-Dimensional Reconstruction of Human Brain Cellular Anatomy Using Advanced Imaging Techniques. Front. Neuroanat. 15, 752234 (2021).
Gao, R. et al. Cortical column and whole-brain imaging with molecular contrast and nanoscale resolution. Science 363, eaau8302 (2019).
Lillvis, J. L. et al. Rapid reconstruction of neural circuits using tissue expansion and light sheet microscopy. eLife 11, e81248 (2022).
Dunsby, C. Optically sectioned imaging by oblique plane microscopy. Opt. Express 16, 20306 (2008).
Kumar, S. et al. High-speed 2D and 3D fluorescence microscopy of cardiac myocytes. Opt. Express 19, 13839 (2011).
Lamb, J. R., Cardoso Mestre, M., Lancaster, M. & Manton, J. D. Direct-view oblique plane microscopy. Optica 12, 469 (2025).
Jacquemet, G., Carisey, A. F., Hamidi, H., Henriques, R. & Leterrier, C. The cell biologist’s guide to super-resolution microscopy. J. Cell Sci. 133, jcs240713 (2020).
Schermelleh, L. et al. Super-resolution microscopy demystified. Nat. Cell Biol. 21, 72–84 (2019).
Aristova, D. et al. Nanoscale imaging of biological systems via expansion and super-resolution microscopy. Appl. Phys. Rev. 12, 021311 (2025).
Woo, N. & Brown, C. M. Review of expansion microscopy combined with advanced imaging modalities. Journal of Microscopy jmi.70048 https://doi.org/10.1111/jmi.70048 (2025).
Gustafsson, N. et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 7, 12471 (2016).
Laine, R. F. et al. NanoJ: a high-performance open-source super-resolution microscopy toolbox. J. Phys. D: Appl. Phys. 52, 163001 (2019).
Robb, N. C. Virus morphology: Insights from super-resolution fluorescence microscopy. Biochimica et. Biophysica Acta (BBA) - Mol. Basis Dis. 1868, 166347 (2022).
Chojnacki, J. et al. Maturation-Dependent HIV-1 Surface Protein Redistribution Revealed by Fluorescence Nanoscopy. Science 338, 524–528 (2012).
Laine, R. F. et al. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 6, 5980 (2015).
Mehedi, M. et al. Multicolor Stimulated Emission Depletion (STED) Microscopy to Generate High-resolution Images of Respiratory Syncytial Virus Particles and Infected Cells. BIO-PROTOCOL 7, (2017).
Liu, Q., Chen, L., Aguilar, H. C. & Chou, K. C. A stochastic assembly model for Nipah virus revealed by super-resolution microscopy. Nat. Commun. 9, 3050 (2018).
Kummer, S., Avinoam, O. & Kräusslich, H.-G. IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells. Viruses 11, 548 (2019).
Mäntylä, E. et al. Iterative immunostaining combined with expansion microscopy and image processing reveals nanoscopic network organization of nuclear lamina. MBoC 34, br13 (2023).
Trus, B. L. et al. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19c and VP23 in assembly. J. Mol. Biol. 263, 447–462 (1996).
Norman, R. X. et al. One step 4× and 12× 3D-ExM enables robust super-resolution microscopy of nanoscale cellular structures. J. Cell Biol. 224, e202407116 (2025).
Nanbo, A., Noda, T. & Ohba, Y. Epstein–Barr Virus Acquires Its Final Envelope on Intracellular Compartments With Golgi Markers. Front. Microbiol. 9, 454 (2018).
Gao, R. et al. A highly homogeneous polymer composed of tetrahedron-like monomers for high-isotropy expansion microscopy. Nat. Nanotechnol. 16, 698–707 (2021).
Grove, J. Super-Resolution Microscopy: A Virus’ Eye View of the Cell. Viruses 6, 1365–1378 (2014).
Witte, R., Andriasyan, V., Georgi, F., Yakimovich, A. & Greber, U. F. Concepts in Light Microscopy of Viruses. Viruses 10, 202 (2018).
Castelletto, S. & Boretti, A. Viral particle imaging by super-resolution fluorescence microscopy. Chem. Phys. Impact 2, 100013 (2021).
Arista-Romero, M., Pujals, S. & Albertazzi, L. Towards a Quantitative Single Particle Characterization by Super Resolution Microscopy: From Virus Structures to Antivirals Design. Front. Bioeng. Biotechnol. 9, (2021).
Goonawardane, N., Yin, C., Roberts, G. C., Zothner, C. & Harris, M. A key role for hepatitis C virus NS5A serine 225 phosphorylation revealed by super-resolution microscopy. Sci Rep 15, (2025).
Aho, V. et al. Quantitative Microscopy Reveals Stepwise Alteration of Chromatin Structure during Herpesvirus Infection. Viruses 11, 935 (2019).
Leclerc, S. et al. Herpesvirus-Induced Manipulation of the Nucleolus. Microscopy and Microanalysis 30, (2024).
Scherer, K. M. et al. A fluorescent reporter system enables spatiotemporal analysis of host cell modification during herpes simplex virus-1 replication. J. Biol. Chem. 296, 100236 (2021).
Petrich, A. et al. Expanding Insights: Harnessing Expansion Microscopy for Super-Resolution Analysis of HIV-1–Cell Interactions. Viruses 16, 1610 (2024).
De Souza Cardoso, R., Murakami, T., Jacobovitz, B., Veatch, S. L. & Ono, A. PIP2 promotes the incorporation of CD43, PSGL-1, and CD44 into nascent HIV-1 particles. Sci. Adv. 11, (2025).
Amodio, A. et al. Nanoscale probing and imaging of HIV-1 RNA in cells with a chimeric LNA–DNA sensor. Nanoscale 14, 3049–3061 (2022).
Kumar, A. et al. Influenza virus exploits tunneling nanotubes for cell-to-cell spread. Sci Rep 7, (2017).
Cong, Y. et al. Influenza A virus subverts the LC3-pericentrin dynein adaptor complex for host cytoplasm entry. Sci. Adv. 11, (2025).
Wang, Y. et al. Single cell analysis of PANoptosome cell death complexes through an expansion microscopy method. Cell. Mol. Life Sci. 79, (2022).
Nijenhuis, W. et al. Optical nanoscopy reveals SARS-CoV-2-induced remodeling of human airway cells. Preprint at https://doi.org/10.1101/2021.08.05.455126 (2021).
Scherer, K. M. et al. SARS-CoV-2 nucleocapsid protein adheres to replication organelles before viral assembly at the Golgi/ERGIC and lysosome-mediated egress. Sci. Adv. 8, eabl4895 (2022).
Murigneux, E. et al. Proteomic analysis of SARS-CoV-2 particles unveils a key role of G3BP proteins in viral assembly. Nat Commun 15, (2024).
Zhao, Y. et al. Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nat. Biotechnol. 35, 757–764 (2017).
Bucur, O. et al. Nanoscale imaging of clinical specimens using conventional and rapid-expansion pathology. Nat. Protoc. 15, 1649–1672 (2020).
Park, H. et al. Scalable and isotropic expansion of tissues with simply tunable expansion ratio. Adv. Sci. 6, (2019).
Cheng, Z. et al. MicroMagnify: a multiplexed expansion microscopy method for pathogens and infected tissues. Adv. Sci. 10, (2023).
Matschke, J. et al. Inefficient tissue immune response against MPXV in an immunocompromised mpox patient. J. Med. Virol. 96, (2024).
Acknowledgements
K.M.S., L.Ö. and M.B. received funding from the returning scholars programme of the Ministry of Culture and Science of the State of North Rhine-Westphalia, Germany. K.B. was supported by the Erasmus+ programme of the EU and the NeurotechEU internship programme (Horizon 2020 Framework Programme, grant ID: 101035817).
Author information
Authors and Affiliations
Contributions
K.M.S. designed the review outline and acquired funding. L.Ö., M.B. and K.M.S. discussed the content of the sections and reviewed the literature. R.B. and K.B. provided the images for Figure 5. L.Ö. and M.B. designed the figures. K.M.S. wrote the paper. All authors reviewed and revised the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Övermöhle, L., Baum, M., Bhatia, R. et al. Visualising viral interactions and mechanisms at the nanoscale with expansion microscopy. npj Viruses 4, 1 (2026). https://doi.org/10.1038/s44298-025-00169-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44298-025-00169-y
