
Abstract
Genomic epidemiology has transformed the way public health scientists detect, monitor, and respond to infectious disease threats such as SARS-CoV-2 on a global scale. Early in the COVID-19 pandemic, vast inequities in whole-genome sequence data availability between high- and low-income countries were highlighted, but the persistence of these disparities five years into a global pandemic has not been quantified. Also, while it is generally known that genomic surveillance of SARS-CoV-2 largely declined following the end of the COVID-19 public health emergency, this has not been formally measured, and how it impacts our ability to detect and characterize new variants, remains unknown. Therefore, we performed an analysis of SARS-CoV-2 sequence submissions on the Global Initiative for Sharing All Influenza Data (GISAID) platform from 2020 to 2025, by country and World Bank income classification. There were large differences in SARS-CoV-2 sequence submissions by income classification, indicating a disparity in our ability to monitor SARS-CoV-2 evolution worldwide, which has important consequences for preventative measures such as vaccine strain selection. Nevertheless, there are important barriers to sustainable sharing of SARS-CoV-2 sequence data, which we discuss in detail, along with their relevance to other pathogens of public health importance. Also, the decrease in sequence submissions in high income countries from 577 million at the peak of the pandemic, to under 50 million in 2024, represents a loss of capacity to monitor SARS-CoV-2 evolution in countries with known capabilities. Ultimately, data drive the impact of genomic epidemiology, and long-term investments in genomic surveillance programs, as well as incentives for timely data sharing, are urgently needed to detect and characterize new viral variants worldwide.
Similar content being viewed by others

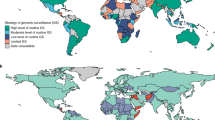
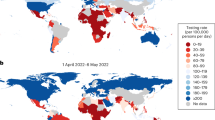
Introduction
Whole-genome sequence (WGS) data are critical to detect and monitor the emergence and spread of pathogens and have catalyzed the development of diagnostics, vaccines, and therapeutics needed for epidemic preparedness and response. SARS-CoV-2 is the most sequenced virus in history; the abundance of genomic data generated during the COVID-19 pandemic has transformed the way public health scientists monitor pathogen evolution and respond to emerging variants. Publicly accessible tools such as Nextstrain, UShER, and Cov-spectrum, which automatically ingest and analyze SARS-CoV-2 WGS data, have facilitated rapid detection of novel variants on an unprecedented scale1,2,3. The relevance of these tools to global public health depends on a comprehensive dataset from the Global Initiative on Sharing All Influenza Data (GISAID) platform, which relies on submissions of sequence data and metadata from scientists worldwide4.
In the early stages of the COVID-19 pandemic, the availability of SARS-CoV-2 sequence data on GISAID was heavily skewed toward high-income countries, with underrepresentation from low- and middle-income countries (LMICs)4,5,6. The recognition of this disparity accelerated the formation of public-private partnerships and academic collaborations dedicated to capacity-building for pathogen genomics in low-resource settings7,8,9. By May 2023, when the World Health Organization (WHO) declared the end of the COVID-19 public health emergency, more than 15 million SARS-CoV-2 sequences had been submitted to GISAID from more than 200 countries and territories, an accomplishment that cannot be overstated4,10. Here, we evaluate global disparities in SARS-CoV-2 sequence data available on GISAID over time, which affects efforts to detect, characterize, and respond to new variants. Additionally, we discuss barriers to sustainable and timely genomic data sharing, which are broadly applicable to all pathogens that threaten global public health.
Inequities in sequence submissions hinder monitoring of viral evolution
To assess the global distribution of sequences in GISAID, we conducted analyses using SARS-CoV-2 monthly submission totals from January 2020 through June 20254,11. A total of 17,407,588 SARS-CoV-2 sequences were available from 201 countries and territories in GISAID during this period, as of September 28, 2025. Among the 17,351,199 sequences originating from countries and territories with available 2025 World Bank income-level classifications and population information, 15,878,350 (91.5%) originated from high-income countries, 1,000,310 (5.8%) from upper-middle income countries, 454,794 (2.6%%) from lower-middle income countries, and 17,745 (0.1%) from low-income countries, representing a vast disparity in sequence submissions by income classification (Table 1)12,13.
To account for the impact of population size on these comparisons, we calculated sequence submissions per million population, using population data from 2025 from Worldometer, by year and income classification (Table 2, Figure 1). During 2020–2022, high income countries submitted more than ten times the number of sequences per million population than low income, lower-middle income, and upper-middle income countries combined. That disparity lessened slightly from 2023–2025, largely due to a major drop-off in sequence submissions from high-income countries rather than increased submissions from LMICs. Overall, the limited availability of sequences from LMICs represents a major impediment to monitoring SARS-CoV-2 evolution, adversely affecting national public health responses in LMICs as well as international efforts to track emerging threats worldwide14,15. Without timely sharing of more sequences, LMICs are also underrepresented in data that guides decision-making about vaccine strain selection and use of monoclonal antibodies. Additionally, shortly after the emergence of a new variant while it is still at low prevalence, laboratories are able to generate relevant data on factors such as transmissibility and immune escape, providing guidance to public health programs before the variant becomes dominant in the population16,17. Without sufficient volume of sequence data, knowledge of emerging variants, their rate of spread, and potential public health impact will remain unknown until those variants are detected in high-income settings.
Barriers to sustained sequencing and data sharing persist in LMICs
It is important to distinguish the lack of capacity for SARS-CoV-2 sequencing from a lack of continued interest and financial support. Prior studies have determined that several countries of lower-middle income classification in Asia have moderate or high pathogen genomic surveillance capacity, yet those countries did not have any recent sequence submissions to GISAID18,19. Similarly, scientists from LMICs have published studies recently that indicate sequencing capacity, but the data were delayed, the variants were no longer in circulation, and the opportunity for those data to inform global surveillance and response had passed20,21,22,23. More detailed analyses are needed to elucidate, at the country level, specific barriers to sustained sequencing and timely sharing of data, particularly in LMICs with existing capacity for genomic surveillance.
It is also possible that more SARS-CoV-2 sequence data exist in LMICs than are shared with the global scientific community on GISAID. Travel restrictions, such as those imposed on South Africa following the emergence of the Omicron variant, may serve as disincentives to the timely sharing of novel sequence data relevant to public health24. Hesitance to share sequencing data may be further exacerbated by the inequalities in data sharing versus benefit sharing, as LMICs that shared SARS-CoV-2 sequence data used to inform COVID-19 vaccine development did not have equitable access to those vaccines once they were developed25. The recently adopted WHO Pandemic Agreement, which outlines a “pathogen access and benefit sharing” system, may be an initial step towards advancing equitable distribution of products and technology developed using data from LMICs26.
Diminishing surveillance in high-income countries has global repercussions
While high-income countries have submitted vastly more sequence data to GISAID than LMICs overall, the number of sequence submissions by high-income countries has declined substantially in recent years. From 2022 to 2023, there was a 5-fold decrease in sequence submissions per million population by high-income countries, which was far greater than decreases in submissions from LMICs during the same time period (Table 2). The difference was even greater from 2022 to 2024, which had a 25-fold decrease in sequence submissions per million population by high-income countries. This demonstrates a major loss of capacity to monitor SARS-CoV-2 evolution in countries with known resources to detect and respond to emerging variants.
Geographically representative genomic data is important for estimates of the global burden and resource allocation for public health response. However, knowledge of how quickly a new variant is spreading is entirely dependent of large volumes of data, which has historically come from high-income countries, regardless of where the variant was initially detected. Recent examples include BA.2.86 and BA.3.2, both of which are thought to have origins on the African continent prior to worldwide dissemination, which eventually precipitated risk assessment by WHO27,28. Without large volumes of sequence data, there may be delays in designating potential variants of interest and evaluation of potential public health impacts.
It is likely that the shift from diagnostic polymerase chain reaction tests for COVID-19 to rapid, at-home testing has decreased the number of clinical samples available for laboratories to sequence. To overcome this challenge, many public health systems, both in high-income countries and LMICs, have successfully implemented wastewater-based surveillance to monitor SARS-CoV-2 variant abundance in the community29,30,31. This approach may also be favorable for surveilling populations that have more limited access to healthcare facilities. However, estimating the prevalence of different variants in wastewater relies on a priori knowledge of what those variants are, which are typically designated from WGS data based on clinical samples32,33. Therefore, wastewater-based surveillance should complement, not replace, WGS-based surveillance.
Data drive the impact of genomic epidemiology
Building and sustaining investment in genomic surveillance capacity is essential to ensure early detection and response to SARS-CoV-2 and other pathogens with epidemic potential. However, SARS-CoV-2 sequence submissions have decreased dramatically since the emergence of the Omicron variant in January 2022, and many large-scale efforts to analyze SARS-CoV-2 data have ended, indicating a diminishing interest in SARS-CoV-2 genomic surveillance1,34,35,36. Nevertheless, viral evolution is constant, and new SARS-CoV-2 variants will continue to emerge regardless of continued global commitment or funding to detect them. The reactive, crisis-driven nature of funding public health is not a new problem, but a persistent one with inequitable impact. By the time variant X has spread extensively enough to garner attention, it may be too late to use genomic data to inform timely public health action that could reduce worldwide morbidity and mortality.
The question, “how much sequence data is enough?” does not have a simple answer, as it is largely dependent on the sampling strategy, purpose of sequencing, quality of the resulting sequences, and granularity of the associated metadata. For example, the sample size needed to detect a new SARS-CoV-2 variant will differ from the sample size needed to accurately estimate variant prevalence37. Public health laboratories also typically rely on convenience samples from healthcare facilities, which may be biased towards individuals experiencing severe disease, as well as geographic regions with SARS-CoV-2 testing capacity. The complexities in determining appropriate sampling and sequencing strategies are tremendous, yet critical for optimal allocation of resources to monitor a virus with dire public health consequences Fig. 1.

Month is represented on the x-axis. Different colors represent different World Bank income classifications. a The y-axis represents the raw average of sequences per million population. b The y-axis represents the log of the average sequences per million population, using the following formula: y = log(x) + 1.
Importantly, the analyses described here relied solely on data from GISAID, which represents just one of several data-sharing platforms used for SARS-CoV-2 genomic surveillance. However, GISAID has consistently had the most geographically representative genomic dataset for SARS-CoV-2 throughout the pandemic, which better enabled comparisons across countries. Other platforms, such as those within the International Nucleotide Sequence Database Collaboration (INSDC), which are fully public and considered more robust in terms of transparency and interoperability with downstream analytical tools, have much fewer sequences and could not be leveraged in the same way for such a comparison. Since these analyses were performed, information has become available indicating that GISAID may not be abiding by best practices, compromising many global efforts to monitor SARS-CoV-238,39. The use of GISAID data here is not an endorsement of the platform, but an emphasis on the need for continued genomic surveillance of SARS-CoV-2 despite the lack of a perfect, one-size-fits-all solution for data-sharing.Many of the barriers to continued sequencing and sharing of SARS-CoV-2 data are broadly applicable to other pathogens. For example, over the past few years, we have seen the emergence of a new mpox virus clade and reassortment of influenza viruses resulting in spillover into a new mammalian host40,41. Genomic data remain critical to providing insight on the origins and spread of newly emergent and re-emergent viruses, and will continue to play a pivotal role in the global response to pathogens with epidemic and pandemic potential. The WHO published attributes and principles of genomic data-sharing platforms in 2025, which need to be universally adopted and applied42. Specific sampling frameworks and sample size estimates for sequencing SARS-CoV-2 and other pathogens with pandemic potential are urgently needed, as well as guidelines for incorporating pathogen genomic data and metadata from animal hosts. Improved advocacy, political will, and long-term investment will be essential to sustain and expand sequencing of SARS-CoV-2 and other pathogens of public health importance. Genomic epidemiology is an incredibly powerful tool, but only as powerful as the data we provide.
Data availability
All data used for the analysis were downloaded from GISAID on September 28, 2025 using the “Global by month” Excel spreadsheet available on the platform.
References
Chen, C. et al. CoV-Spectrum: analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. https://doi.org/10.1093/bioinformatics/btab856.
Turakhia, Y. et al. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet 53, 809–816 (2021).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. https://doi.org/10.1093/bioinformatics/bty407.
Shu, Y. & McCauley, J. GISAID: Global initiative on sharing all influenza data—from vision to reality. Eurosurveillance 22, 30494 (2017).
Brito, A. F. et al. Global disparities in SARS-CoV-2 genomic surveillance. Nat. Commun. 13, 1–13 (2022).
Ohlsen, E. C. et al. Determining gaps in publicly shared SARS-CoV-2 genomic surveillance data by analysis of global submissions. Emerg. Infect. Dis. 28, S85 (2022).
Knyazev, S. et al. Unlocking capacities of genomics for the COVID-19 response and future pandemics. Nat. Methods 19, 374–380 (2022).
Tegally, H. et al. The evolving SARS-CoV-2 epidemic in Africa: Insights from rapidly expanding genomic surveillance. Science https://doi.org/10.1126/science.abq5358 (2022).
Mboowa, G. et al. Whole-genome sequencing of SARS-CoV-2 in Uganda: implementation of the low-cost ARTIC protocol in resource-limited settings. F1000Research 10, 598 (2021).
Statement on the fifteenth meeting of the International Health Regulations (2005) Emergency Committee regarding the coronavirus disease (COVID-19) pandemic. https://www.who.int/europe/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(covid-19)-pandemic.
Submissions and Variant statistics: Global by month. (2025).
World Bank Country and Lending Groups – World Bank Data Help Desk. https://datahelpdesk.worldbank.org/knowledgebase/articles/906519-world-bank-country-and-lending-groups.
Population by Country (2025) - Worldometer. https://www.worldometers.info/world-population/population-by-country/.
Wickham, H., François, R., Müller, K. & Vaughan, D. dplyr: A Grammar of Data Manipulation. R package version 1.1. (2023). https://dplyr.tidyverse.org/.
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2024).
Kaku, Y. et al. Virological characteristics of the SARS-CoV-2 KP.3, LB.1, and KP.2.3 variants. Lancet Infect. Dis. 24, e482–e483 (2024).
Liu, J. et al. Virological and antigenic characteristics of SARS-CoV-2 variants LF.7.2.1, NP.1, and LP.8.1. Lancet Infect. Dis. 25, e128–e130 (2025).
Chen, Z. et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat. Genet 54, 499–507 (2022).
Getchell, M. et al. Pathogen genomic surveillance status among lower resource settings in Asia. Nat. Microbiol 9, 2738–2747 (2024).
Kia, P. et al. Genomic characterization of SARS-CoV-2 from Uganda using MinION nanopore sequencing. Sci. Rep. 13, 1–8 (2023).
Hailu, G. et al. SARS-CoV-2 genetic variants identified in selected regions of ethiopia through whole genome sequencing: insights from the fifth wave of COVID-19. Genes 16, 351 (2025).
Shempela, D. M. et al. Detection and characterisation of SARS-CoV-2 in eastern province of Zambia: a retrospective genomic surveillance study. Int. J. Mol. Sci. 25, 6338 (2024).
Sow, M. S. et al. Genomic characterization of SARS-CoV-2 in Guinea, West Africa. PLoS One 19, e0299082 (2024).
Moodley, K. et al. Ethics and governance challenges related to genomic data sharing in southern Africa: the case of SARS-CoV-2. Lancet Glob. Health 10, e1855–e1859 (2022).
Pilkington, V., Keestra, S. M. & Hill, A. Global COVID-19 vaccine inequity: failures in the first year of distribution and potential solutions for the future. Front. Public Health 10, 821117 (2022).
World Health Assembly adopts historic Pandemic Agreement to make the world more equitable and safer from future pandemics. https://www.who.int/news/item/20-05-2025-world-health-assembly-adopts-historic-pandemic-agreement-to-make-the-world-more-equitable-and-safer-from-future-pandemics.
Zhang, L. et al. Epidemiological and virological update on the emerging SARS-CoV-2 variant BA.3.2. Lancet Infect. Dis. 26, e1–e2 (2026).
Khan, K. et al. Evolution and neutralization escape of the SARS-CoV-2 BA.2.86 subvariant. Nat. Commun. 14, 8078 (2023).
Fontenele, R. S. et al. High-throughput sequencing of SARS-CoV-2 in wastewater provides insights into circulating variants. medRxiv 2021.01.22.21250320 https://doi.org/10.1101/2021.01.22.21250320 (2021).
Yousif, M. et al. SARS-CoV-2 genomic surveillance in wastewater as a model for monitoring evolution of endemic viruses. Nat. Commun. 14, 1–9 (2023).
Barnes, K. G. et al. Utilizing river and wastewater as a SARS-CoV-2 surveillance tool in settings with limited formal sewage systems. Nat. Commun. 14, 1–11 (2023).
O’Toole, Á. et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. https://doi.org/10.1093/ve/veab064.
Karthikeyan, S. et al. Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature 609, 101–108 (2022).
Padhiar, N. H. et al. SARS-CoV-2 CoCoPUTs: analyzing GISAID and NCBI data to obtain codon statistics, mutations, and free energy over a multiyear period. https://doi.org/10.1093/ve/veae115.
COVID-19 Genomics UK Consortium. https://www.cogconsortium.uk/.
The Search For Covid-19 Variants. Johns Hopkins Coronavirus Resource Center https://coronavirus.jhu.edu/data/variant-data.
Wohl, S., Lee, E. C., DiPrete, B. L. & Lessler, J. Sample size calculations for pathogen variant surveillance in the presence of biological and systematic biases. CR Med. 4, 101022 (2023).
Nextstrain: Interruption to GISAID-based SARS-CoV-2 sequence analyses. Nextstrain https://nextstrain.org/blog/2025-11-06-gisaid-based-ncov-analyses (2025).
Fresh conflicts erupt around giant database for flu and COVID-19 sequences. https://www.science.org/content/article/fresh-conflicts-erupt-around-giant-database-flu-and-covid-19-sequences.
Masirika, L. M. et al. Ongoing mpox outbreak in Kamituga, South Kivu province, associated with monkeypox virus of a novel Clade I sub-lineage, Democratic Republic of the Congo, 2024. Eurosurveillance 29, 2400106 (2024).
Caserta, L. C. et al. Spillover of highly pathogenic avian influenza H5N1 virus to dairy cattle. Nature 634, 669–676 (2024).
Attributes and Principles of Genomic Data-Sharing Platforms Supporting Surveillance of Pathogens with Epidemic and Pandemic Potential. https://pandemichub.who.int/publications/attributes-and-principles-of-genomic-data-sharing-platforms-supporting-surveillance-of-pathogens-with-epidemic-and-pandemic-potential (2025).
Acknowledgements
This work was supported by the Coalition for Epidemic Preparedness Innovations (CEPI) grant awarded to the University of Minnesota Center for Infectious Disease Research and Policy, “Monitoring Progress of R&D for Broadly Protective Coronavirus Vaccines.” CEPI had no role in the writing of this manuscript or decision to submit it for publication. We gratefully acknowledge all data contributors, i.e., the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based.
Author information
Authors and Affiliations
Contributions
E.S. conceived the idea for the manuscript, performed all analyses, and wrote the original draft. D.F. and A.U. reviewed the manuscript draft and provided critical feedback on content and analysis. E.L. provided oversight for all aspects of this work. All authors discussed and approved the final version of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Smith, E.A., Fleming, D.F., Lackritz, E.M. et al. Inequities and global declines in SARS-CoV-2 genomic data availability hinder response to emerging variants. npj Viruses 4, 13 (2026). https://doi.org/10.1038/s44298-026-00176-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44298-026-00176-7
