
Abstract
Chronic neuroinflammation plays a key role in the progression of Alzheimer’s disease (AD), and the cytosolic calcium-dependent phospholipase A2 (cPLA2) enzyme is a critical mediator of inflammatory lipid signaling pathways. Here we investigate the therapeutic potential of novel cPLA2 inhibitors in modulating neuroinflammation in AD. By leveraging the giga-scale V-SYNTHES2 virtual screening in on-demand chemical space and conducting two rounds of optimization for potency and selectivity, we have identified BRI-50460, achieving an IC50 of 0.88 nM in cellular assays that measure cPLA2-mediated arachidonic acid release. In vivo studies revealed favorable brain-to-plasma ratios, highlighting the ability of BRI-50460 to penetrate the central nervous system, modulating neuroinflammatory pathways, and restoring lipid homeostasis. In astrocytes and neurons derived from human induced pluripotent stem cells, BRI-50460 mitigates the effects of amyloid beta 42 oligomers on cPLA2 activation, tau hyperphosphorylation, and synaptic loss. Our results support that small molecule inhibitors of cPLA2 can modulate the downstream inflammatory signaling, offering a promising therapeutic strategy for neurodegenerative diseases.
Similar content being viewed by others

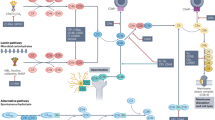

Introduction
The lack of effective treatments for neurodegenerative diseases such as Alzheimer’s disease (AD) underscores the challenge in identifying the critical pathways that influence neurodegeneration. Neuroinflammation is a hallmark of AD and other neurodegenerative disorders. Inflammation-related targets constitute one of the largest growing categories (approximately 16%) in the AD drug development pipeline1. Understanding the complex role of neuroinflammation in disease pathogenesis unveils more effective therapeutic approaches and druggable targets. Among the inflammatory pathways driving chronic, unresolved inflammation are pro-inflammatory mediators generated from arachidonic acid (AA) metabolism2,3,4. The enzyme cytosolic phospholipase A2 (cPLA2) plays a critical role in this process by hydrolyzing membrane phospholipids to release AA, which is subsequently converted to both pro-inflammatory and anti-inflammatory eicosanoids, with greater representation of proinflammatory pathways.
One of the key lipoproteins regulating brain lipid metabolism is apolipoprotein E (ApoE), which exists in three common variants: ApoE2, ApoE3, and ApoE4. The APOE4 allele is not only the strongest genetic risk factor for late onset AD but also has been shown to exacerbate neuroinflammation5. Our group has reported increased cPLA2 activity in APOE4 cellular and animal models6, as well as in postmortem brain tissues7, contributing to lipid dysregulation and neuronal damage. In this context, cPLA2 inhibitors may offer a novel therapeutic approach by addressing both unresolved inflammation and lipid homeostasis. cPLA2 catalyzes the hydrolysis of phospholipids, resulting in the production of free fatty acids and lysophospholipids8. As APOE4 enhances the activation of cPLA2 6in the brain, the ω-6 to ω-3 balance is disrupted. Reducing cPLA2 activity realigns the ω-6 to ω-3 balance9, suggesting an attractive therapeutic strategy.
The development of selective cPLA2 inhibitors has proven challenging due to off-target effects and difficulties with isoform specificity, with the additional challenge of blood-brain barrier (BBB) penetrance. After a decade of developing these compounds, only one has gone through human trials (AK106-001616). A Phase 2 clinical trial explored AK106-001616 in patients with rheumatoid arthritis (RA)10. While clinical safety for AK106-001616 has been established in RA patients, its ability to penetrate the BBB is likely to be limited. Therefore, new, specific, and brain-penetrant cPLA2 inhibitors are needed.
In this study, we employed V-SYNTHES2, a novel approach to virtually screen the Enamine REAL (REadily AvailabLe) Space of 36 billion on-demand synthesizable compounds for potential cPLA2 inhibitors. Optimization of the initial hits in REAL Space resulted in several lead compounds that merit further development. The most promising candidate BRI-50460 potently and selectively inhibits cPLA2 in vitro and in our cellular experiments; it also mitigates the effects of amyloid beta 42 oligomers (Aβ42O) on cPLA2 activation, tau hyperphosphorylation, and synaptic and dendritic reduction in induced pluripotent stem cells (iPSCs). These results offer a therapeutic strategy that not only targets the enzyme itself but also modulates the downstream lipid signaling pathways associated with AD pathology.
Results
Giga-Scale virtual screen for BBB penetrant cPLA2 inhibitors with V-SYNTHES2
We used the V-SYNTHES2 platform to virtually screen the Enamine REAL chemical space to identify potential small-molecule inhibitors of cPLA2. Application of the V-SYNTHES2 enabled effective screening of the 36-billion compound chemical space, while performing the docking of only a small fraction of full molecules (~2 million). The algorithm starts with the docking of a pre-generated Minimal Enumeration Library (MEL) to the cPLA2 binding pocket (Supplementary Fig. S1). MEL represents the whole chemical space, with ~1.8 million fragment-like combinations of every reaction scaffold with one real synthon from the chemical space, and minimal “caps” that substitute the real counter-synthons. The best MEL compounds are selected based on the cPLA2 docking score and molecule pose in the binding pocket and enumerated with the second real synthon. For 2-component reaction sub-space, full molecules are generated after one round of enumeration: for 3-component reactions sub-space – after two rounds of enumeration. The full molecules are then docked into the cPLA2 binding pocket, and the best-scoring candidates are analyzed to make the final selection of compounds for synthesis and testing.
A giga-scale virtual screening was performed using a ligand-optimized cPLA2 structural model based on the apo-enzyme structure (PDB ID 1CJY, Fig. 1). Candidate hit compounds were selected based on predicted docking scores and binding poses (Supplementary Fig. S2), chemical novelty, and diversity, and filtered for low PAINS and toxicity scores, as well as high BBB penetration properties. A total of 127 top-scoring compounds (Supplementary Table S1) were selected for synthesis, out of which 117 compounds were synthesized by Enamine in less than 6 weeks.

a Best hits from the 1st Generation of cPLA2 screening. b Predicted binding poses of best hits in catalytic binding site of cPLA2 apo crystal structure (PDB ID 1CJY). The residues of cPLA2 active site are shown as gray sticks. The catalytic serine S228 and its catalytic partner D549 are shown in magenta. Hydrogen bonds are shown as blue dots.
Identification of the Initial High-quality cPLA2 Inhibitor Hits
To test the predicted hit candidates in vitro, we used a cPLA2 inhibition fluorescence assay (Fig. 2a), with commercially available cPLA2 inhibitor ASB14780 serving as the positive control. This initial screen identified 19 compounds with >40% inhibition of cPLA2 at 10 μM for a prospective screening rate of 16%. Dose-response curves measured for the best 7 new compound chemotypes (Supplementary Table S2) showed IC50 values below 10 μM, comparable to our reference inhibitor ASB14780 with a determined IC50 = 6.66 μM in the same assay. The discrepancy in the IC50 value measured in this assay for ASB14780 from the previously reported value of 20 nM from a radiometric assay11 is to be expected, as IC50 values for cPLA2 are influenced by lipid content due to the enzyme’s surface dilution kinetics, where catalytic activity depends on membrane binding12,13. Assays with higher lipid surface area or detergent-based micelles (as in radiometric formats) typically yield lower apparent IC50 values than fluorescence assays with soluble substrates14. Despite these differences, relative potency rankings remain valid, as structurally related inhibitors show consistent trends across formats.

A A fluorescence-based assay was used to determine the activity of recombinant human cPLA2 incubated with 10 μM test compound relative to a control (substrate, enzyme, no compound) in our initial screen. ASB14780 was also included as an inhibitor positive control. Compounds BRI-50006, BRI-50049, BRI-50054, BRI-50077, and BRI-50125 were among the hits found through this initial screen. B The compounds (10 µM) were mixed with stimuli (TIA: TNFa+rIFN+A23187) and added to astrocytes for 30 minutes. The cell lysate was harvested and used to test the protein levels of phosphorylated and total cPLA2 by Western blot. ASB14780 was used as a positive cPLA2 inhibitor control. C BV-2 microglia cells were pre-treated with different concentrations of BRI-50054, BRI-50077, and BRI-50125 for 30 minutes, then treated with LPS (10 ng/mL) for 16 hours. PGE2 levels in the medium were measured using a PGE2 ELISA assay kit.
Confirmed cPLA2 inhibitors have diverse chemical scaffolds with a Tanimoto distance >0.3 between each other and >0.4 to known cPLA2 inhibitors. Figure 1 shows the diversity of predicted docking poses for the 7 hit compounds. Compounds BRI-50006, BRI-50031, and BRI-50077 have similar moieties on one side of the molecule; however, the second synthon adds diversity to the chemical scaffold and ensures a variety of interactions between the ligand and the residues of the binding pocket. In all three molecules, carbonyl oxygen from the ester group and nitrogen from the amide group form hydrogen bond interactions with the backbone of residue A578. The other parts of the 3 hit molecules cover different interactions: for instance, compound BRI-50006 forms hydrogen bonds with residue D549 and backbone of residue G197, BRI-50031 interacts with the backbone of residue G197 and P585, and BRI-50077 with catalytically active residue S228, backbone of residues G197 and P585. Compounds BRI-50026 and BRI-50049 form hydrogen bonds with the backbone of residue A578 and P585. Compound BRI-50054 forms hydrogen bonds with the residue S577 and the backbone of residue A578. BRI-50125 forms hydrogen bonds with residue E589 and the backbone of G197 and G198.
Subsequent cellular screening revealed that BRI-50054 showed potent inhibition and reduced p-cPLA2/cPLA2 levels (Fig. 2B) as well as reduced downstream production of PGE2 in cell models of neuroinflammation (Fig. 2C), with minimal impact on mitogen-activated protein kinase (MAPK) pathways and limited cytotoxicity at up to 40 μM (Supplementary Fig. S3). These in vitro hits were further assessed in mice, where the pharmacokinetics of each compound were evaluated in vivo for their bioavailability, drug distribution in various organs, metabolic stability, and various PK parameters. Compound plasma and brain concentrations were measured at multiple timepoints. This integrative approach provided the basis for selecting compounds for further development and testing in animal models.
Furthermore, pharmacokinetic studies in C57BL/6 mice revealed that the 1st Generation compound BRI-50054 was well-tolerated and exhibited dose-dependent increases in both the plasma and brain. This was accompanied by reduced brain AA levels and elevated levels of ω-3 fatty acids (DHA and EPA, Fig. 3B, D) and their pro-resolving metabolites, such as resolvins. These findings support the ability of cPLA2 inhibitors to engage their target in vivo, restore brain lipid homeostasis, and mitigate the neuroinflammatory environment characteristic of AD.

A C57BL/6J mice (n = 3) treated with vehicle, 1.0 or 3.0 mg/kg/day IP BRI-50054 for 3 days. Brain levels of DHA (B), DPA (C) and EPA (D) were measured using a validated LC-MS/MS lipidomic assay. *p < 0.05, **p < 0.01, ***p < 0.001.
SAR-by-catalog and the first round of hit optimization
Hit compounds identified using the V-SYNTHES2 algorithm can be effectively optimized using on-demand synthesis, as the combinatorial nature of REAL space contains thousands of readily available close analogs. Based on the in vitro fluorescence assays, molecules BRI-50054, BRI-50077, and BRI-50125 were nominated as the most promising hits for optimization. The structure-activity relationships (SAR)-by-catalog was performed using an in-house developed Synthon Based Space Search algorithm that emerged as a stand-alone tool for chemical search in REAL space. Like V-SYNTHES, this search algorithm effectively leverages the combinatorial nature of REAL Space. The search algorithm decomposes hit compounds into a reaction scaffold and the synthons from which the compound was generated (Supplementary Fig. S4) and performs a search of synthon analogs in the REAL space. Then, full analog molecules are generated by enumeration of identified synthon analogs according to the connection rules specified in REAL Space. This synthon search algorithm avoids full enumeration of the chemical space and takes only minutes to find tens of thousands of hit analogs among >36 billion REAL Space compounds. Based on docking scores and predicted physico-chemical properties like solubility, PAINS, and BBB penetration, a total of 100 analogs for hit compounds BRI-50054, BRI-50077, and BRI-50125 were selected for synthesis (Supplementary Table S3). Out of 100 requested, 85 molecules were synthesized by Enamine in under 6 weeks and experimentally tested.
Using a cellular assay in astrocytes stimulated with TNFα + IFNγ + A23187 (TIA) to induce cPLA2 phosphorylation, inhibitory effects of these 2nd Generation analogs were assessed via Western blot (Supplementary Fig. S5A). From the initial screen at 10 μM, 39 compounds were selected for further evaluation at 5 μM (Supplementary Fig. S5B). Subsequently, 10 compounds were tested at 1 μM (Supplementary Fig. S5C), identifying five most promising candidates: BRI-50202, BRI-50206, BRI-50229, BRI-50281 and BRI-50286. These compounds demonstrated significant inhibition of cPLA2 activation, validating their potential as optimized leads.
Second round: lead optimization
Based on the results from the initial screening and SAR-by-catalog, two scaffolds were selected for further optimization: first represented by BRI-50054 and BRI-50202 compounds, and second – by BRI-50125 and BRI-50281 compounds. Priority was given to the scaffold represented by hits BRI-50054 and BRI-50202 (Fig. 4C). The search for analogs was performed in REAL space of 36 billion molecules using the developed in-house Synthon Based Space Search engine. After analysis of docking results for generated analogs, 113 analogs were requested for synthesis from Enamine. Of the 113 requested compounds, 103 (Supplementary Table S4) were synthetized in 6 weeks and experimentally tested.

In vitro Inhibition A Schematic illustrating cPLA2-mediated hydrolysis of membrane phospholipids to release arachidonic acid (AA), which is subsequently converted to PGE2 by cyclooxygenase (COX) enzymes; this cellular cascade provides the basis for our functional inhibition assays. Created in BioRender. Duro, M. (2025) https://BioRender.com/5k81be8. B Preliminary cPLA2 in vitro activity screens of the first and third generation of small molecules. Each square is the cPLA2 in the presence of inhibitor relative to control (no inhibitor). C Progression of iterative structure optimization and potency screening of cPLA2 inhibitors. D Dose-dependency curve for the inhibition of AA efflux by ASB14780 and our two 3rd Gen leads upon administration of A23187 (Ca2+ ionophore). E IC50 estimates for the inhibition of AA efflux into cells and the inhibition of PGE2 production. F Selectivity profile assessed by in vitro iPLA2 inhibition assays for BRI-50460 and previously reported cPLA2 inhibitors (ASB14780, pyrrophenone, and AACOCF3. Assays were performed at 0–50 μM (except for pyrrophenone, which was limited to 12 μM), and activities are expressed as percent relative activity from the control with no inhibitor.
BRI-50460 demonstrates potent and selective cPLA2 inhibition in vitro
The second round of SAR-by-catalog and in vitro screening with a 2 μM threshold (Fig. 4B) yielded 3rd Generation compounds BRI-50460 and BRI-50469, which exhibited superior potency compared to earlier candidates. To validate the computational predictions and prioritize compounds for optimization, we employed cellular assays that measure cPLA2-mediated arachidonic acid release under physiologically relevant conditions. Both compounds demonstrated high efficacy in inhibiting AA release from cells labeled with 3H-AA and treated with a calcium ionophore (A23187). This ³H-AA efflux assay, which captures the immediate product of cPLA2 catalytic activity: the release of AA (Fig. 4A) from membrane phospholipids, was prioritized as the primary measure of cPLA2 inhibitory potency due to its direct measurement of enzymatic activity in the native membrane environments. Among them, BRI-50460 emerged as the most potent inhibitor, with an IC50 of 0.88 nM for cellular AA release and an IC50 of 4.1 nM for downstream PGE2 production, representing a > 20-fold improvement over ASB14780 (Fig. 4D, E, Supplementary Table S5). Compared to other known cPLA2 inhibitors such as ASB14780, pyrrophenone, and ATK, our lead compound BRI-50460 also demonstrated high selectivity, exhibiting negligible inhibition of iPLA2 in our in vitro fluorescence assays at concentrations up to 50 μM (Fig. 4F)—indicating an >100-fold selectivity window for cPLA2. BRI-50460 showed no cytotoxicity in HEK293 and HMC3 cells at concentrations up to 1 µM (Supplementary Fig. S6), >1000-fold above the functional IC₅₀ values, confirming that the observed effects on AA release and PGE2 production were not confounded by cytotoxicity effects.
Experimental validation using fluorescence thermal shift assays—designed to detect ligand-induced changes in protein denaturation temperature—was inconclusive, likely due to the unexpectedly high thermal stability of the cPLA2 protein, which impeded detection of subtle thermal shifts in the presence of BRI-50460. The AA efflux assay however provides the most direct and physiologically relevant measure of cPLA2 inhibitory activity, as it captures the enzyme’s membrane-dependent catalytic function in intact cells with native lipid environments. This assay incorporates all essential elements for cPLA2 activity including membrane association, substrate availability, calcium signaling, and the native cellular context. The sub-nanomolar IC50 observed in this assay represents functional inhibition under conditions that most closely mimic in vivo cPLA2 activity.
Brain delivery and pharmacokinetics of BRI-50460
In an acute pharmacokinetic study, BRI-50460 was administered to C57BL/6J mice (3 mg/kg body weight) either by a subcutaneous (SQ) injection or by oral gavage. Analysis of plasma and brain tissues harvested post-dosing revealed a brain-to-plasma (B/P) ratio exceeding 40% from subcutaneous delivery, more than double that of the 1st Generation compound BRI-50054 (Fig. 5B, C). The absolute bioavailability (F) of 38% (based on AUC0-inf, Fig. 5B, C) was demonstrated using our oral formulation. When compared to SQ, brain AUC0-6 exposure using this oral formulation was 87% of that of SQ (8.9/10.181) which corresponded when Cmax is 84%. The oral route also exhibited a longer plasma half-life in plasma (t1/2 = 4.2 hours oral vs 2.3 hours SC), and comparable brain t1/2 (3.95 vs. 3.53 hours), suggesting the enhanced large intestinal absorption can prolong and sustain systemic and central nervous system (CNS) exposure.

A BRI-50460 (3 mg/kg SQ or Oral) in an acute PK study using C57BL/6 J mice, where plasma and brain tissue were harvested at 0.5, 1, 3, and 6 hours post-dosing. Created in BioRender. Duro, M. (2025) https://BioRender.com/qu6a1hl. B Plasma and brain time-concentration data. C Cmax, Tmax, AUC, and half-life (t1/2) after 3 mg/kg BRI-50460 compared with zolpidem (Ambien®). D Free plasma and brain levels of BRI-50460 expressed in percentage unbound. E Kp,uu of BRI-50460 compared to other CNS penetrating agents.
Free fractions of BRI-50460 independently quantified in the brain (3.28%) and plasma (0.39%) yielded a calculated Kp,uu of 2.94, indicative of robust CNS penetration. This brain penetration surpassed many known CNS-penetrating drugs, including zolpidem (Ambien) (Fig. 5D, E). Overall, these findings confirm that the orally administered formulation of BRI-50460 delivers nearly equivalent brain exposure, supporting its development as an orally available, brain-penetrant therapeutic.
BRI-50460 reduces Aβ42O-induced cPLA2 levels and cPLA2 phosphorylation in human iPSC-derived astrocytes
To assess whether BRI-50460 has an inhibitory effect on cPLA2 phosphorylation induced by AD pathology in vitro, human iPSC-derived astrocytes were first treated with 0.1 or 1 µM of BRI-50460 for 30 minutes, followed by 2.5 µM of Aβ42O for 72 hours. Results show that 1 μM of BRI-50460 can reduce amyloid beta (Aβ)-related increases in cPLA2 levels (Fig. 6), as well as cause a reduction in phosphorylated cPLA2. These data indicate that in parallel with our initial in vitro screening performed in immortalized mouse astrocytes, BRI-50460 also has an inhibitory effect on cPLA2 and phosphorylated cPLA2 in human iPSC-derived astrocytes challenged with Aβ.

Phosphorylated cPLA2 and total cPLA2 (indicated by arrow) levels in cell extracts from iPSC-derived astrocytes were detected by Western blot. Quantification of total cPLA2 and p-cPLA2 protein levels normalized to Ponceau stain. Data are represented as mean ± SD and analyzed using one-way ANOVA followed by Tukey’s test. *p < 0.05, **p < 0.01, ****p < 0.0001.
BRI-50460 mitigates the effects of Aβ42O on cPLA2 activation, tau hyperphosphorylation, and synaptic loss in neurons derived from human iPSCs/NPCs
To examine BRI-50460’s treatment effects on AD pathologies, we first examined its impact on Aβ42O-induced tau hyperphosphorylation in a mixed-culture of neurons and astrocytes derived from human iPSCs carrying APOE3/3 genotype. Abnormal hyperphosphorylated tau leads to the formation of neurofibrillary tangles and neurodegeneration, a pathological feature of AD15. After neural progenitor cells (NPCs) were differentiated for five weeks16, the cells were exposed to 2.5 µM of Aβ42O, with or without 1 µM BRI-50460 for 72 hours. Immunofluorescence staining revealed that BRI-50460 reduced Aβ42O-induced increases in phospho-cPLA2α(Ser505) (p-cPLA2α, Fig. 7A, B) and phospho-tau (Ser202/Thr205) (p-Tau, Fig. 7A, C) in human neurons.

A Representative images of the immunofluorescent staining of p-cPLA2α and p-Tau in human iPSC-derived neurons treated with 2.5 μM Aβ42 oligomers (Aβ42O) with or without 1 μM BRI-50460 for 72 hours. Quantification revealed that BRI-50460 (460) reduced Aβ42O-induced increases in p-cPLA2α (B) and p-tau (C) intensity. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Analyzed image numbers include CTRL (n = 13), Aβ42 O (n = 14), 460 (n = 14).
BRI-50460 reverses synaptic loss in Aβ42O-challenged neurons derived from human iPSCs
Since synaptic loss predicts early memory impairment and correlates with cognitive decline in AD17 and is associated with neuroinflammation18, we next examined whether BRI-50460 has treatment effects on Aβ42O-induced synaptic loss in the mixed-culture of neurons and astrocytes derived from human iPSCs carrying APOE4/4 genotype. After neural progenitor cells (NPCs) were differentiated for four weeks, the cells were exposed to 2.5 µM of Aβ42 O, with or without 1 µM BRI-50460 for 72 hours. Immunofluorescence staining revealed that BRI-50460 protected the cells from Aβ42O-induced reductions of synaptic puncta stained by presynaptic protein synapsin (Fig. 8A, C), which regulates neurotransmitters release at synapses19, and dendritic density (occupied area) stained by postsynaptic protein CamKIIα (Fig. 8A, D) and MAP2 (Fig. 8A, E); CaMKIIa regulates long-term potentiation, a cellular mechanism of synaptic strength underlying learning and memory20,21,22.

A Representative images of the immunofluorescent staining of synapsin (green), CamKII (red), and MAP2 (white) in iPSC-derived neurons treated with 2.5 μM Aβ42 oligomers (Aβ42O) with or without 1 μM BRI-50460 for 72 hours. B Enlarged images of the boxed dendrites in A. BRI-50460 (460) reduced Aβ42O-induced reduction of synapsin puncta (C), CamKIIa (D) and MAP2 (E) density area occupied in neurons derived from human iPSCs. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Analyzed image numbers for CamKIIα and MAP2 include CTRL (n = 12), Aβ42 O (n = 13), 460 (n = 13), for synapsin puncta include CTRL (194), Aβ42O (n = 68), 460 (n = 167).
Discussion
Our structure-based virtual screening and SAR-by-catalog optimization efforts have identified BRI-50460 as a highly promising cPLA2 inhibitor, demonstrating significant potential as a therapeutic candidate for addressing neuroinflammation and pathology of Alzheimer’s disease (AD). Among the compounds derived from our structure-based cPLA2 virtual screening, BRI-50460 exhibits high potency, a brain penetrant pharmacokinetic profile, and an ability to modulate key mediators involved in neuroinflammation and AD pathology in vitro.
BRI-50460 exhibited exceptional potency, with an IC50 of 0.88 nM for inhibiting cellular AA release and of 4.1 nM for blocking downstream PGE2 synthesis, representing a greater than 20-fold improvement over ASB14780, a previously studied cPLA2 inhibitor, while also demonstrating high selectivity over iPLA2 in vitro. This enhanced efficacy underscores the success of our optimization strategy, which focused on improving both binding affinity and functional inhibition of cPLA2 in relevant cellular models. BRI-50460 demonstrated robust inhibition in cell-based assays without significantly affecting MAPK pathways, suggesting a targeted action on cPLA2 without disrupting other critical signaling cascades, which could be important for minimizing off-target effects in clinical applications.
While inhibition of cPLA2 phosphorylation was observed in inhibitors like BRI-50054 and has been used as a readout of the activation of the enzyme23, it is not a direct surrogate for catalytic inhibition of the enzyme. Phosphorylation reflects upstream signaling (e.g., MAPK activation) and may modulate membrane localization or activity, but inhibition of phosphorylation is not required for enzymatic inhibition. We have assessed whether phosphorylation sites could influence compound binding by modeling full-length cPLA2 using AlphaFold324 (Supplementary Fig. S8) and mapped key phosphorylated residues (T268, S437, S454, S505, S515, S727). All six sites are in flexible, unresolved regions and are >20 Å from the predicted binding pocket of BRI-50054, indicating they are unlikely to affect ligand binding. Given this, we did not rely solely on p-cPLA2 levels to assess potency. Instead, our evaluation integrated direct enzymatic assays, AA and PGE2 quantification, and target engagement in human iPSC-derived cells, providing a more accurate assessment of inhibitor activity.
One limitation of our current study is the incomplete characterization of the direct binding of the lead compounds to the catalytic domain of cPLA2. A critical consideration in cPLA2 drug discovery is that the enzyme’s catalytic activity is fundamentally dependent on membrane association. As an interfacial enzyme, cPLA2 activity follows surface dilution kinetics where apparent IC₅₀ values are strongly influenced by membrane composition12,13,14. Cell-free binding assays (SPR, ITC, thermal shift) often fail to correlate with functional inhibition because cPLA2 immobilization or solubilization eliminates the interfacial activation mechanism12,25. Given these constraints, we prioritized functional assays that capture physiologically relevant cPLA2 activity in native membrane environments. This convergent evidence from diverse functional assays spanning in vitro biochemical activity, cellular phenotypes in disease-relevant models, and in vivo pharmacodynamic effects provides strong support for BRI-50460 as a specific and selective cPLA2 inhibitor acting through on-target engagement. While future structural studies employing co-crystallography or cryo-EM with membrane-mimetic environments would be valuable for visualizing inhibitor-enzyme interactions, the extensive functional validation presented here demonstrates that our structure-based virtual screening approach successfully identified compounds that effectively inhibit cPLA2 in its native, membrane-associated context.
In addition to its potent inhibition of cPLA2, BRI-50460 showed promising pharmacokinetic properties that are critical for CNS drug development. Upon subcutaneous administration to C57BL/6J mice, BRI-50460 achieved a brain-to-plasma (B/P) ratio exceeding 40%, more than double that of the first-generation compound BRI-50054. This indicates that BRI-50460 has superior brain penetration, a crucial feature for targeting neuroinflammation in AD. Furthermore, BRI-50460 exhibited a free fraction of 3.28% in the brain and 0.39% in plasma, yielding a calculated Kp,uu of 2.94, which further supports its potential for effective CNS targeting. These values surpass those of several known CNS-penetrating drugs, including zolpidem, highlighting BRI-50460’s promising CNS bioavailability.
The ability of the 1st Generation compound BRI-50054 to reduce brain AA levels and increase ω-3 fatty acids (DHA and EPA), as well as their pro-resolving metabolites like resolvins, is particularly noteworthy. This shift in lipid homeostasis could help mitigate the neuroinflammatory environment characteristic of AD, offering a therapeutic strategy that not only targets the enzyme itself but also modulates the downstream lipid signaling pathways associated with AD pathology. These findings provide strong evidence that BRI-50460 may also effectively engage its target in vivo, restore lipid homeostasis in the brain, and potentially alleviate neuroinflammation—a key hallmark of Alzheimer’s disease. Additionally, BRI-50460 counteracts the impact of Aβ42O on cPLA2 activation, tau phosphorylation, and synaptic loss in astrocytes and neurons derived from human iPSCs. This is especially interesting for restoring CaMKIIα, an essential postsynaptic protein that regulates learning and memory20,22.
The enzyme cPLA2 is notable for releasing AA, which plays an important role in neurotransmission and serves as a precursor for eicosanoids26,27. However, excessive activation of cPLA2 can accelerate the cyclooxygenase (COX) and lipoxygenase (LOX)-mediated metabolism of free AA to the products prostaglandins (PGs) and leukotrienes (LTs), respectively, which are potent inflammatory lipid mediators8,28. The activity of cPLA2 is regulated by phosphorylation and is influenced by the MAPK, particularly through phosphorylated p3829,30.
We have identified the activation of cPLA2 as a critical driver of neuroinflammation in AD, particularly in APOE4 carriers23. This highlights the therapeutic potential of targeting cPLA2 to mitigate the harmful effects of chronic inflammation in the AD brain. Other groups have reported elevated levels of cPLA2 protein and its phosphorylated form in astrocytes surrounding Aβ plaques compared to healthy controls31,32,33. Increased activation of cPLA2 is also observed in the hippocampus of human amyloid precursor protein transgenic mice32. In addition, Aβ oligomers can activate cPLA2, thereby promoting neurodegeneration34,35. Importantly, the Mucke group reported that cPLA2 genetic deficiency ameliorates memory impairment and hyperactivated glial cells observed in an AD mouse model (Fig. 2)32,36. Knock-out of cPLA2 in microglia can decrease lipopolysaccharide-induced inflammatory response37. In APOE-targeted replacement mice and human brains, we observed that APOE4 induces greater MAPK p38-mediated activation of cPLA2. Importantly, inhibiting cPLA2 reduced the release of inflammatory neurotoxic lipids and eicosanoids23, corresponding to reduction in neuroinflammation and synaptic loss.
Our findings reveal a successful application of fast structure-based giga-scale V-SYNTHES2 screening and optimization to discover new potent cPLA2 inhibitor chemotypes. This includes BRI-50460, which exhibits enhanced potency, selectivity, pharmacodynamic, and pharmacokinetic profiles compared to the previously developed inhibitor ASB14780. BRI-50460 demonstrated strong in vitro inhibition of cPLA2-mediated AA release, with sub-nanomolar IC50 value and improved BBB penetration measures. By inhibiting cPLA2, BRI-50460 has the potential to restore lipid homeostasis, reduce AD pathology and synaptic dysfunction, particularly in APOE4 models, making it a promising candidate for further clinical development.
This study underscores the potential of advanced virtual screening platforms like V-SYNTHES2 in accelerating the discovery of CNS-targeting therapeutics and lays the groundwork for further exploration of BRI-50460 in chronic treatment paradigms with a prominent cPLA2 activation profile. Ongoing efforts will focus on refining its pharmacokinetics and evaluating its therapeutic effects in preclinical AD models, with the goal of advancing it toward clinical evaluation for the treatment of neuroinflammatory conditions in AD.
Methods
Drug discovery using V-SYNTHES2 platform
cPLA2 Model Preparation: The prospective screening was performed using a structural model of human cPLA2 refined using a ligand-guided approach38. A 2.5 Å resolution cPLA2 crystal structure39 that includes a N-terminal calcium-dependent lipid-binding/C2 domain, and a catalytic unit was downloaded from PDB (PDBID:1CJY). The crystal structure of cPLA2 was converted from PDB coordinates into an ICM molecular object. The conversion algorithm adds missing heavy side-chain atoms, performs global optimization of hydrogens for best hydrogen bonding network, optimizes His, Asn, Gln, and Cys residues, and assigns partial charges. To validate the model, 50 diverse, high affinity cPLA2 ligands available in ChEMBL (Target: CHEMBL3816, pAct>5) were docked into the binding site of the catalytic domain of cPLA2, and the best predicted docking scores and poses were saved for each ligand. The docking results showed reproducible binding poses and docking scores of about -25 for the known ligand scaffolds. The pose was considered reproducible if the best-predicted binding pose was the same (RMSD < 1 Å) between at least 3 out of 5 docking runs. To further improve the docking performance, the hydrogens of amino acids side chains in the binding pocket in 8 Å radius from the ligand were optimized in the presence of docked high affinity cPLA2 ligands (Supplementary Fig. S1). Performance of each model was evaluated based on the docking score and pose reproducibility, the value of the best docking score and an average docking score for 20 best ligands. The model optimized with molecule CHEMBL370113 showed the best reproducible results, with the best docking scores as good as –29 and was selected for prospective screening. Minimal structural changes were introduced during the optimization process with RMSDheavy atoms = 0.22 Å between the structure and the model for whole cPLA2 protein and RMSDheavy atoms = 0.19 Å for residues within 8 Å from the BRI-50054 compound (Supplementary Fig. S1).
Application of V-SYNTHES2: The giga-scale screening in the REAL chemical space of 36 billion molecules was performed using the V-SYNTHES2, a completely automated version of the V-SYNTHES platform described previously40. Docking of the pre-generated Minimal Enumeration Library (MEL), partially enumerated, and final molecules was performed with thoroughness set to 2. The docking and cheminformatic steps of V-SYNTHES2 were performed in ICM-Pro (Molsoft LLC) molecular modeling software (version 3.9-2b). Docking steps were performed in the USC CARC (Center for Advanced Research Computing at the University of Southern California) facility. Docking of MEL, partially and fully enumerated compounds from 2-component and 3-component subspaces of REAL took around 45 hours to complete using 320 cores. The cheminformatic steps of V-SYNTHES2 were performed on Linux workstation (AMD Ryzen 9 7950×16-Core Processor); 40 GB of space was needed to complete the project.
Selection and Synthesis of Candidate Hits for cPLA2: The selection of the most promising docked molecules for synthesis included the following standard post-processing steps: (i) filtering out molecules with undesirable physical-chemical properties, such as low drug-likeness, potential toxicity, and pan-assay interference (PAINS) predicted by ICM-Pro41 (ii) prioritizing molecules with high predicted solubility (molLogS) and BBB penetration predicted by ICM-Pro42 (iii) ensuring chemical novelty of selected molecules as compared to known high affinity ligands, using Tanimoto distance as a quantitative parameter for similarity. Also, the interactions between the predicted hits and binding pocket were analyzed and molecules with H-bond to R200, S228, T680 were given priority in the selection process. To ensure inhibition of the enzyme, molecules with close distances to catalytically active residue S228 at the bottom of the binding pocket were considered in the selection process: d < 6 Å, or d = 6–8 Å with H-bonds to R200, T680, or A578. Even though the whole REAL space was screened in this work, molecules from 2-comp reactions sub-space were predicted to have the best binding due to the small and shallow nature of the cPLA2 binding pocket and were prioritized for synthesis and testing. A total of 127 compounds were selected for synthesis, out of which 117 compounds were synthesized with >90% purity confirmed by LC-MS (Supplementary Table S1, Supplementary Fig. S7) and delivered by Enamine in 6 weeks.
Synthon Based Space Search for Analog Generation: The search for analogs of confirmed hits was performed in up-to-date versions of REAL Space. The analog search was performed using an in-house developed Synthon Based Space Search algorithm that takes advantage of the combinatorial nature of REAL chemical space, avoiding full enumeration of all molecules and working in the space of synthons (Supplementary Fig. S4). The algorithm starts with the decomposition of a hit molecule into the reaction scaffold and the synthons from which the compound was generated. In the next step, the algorithm searches for analogs of each of the synthons in the REAL Space using a specific Tanimoto distance (set to 0.4 in this case) as a cutoff to a molecule’s similarity. In the next step, identified synthon analogs are enumerated with each other according to the connection rules specified in REAL Space, considering physico-chemical properties to generate full molecules that comply with Lipinski’s rule of five. The Synthon Based Space Search algorithm finds tens of thousands of analogs in the Enamine REAL Space of 36 billion molecules in minutes. Generated analogs of confirmed hits from REAL Space were docked into the binding pocket of cPLA2, with increased effort equal to 5 to ensure comprehensive sampling of small molecules in the binding pocket. The combination of structure- and ligand-based approaches was used to select the analogs on each round of optimization. The analogs with the lowest predicted docking scores, docking poses similar to the initial hit compound, and lowest Tanimoto distances were prioritized for synthesis and testing.
cPLA2 biochemical testing
Testing using 10 μM Thresholds: Recombinant human cPLA2 (49 nmol, Abcam ab198469) was first pre-incubated with test compound (Supplementary Table S1, dissolved in DMSO) in 50 μL 1X PLA2 buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM CaCl2, pH 8.9, EnzChek™ Phospholipase A2 (PLA2) Assay Kit, Invitrogen E10217) at room temperature for 30 min. To this, 50 μL of the substrate-liposome solution [1-O-(6-BODIPY 558/568-aminohexyl)-2-BODIPY FL C5-sn-glycero-3-phosphocholine (Red/Green BODIPY PC-A2), dioleoylphosphatidylglycerol (DOPG), and dioleoylphosphatidylcholine (DOPC)], prepared according to manufacturer’s instructions, was added. Final concentrations were 10 μM of test compound, 600 ng/mL enzyme, and 1.6 μM fluorogenic substrate. Fluorescence (excitation at 460 nm, emission at 515 nm) was measured at 60 min from the start of the reaction and was corrected for the background fluorescence (setup with substrate but no enzyme). Enzyme activity was then calculated relative to the positive control (setup with substrate and enzyme but no inhibitor).
Testing using 2 μM Thresholds: Recombinant human cPLA2 (49 nmol) was first pre-incubated with test compound (Supplementary Table S4, dissolved in DMSO) in 50 μL 1X PLA2 buffer (same as above) at 37°C for 40 min. To the recombinant cPLA2-test compound mix was added 50 μL of the substrate-liposome solution (Red/Green BODIPY PC-A2, DOPC, and DOPG), prepared according to the manufacturer’s instructions. Final concentrations were 10 μM of test compound, 800 ng/mL enzyme, and 0.55 μM fluorogenic substrate. Fluorescence (excitation at 460 nm, emission at 515 nm) was measured at 0–90 min from the start of the reaction and was corrected for background fluorescence (set up with substrate but no enzyme). The slope of the fluorescence at 30–70 min was used to calculate enzyme activity relative to the positive control (setup with substrate and enzyme but no inhibitor).
iPLA2 Biochemical Testing: Because there is no commercial source of human recombinant calcium-independent phospholipase A2 (iPLA2), the enzyme was prepared from an overexpression cell line. Cell lysate was first pre-incubated with test compound (BRI-50460, ASB14780, pyrrophenone, or AACOCF3 dissolved in DMSO) in iPLA2-specific buffer43 (100 mM Tris-HCl, pH 7.0, and 4 mM EGTA). To the recombinant iPLA2-test compound mix was added 50 μL of the substrate-liposome solution (Red/Green BODIPY PC-A2, DOPC, and DOPG), prepared according to the manufacturer’s instructions but replacing the buffer with 1X iPLA2-specific buffer. Final concentrations were 0–50 μM of test compound, 20 μg/mL lysate protein, and 0.55 μM substrate. Fluorescence (excitation at 460 nm, emission at 515 nm) was measured every 10 min from the start of the reaction and was corrected for background fluorescence (set up with substrate but no enzyme). The slope of the fluorescence at 10–30 min was used to calculate enzyme activity relative to the positive control (setup with substrate and enzyme but no inhibitor).
Neuroinflammation cell model
Immortalized mouse astrocytes (gift from Dr. David Holtzman) were grown in DMEM/F12 (Corning, MT10090CV) containing 10% FBS, 1 mM sodium pyruvate (Thermo Fisher, 11360070), 1 mM geneticin (Thermo Fisher, 10131-035), and 1% anti-anti. Cells were seeded in a 24-well plate (0.25×105 cells/500 μL) and cultured overnight. The cells were treated with cPLA2 inhibitors plus TIA [TNFα (10 ng/mL) + IFNγ (10 ng/mL) + A23187 (1 μM)] in FBS-free medium for 30 min. After washing twice with cold PBS, cells were lysed with 1X sample buffer. Phosphorylated cPLA2 protein levels in cell lysate were detected by Western blot.
3H-AA efflux assay
AA released from cells was measured as described previously44. Tritium-labeled AA (3H-AA) was incorporated into the cell membrane phospholipids of BV2 cells by coculturing overnight. Labeled AA was released by stimulating the cells with Ca2+ ionophore (A23187) (Sigma-Aldrich) at a final concentration of 0.2 μM. Radioactivity of the culture supernatant was measured with a liquid scintillation counter (LS6500, Beckman Coulter) 30 min after the stimulation by A23187. The cPLA2 inhibitors were added 3 hr before stimulation by A23187 to examine their inhibitory effect on AA release.
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Southern California. The C57BL/6J mice used in the study were bred in the USC animal facility. Animals were housed with standardized 12 h light and dark cycles and had access to food and water ad libitum. Vivarium temperature was maintained between 22 °C and 24 °C, and humidity was maintained between 50 and 60%.
BRI-50460 administration and tissue collection
C57BL/6J mice were randomly assigned to 8 groups (n = 4 each group) and treated with one dose of BRI-50460 (3 mg/kg) formulated in an aqueous solution with 22.5% hydroxypropyl-β cyclodextrin—16 mice were treated via subcutaneous injection and the other 16 via oral gavage. After the treatment, animals at the designated time point (0.5, 1, 3, and 6 hr; 4 mice per time point) were euthanized with isoflurane. The blood was collected into EDTA-coated tubes via cardiac puncture, and plasma was isolated by centrifugation. Remaining blood was removed by transcardial perfusion with PBS, after which the mouse brains were harvested and flash frozen along with plasma samples for further processing.
BRI-50460 quantification by LC-MS/MS
BRI-50460 was extracted from mouse plasma (100 µL) and brain tissue (150–200 mg) and quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using multiple reaction monitoring (MRM). Mouse brains were cut in half through the midsagittal plane prior to analysis, and the left brain was used for quantitative analysis. A standard curve was prepared by spiking 3X stripped C57BL mouse plasma (BioIVT) aliquots with increasing concentrations of BRI-50460. To extract BRI-50460 and to prevent lipid oxidation, methanol (MeOH) containing 0.05% butylated hydroxytoluene and triphenylphosphine was added to plasma samples (200 µL) and brain tissues (500 µL). Each sample was also spiked with 50 µL of a structurally similar synthetic analog BRI-50469 (50 ng/mL), which was used as the internal standard. BRI-50460 was extracted from plasma samples by vortexing for 10 seconds, followed by resting on ice for 10 minutes while brain tissues were homogenized using a TissueLyser II (Qiagen) at 30 Hz for 1.5-min intervals. Subsequently, plasma and brain samples were centrifuged at 10,000 × g for 10 min at 4 °C, after which, the supernatant was collected and diluted with water to 10% MeOH. Samples were then purified by solid-phase extraction using Strata-X 33 μm Polymeric Reversed Phase cartridges (Phenomenex), dried, and prepared for LC-MS/MS analysis as previously described45.
BRI-50460 and the internal standard (BRI-50469) were separated and quantified in an Agilent 1290 UPLC system coupled to a Sciex API4000 system using electrospray ionization (ESI) in negative ion mode. A Poroshell 120 EC-C18 column (2.7 μm, 4.6 × 100 mm, Agilent) was used with the mobile phases: A, 0.1% formic acid in H2O; and B, 0.1% formic acid in MeOH. The gradient was as follows: 0–2 min, 20% B; 2–6 min, 20-95% B; 6–11 min, 95% B; 11–12 min, 95-20% B; 12–15 min, 20% B. An injection volume of 20 μL was used, the flow rate was set to 0.5 mL/min, and the column temperature was maintained at 40°C. Data analysis and quantification were performed using Skyline 23.1.3. The MRM transitions used to quantify BRI-50460 and its internal standard are detailed in Supplementary Table 6. The quantification of PUFAs and oxylipin metabolites was performed according to our previously described protocol7.
Free and protein-bound fractions of BRI-50460 were separated using Single-Use Rapid Equilibrium Dialysis Plates (Thermo Scientific) and quantified using the previously described LC-MS/MS method. To assess brain uptake, the Kpuu was calculated as the ratio of unbound drug concentration in the brain divided by unbound drug concentration in the plasma46.
iPSC AD models
Human iPSC-derived astrocytes: APOE isogenic human iPSCs were purchased from Jackson Laboratory (KOLF2.1 J). Dissociated forebrain NPCs were differentiated into astrocytes in astrocyte medium (#1801, ScienCell), as previously described47,48. Briefly, forebrain NPCs were maintained at a high density on poly L-ornithine hydrobromide (#P3655-50MG, Sigma) and laminin (#23017015, Thermo Fisher)- coated plates and cultured in NPC medium [DMEM/F12 (Corning, MT10090CV), 1 x N2 supplement (Thermo Fisher, 17502048), 1xB27 supplement (Thermo Fisher, 12587010), 1 mg/mL laminin, and 20 ng/mL FGF2 (Thermo Fisher 13256029)]. The cells were split at approximately 1:3 to 1:4 every week with Accutase (Sigma, SCR005). NPCs were differentiated into astrocytes by seeding dissociated single cells at a density of 15,000 cells/cm2 on Matrigel (Corning, 356255) -coated plates and cultured in complete astrocyte medium (#1801, ScienCell). After 30 days of differentiation, the astrocytes were ready for further experiments. Mature APOE ε3/ε3 astrocytes were seeded at 0.5 × 105 cells/well in a 24-well plate and grown to 70-80% confluency. Cells were washed with FBS-free medium and treated with or without 0.1 or 1 µM BRI-50460 for 30 minutes. Then, cells were treated with or without 2.5 µM amyloid beta 1-42 (Anaspec) for 72 hours. After treatment, cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (9806, Cell Signaling Technology, CST), and total cPLA2 and phosphorylated cPLA2 protein levels were detected by western blotting.
Human iPSC-derived neural progenitor cells (NPCs): Neural progenitor cells (NPCs) were differentiated from iPSCs using the StemCell Technologies STEMdiff™ SMADi Neural Induction Kit (Catalog # 08581) following manufacturer’s protocol. Briefly, JAX IPSC 1162 (APOE3/3) and JAX IPSC 1150 (APOE4/4) iPSCs were grown on Matrigel (Corning Catalog # 354230)-coated dishes in mTeSR™ Plus (StemCell Technologies Catalog # 100-0276). To initiate NPC induction, iPSCs were passaged to either Matrigel- or Poly-L-Ornithine hydrobromide (PORN; Sigma Aldrich P3655)/laminin (ThermoFisher 23017015)-coated plates and were fed with STEMdiff™ Neural Induction Medium + SMADi. At passage 3, cells were switched to STEMdiff™ Neural Progenitor Medium (StemCell Technologies Catalog #05833) and were maintained in that medium. For neuronal differentiation, NPCs were seeded on PORN/laminin-coated chamber slides at 20,0000 cells/cm2. For neuronal induction, cells were fed with the neuronal medium as described in Bardy et al16. At four to five weeks of neuronal differentiation, the cells were treated with 2.5 µM amyloid-beta 42 oligomers with or without BRI-50460 for 72 hours, then the cells were fixed in 4% PFA for 15 mins and stained with antibodies: phospho-cPLA2Ser505 (1:100, Sigma Aldrich Catalog # SAB4503812), phospho-tau Ser202/Thr205 (AT8) (1: 300, ThermoFisher Catalog # MN1020), CaMKIIα (1: 300, Catalog # 50049, Cell Signaling), synapsin I (1: 300, Sigma Aldrich Catalog # AB1543), and MAP2 (1: 1000, ThermoFisher Catalog # PA1-10005). All images were taken under a Leica TCS SP8 confocal microscope. The images were processed and analyzed using ImageJ and CellProfiler.
Data availability
All data will be available upon request to the corresponding authors after appropriate institutional approvals.
Code availability
Not applicable.
References
Cummings, J. et al. Alzheimer’s disease drug development pipeline: 2023. Alzheimers Dement 9, e12385 (2023).
Arellanes, I. C. et al. Brain delivery of supplemental docosahexaenoic acid (DHA): A randomized placebo-controlled clinical trial. EBioMedicine 59, 102883 (2020).
Tomaszewski, N. et al. Effect of APOE Genotype on Plasma Docosahexaenoic Acid (DHA), Eicosapentaenoic Acid, Arachidonic Acid, and Hippocampal Volume in the Alzheimer’s Disease Cooperative Study-Sponsored DHA Clinical Trial. J. Alzheimers Dis. 74, 975–990 (2020).
Yassine, H. N. et al. The effect of APOE genotype on the delivery of DHA to cerebrospinal fluid in Alzheimer’s disease. Alzheimers Res. Ther. 8, 25 (2016).
Asante, I., Louie, S. & Yassine, H. N. Uncovering mechanisms of brain inflammation in Alzheimer’s disease with APOE4: Application of single cell-type lipidomics. Ann. N. Y Acad. Sci. 1518, 84–105 (2022).
Wang, S. et al. Calcium-dependent cytosolic phospholipase A2 activation is implicated in neuroinflammation and oxidative stress associated with ApoE4. Mol. Neurodegen. 17, 42 (2022).
Ebright, B. et al. Eicosanoid lipidome activation in post-mortem brain tissues of individuals with APOE4 and Alzheimer’s dementia. Alzheimers Res Ther. 14, 152 (2022).
Six, D. A. & Dennis, E. A. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys. Acta. 1488, 1–19 (2000).
Hayashi, D., Mouchlis, V. D. & Dennis, E. A. Omega-3 versus Omega-6 fatty acid availability is controlled by hydrophobic site geometries of phospholipase A(2)s. J. Lipid Res. 62, 100113 (2021).
Kozaki, T. et al. Evaluation of drug–drug interaction between the novel cPLA2inhibitor AK106-001616 and methotrexate in rheumatoid arthritis patients. Xenobiotica. 45, 615–624 (2015).
Tomoo, T. et al. Design, synthesis, and biological evaluation of 3-(1-Aryl-1H-indol-5-yl)propanoic acids as new indole-based cytosolic phospholipase A2alpha inhibitors. J. Med. Chem. 57, 7244–7262 (2014).
Mouchlis, V. D. & Dennis, E. A. Membrane Association Allosterically Regulates Phospholipase A(2) Enzymes and Their Specificity. Acc. Chem. Res. 55, 3303–3311 (2022).
Dennis, E. A. Phospholipase A2 activity towards phosphatidylcholine in mixed micelles: surface dilution kinetics and the effect of thermotropic phase transitions. Arch. Biochem. Biophys. 158, 485–493 (1973).
McKew, J. C. et al. Inhibition of cytosolic phospholipase A2alpha: hit to lead optimization. J. Med. Chem. 49, 135–158 (2006).
Wang, J. Z., Xia, Y. Y., Grundke-Iqbal, I. & Iqbal, K. Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis. 33, S123–139 (2013).
Bardy, C. et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc. Natl. Acad. Sci. USA 112, E2725–2734 (2015).
Griffiths, J. & Grant, S. G. N. Synapse pathology in Alzheimer’s disease. Semin Cell Dev. Biol. 139, 13–23 (2023).
Ma, Q. L. et al. The Novel Omega-6 Fatty Acid Docosapentaenoic Acid Positively Modulates Brain Innate Immune Response for Resolving Neuroinflammation at Early and Late Stages of Humanized APOE-Based Alzheimer’s Disease Models. Front. Immunol. 11, 558036 (2020).
Maiole, F. et al. Synapsins are expressed at neuronal and non-neuronal locations in Octopus vulgaris. Sci. Rep. 9, 15430 (2019).
Lisman, J., Yasuda, R. & Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182 (2012).
Chang, J. Y., Nakahata, Y., Hayano, Y. & Yasuda, R. Mechanisms of Ca(2+)/calmodulin-dependent kinase II activation in single dendritic spines. Nat. Commun. 10, 2784 (2019).
Yasuda, R., Hayashi, Y. & Hell, J. W. CaMKII: a central molecular organizer of synaptic plasticity, learning and memory. Nat. Rev. Neurosci. 23, 666–682 (2022).
Wang, S. et al. Calcium-dependent cytosolic phospholipase A(2) activation is implicated in neuroinflammation and oxidative stress associated with ApoE4. Mol. Neurodegener. 17, 42 (2022).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Leslie, C. C. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot. Ess. Fat. Acids 70, 373–376 (2004).
Berk, P. D. & Stump, D. D. Mechanisms of cellular uptake of long chain free fatty acids. Mol. Cell. Biochem. 192, 17–31 (1999).
Gijon, M. A. & Leslie, C. C. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J. Leukoc. Biol. 65, 330–336 (1999).
Leslie, C. C. Cytosolic phospholipase A(2): physiological function and role in disease. J. Lipid Res. 56, 1386–1402 (2015).
Lin, L. L. et al. cPLA2 is phosphorylated and activated by MAP kinase. Cell 72, 269–278 (1993).
Xu, J. et al. Role of PKC and MAPK in cytosolic PLA2 phosphorylation and arachadonic acid release in primary murine astrocytes. J. Neurochem. 83, 1239–1239 (2002).
Colangelo, V. et al. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 70, 462–473 (2002).
Sanchez-Mejia, R. O. et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 11, 1311–1318 (2008).
Stephenson, D. T., Lemere, C. A., Selkoe, D. J. & Clemens, J. A. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer’s disease brain. Neurobiol. Dis. 3, 51–63 (1996).
Palavicini, J. P. et al. Oligomeric amyloid-beta induces MAPK-mediated activation of brain cytosolic and calcium-independent phospholipase A(2) in a spatial-specific manner. Acta Neuropathol. Commun. 5, 56 (2017).
Sun, G. Y. et al. Integrating cytosolic phospholipase A(2) with oxidative/nitrosative signaling pathways in neurons: a novel therapeutic strategy for AD. Mol. Neurobiol. 46, 85–95 (2012).
Qu, B., Gong, Y., Gill, J. M., Kenney, K. & Diaz-Arrastia, R. Heterozygous knockout of cytosolic phospholipase A(2alpha) attenuates Alzheimer’s disease pathology in APP/PS1 transgenic mice. Brain Res. 1670, 248–252 (2017).
Chuang, D. Y., Simonyi, A., Kotzbauer, P. T., Gu, Z. & Sun, G. Y. Cytosolic phospholipase A2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. J. Neuroinflamm. 12, 199 (2015).
Katritch, V., Rueda, M. & Abagyan, R. Ligand-guided receptor optimization. Methods Mol. Biol. 857, 189–205 (2012).
Dessen, A. et al. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell 97, 349–360 (1999).
Sadybekov, A. A. et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459 (2022).
https://www.molsoft.com/icmpro/calculate-properties.html#toxscore.
https://www.molsoft.com/icmpro/calculate-properties.html#bbb.
Jenkins, C. M., Han, X., Mancuso, D. J. & Gross, R. W. Identification of calcium-independent phospholipase A2 (iPLA2) beta, and not iPLA2gamma, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J. Biol. Chem. 277, 32807–32814 (2002).
Shimizu, H. et al. AK106-001616, a Potent and Selective Inhibitor of Cytosolic Phospholipase A(2): In Vivo Efficacy for Inflammation, Neuropathic Pain, and Pulmonary Fibrosis. J. Pharm. Exp. Ther. 369, 511–522 (2019).
Duro, M. V. V. et al. Synthesis and Preclinical Evaluation of 22-[(18)F]Fluorodocosahexaenoic Acid as a Positron Emission Tomography Probe for Monitoring Brain Docosahexaenoic Acid Uptake Kinetics. ACS Chem. Neurosci. 14, 4409–4418 (2023).
de Lange, E. C. Utility of CSF in translational neuroscience. J. Pharmacokinet. Pharmacodyn. 40, 315–326 (2013).
Tcw, J. et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 9, 600–614 (2017).
Wang, S. et al. Cellular senescence induced by cholesterol accumulation is mediated by lysosomal ABCA1 in APOE4 and AD. Mol. Neurodegener. 20, 15 (2025).
Acknowledgements
The authors acknowledge the Center for Advanced Research Computing (CARC) at the University of Southern California (USC) for providing computing resources that have contributed to the research results reported within this publication. URL. https://carc.usc.edu. This work was supported by the National Institute on Aging (RF1AG076124, R01AG055770, R01AG067063, R01AG054434, R21AG056518, and P30AG066530 to H.N.Y.; the Alzheimer’s Drug Discovery Foundation (ADDF) (GC-201711–2014197 to H.N.Y.), and donations from the Vranos and Tiny Foundations and Ms. Lynne Nauss to H.N.Y., R01GM147537 to VK, and USC CTSI KL2 (UL1 TR000004) to IA. HNY, VK, SL are supported by U01AG094622 from NIA.
Author information
Authors and Affiliations
Contributions
Anastasiia V. Sadybekov ran the V-SYNTHES2 virtual screening in on-demand chemical space and conducting two rounds of optimization for potency and selectivity, Marlon Vincent Duro did the cPLA2 screening assays, Shaowei Wang designed and performed the cell assays, Brandon Ebright formulated the small molecules and measures levels using LCMS, Dante Dikeman formulated the small molecules and measures levels using LCMS, Cristelle Hugo completed the iPSC derived astrocyte experiment, Bilal Ersen Kerman supervised and helped with iPSC derived neurons and astrocytes, Qiu-Lan Ma completed the iPSC derived neuron experiments, Antonina L. Nazarova, Arman A. Sadybekov helped with V-SYNTHES2 virtual screening in on-demand chemical space and optimization for potency and selectivity, Isaac Asante assisted with the formulations, Stan G Louie supervised the formulations, in vivo study design, mass spec measurements, Vsevolod Katritch led the drug discovery team and Hussein N. Yassine led the neuroscience and target validation team. All authors contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
HNY is the founder of PeBRx, which is developing cPLA2 inhibitors. No other authors have any competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sadybekov, A.V., Duro, M.V., Wang, S. et al. Development of potent, selective cPLA2 inhibitors for targeting neuroinflammation in Alzheimer’s disease and other neurodegenerative disorders. npj Drug Discov. 3, 2 (2026). https://doi.org/10.1038/s44386-025-00035-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44386-025-00035-0
This article is cited by
-
Evidence for cPLA2 activation in Alzheimer’s disease synaptic pathology
Acta Neuropathologica Communications (2026)
