
Abstract
Over the past decade, advances in generative modeling, such as generative adversarial networks, masked autoencoders, and diffusion models, have significantly transformed biological research and discovery, enabling breakthroughs in molecule design, protein generation, catalysis discovery, drug discovery, and beyond. At the same time, biological applications have served as valuable testbeds for evaluating the capabilities of generative models. Recently, flow matching has emerged as a powerful and efficient alternative to diffusion-based generative modeling, with growing interest in its application to problems in biology and life sciences. This paper presents the first comprehensive survey of recent developments in flow matching and its applications in biological domains. We begin by systematically reviewing the foundations and variants of flow matching, and then categorize its applications into three major areas: biological sequence modeling, molecule generation and design, and peptide and protein generation. For each, we provide an in-depth review of recent progress. We also summarize commonly used datasets and software tools, and conclude with a discussion of potential future directions.
Similar content being viewed by others

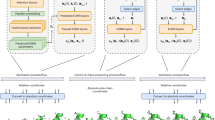
Introduction
Flow matching (FM)1 has recently emerged as a powerful paradigm for generative modeling, offering a flexible and scalable framework applicable across a wide range of domains, such as computer vision1,2, and natural language processing3,4. By constructing a continuous probability trajectory between simple and complex distributions, FM provides an efficient and principled method to model high-dimensional, structured data. While FM has demonstrated strong performance in conventional generative tasks such as image, video, and language synthesis, its potential extends far beyond these domains. In particular, its ability to model diverse modalities while preserving structural and geometric constraints makes it especially well-suited for applications in biology and life sciences.
At the same time, biological and life science applications present a natural testbed for FM (Fig. 1). These tasks, ranging from genomic sequence modeling5,6,7, molecular graph generation8,9,10, and protein structure prediction11,12,13, to biomedical image synthesis14,15,16,17, are often high-dimensional, multimodal, and governed by strict structural, physical, or biochemical constraints. In fact, they have already served as benchmarks for validating the performance of various generative modeling paradigms, such as Generative Adversarial Networks18,19,20, Masked Autoencoders21,22,23,24, and Diffusion Models25,26,27. Compared to traditional rule-based simulations28,29,30,31 and physics-driven models32,33,34,35, which often suffer from limited scalability and reliance on expert-crafted rules, these machine-learning-based generative models offer a data-driven alternative that can scale to complex biological systems, adapt to diverse modalities, and generalize beyond handcrafted constraints36,37,38,39,40,41,42,43,44. By learning directly from empirical data, they enable the generation of biologically plausible outputs while significantly reducing the need for domain-specific assumptions. FM, as a newer yet promising alternative, inherits key advantages from these models such as expressiveness, scalability, and data efficiency, while introducing a more stable training objective based on continuous probability flows. Its ability to generate high-quality samples with fewer inference steps makes it particularly appealing for biological applications, where modeling precision and computational efficiency are both critical.

Flow matching serves as a powerful generative modeling paradigm for a wide range of biological and life science applications. Conversely, these domains offer rich and diverse tasks for evaluating and advancing flow matching techniques. In this survey, we first present state-of-the-art flow matching models and their variants, then categorize their applications into four major areas: sequence modeling, molecule generation, protein design, and other emerging biological applications. The corresponding curated resources are available at https://github.com/Violet24K/Awesome-Flow-Matching-Meets-Biology.
Interest in applying FM to biological problems is growing rapidly. As illustrated in Fig. 2, we have observed a steadily growing trend in the number of FM-related publications, with a visible rise in bio-related applications. The first biological applications appeared at NeurIPS 202345,46, both focusing on molecule generation. This momentum continued with the introduction of FM-based protein generation models at ICLR 202447, followed by further progress in biological sequence and peptide generation at ICML 2024. Beyond these milestones, 2024 and 2025 have seen the emergence of increasingly specialized FM variants, such as categorical FM48, rectified FM49, and non-Euclidean formulations including Riemannian50 and Dirichlet51 FM. Many of these have begun to find applications in structural biology, molecular conformation modeling, and biomedical imaging. More recently, NeurIPS 2025 features over 30 accepted FM papers, and ICLR 2026 received more than 150 FM-related submissions. As of the time this survey is under peer review (Nov 2025), these venues collectively include over 20 new FM-for-biology works. Since their proceedings are not yet public, we only cover the NeurIPS 2025 papers with available preprints and leave full coverage of these emerging results to future iterations. This upward trajectory highlights not only the methodological innovation within FM, but also its growing relevance in life science domains that demand high-dimensional, structure-aware generative modeling.

The blue line indicates the total number of FM papers, while the orange line shows the subset focused on biological applications. Annotations highlight key milestones in FM and its adoption for molecule, sequence, and protein generation, illustrating the rapid growth and expanding interest in this area.
As both FM and its biological applications evolve, the landscape has become increasingly fragmented, making it difficult to keep track of key developments and emerging trends. This survey addresses this gap by providing the first comprehensive review of FM in the context of biology and life sciences. We begin with a systematic overview of FM methods and variants, and then categorize their biological applications into three core areas: biological sequence modeling, molecule generation and design, and protein generation. We also review auxiliary topics such as bioimage modeling and spatial transcriptomics, summarize commonly used datasets and tools, and conclude with open challenges and future directions. Our goal is to offer an accessible entry point for newcomers, while equipping experienced researchers with a clear map of the field’s current trajectory. Our curated resources are publicly available at https://github.com/Violet24K/Awesome-Flow-Matching-Meets-Biology.
Challenges of generative modeling for biology
Biological systems are among the most intricate and multifaceted systems in the natural world52,53,54, shaped by billions of years of evolution and governed by deeply intertwined physical, chemical, and informational processes. Modeling such systems has long been a grand challenge across scientific disciplines, demanding tools that can reconcile precision with flexibility55,56,57,58,59,60. The complexity of biological data and phenomena stems from a confluence of factors, with some of the most formidable challenges including: (1) the necessity to embed rich domain knowledge, ranging from physical laws to biochemical constraints, into generative models in a way that ensures structural and functional validity; (2) the scarcity, incompleteness, and noise characteristic of real-world biological datasets, often resulting from expensive or error-prone experimental procedures; (3) the inherently multi-scale and multi-modal nature of biological processes, which span atomic interactions to cellular behavior, and integrate diverse data types such as sequences, structures, and spatial-temporal signals; (4) the increasing demand for controllable and condition-aware generation, where outputs must satisfy explicit biological properties or therapeutic objectives; and (5) the pressing need for models that are not only accurate but also computationally scalable and sample-efficient, especially in applications such as drug discovery or protein design where inference speed can be critical. Together, these challenges make it challenging for biology models.
FM, as a recently introduced generative modeling paradigm, holds strong potential for addressing the unique challenges of biological data. It learns a deterministic vector field to map a simple base distribution directly to complex target data via continuous probability trajectories. This yields several advantages particularly relevant to biological applications, such as faster and more stable sampling, easier conditioning on structured inputs, and the ability to incorporate geometric or physical priors into the modeling process. Since its introduction, a growing number of studies have explored the use of FM in tackling biological tasks. These early successes demonstrate not only the method’s versatility but also its capacity to model the structured, multimodal, and constraint-rich nature of biological systems, positioning FM as a compelling alternative to conventional generative frameworks in the life sciences.
Our contributions
This survey presents the first comprehensive review of FM and its applications in biology and life sciences. Our key contributions are summarized as follows:
-
A unified taxonomy of flow matching variants: we introduce a structured taxonomy of FM methodologies, spanning general FM, conditional and rectified FM, non-Euclidean and discrete FM, and hybrid variants.
-
In-depth survey of biological applications: we systematically categorize and review the use of FM across three primary biological domains: biological sequence modeling, molecule generation and design, and protein generation. We further explore several other emerging applications beyond this scope.
-
Comprehensive benchmark and dataset survey: we compile and review widely used biological datasets, benchmarks, and software tools adopted in FM research.
-
Trend, challenges, and emerging directions: we contextualize the evolution of FM through bibliometric trends and identify key methodological innovations. We further analyze domain-specific modeling challenges which may motivate new FM research directions.
-
Bridging modeling and biology communities: by mapping methodological advances in FM to diverse biological challenges, we offer a cross-disciplinary bridge that connects the machine learning community developing FM algorithms with the biological sciences community seeking powerful generative tools.
Connection to existing survey
Existing related surveys can be broadly categorized into three groups. The first category focuses exclusively on generative modeling methodologies. These surveys either provide comprehensive overviews of specific classes of generative models61,62,63 or examine their applications within particular domains, such as computer vision64, recommendation systems65, and anomaly detection66. The second category surveys the use of generative models in biology prior to the advent of FM. For example67, reviews generative models for molecular design68, focuses on de novo drug design, and69 provides a broad overview of machine learning methods in both predictive and generative biological modeling. A concurrent survey70 emphasizes practical guidance and open-source tooling, our survey offers a unified taxonomy of flow-matching methodologies with fine-grained links to specific biological problem classes. Table 1 presents a comparison of existing surveys on generative modeling, highlighting their covered model classes and application domains. To the best of our knowledge, this work presents the first comprehensive survey dedicated to FM and its applications in biology and life sciences. By bridging recent developments in generative modeling with their emerging applications in biological domains, this survey aims to fill a critical gap in the literature.
Outline of the survey
To provide a comprehensive understanding of FM in the context of biology and life sciences, this survey is organized into several key sections. We begin by introducing the fundamental concepts and methodologies underlying FM in Section “Flow-matching basics”, establishing a foundation for its application in biological contexts. Next, in Section “Sequence modeling”, we delve into specific areas of application, starting with biology sequence generation, followed by molecule generation and design in Section “Molecule generation”, and then peptide and protein generation in Section “Protein generation”, each highlighting recent advancements and representative studies. In Section “Other bio applications”, we also discuss other emerging applications of FM in biology. Finally, we conclude by outlining future research directions and potential challenges, aiming to inspire further exploration and innovation in this rapidly evolving field. Figure 3 presents the overall structure of this survey, with each section divided into various subtopics for a more detailed exploration.

We begin by introducing the foundations of flow matching, including its core models and variants. Our taxonomy then categorizes flow matching applications into major biological domains and tasks.
Background
Generative modeling seeks to learn a probability distribution pdata(x) from a dataset of examples \({\{{x}_{i}\}}_{i=1}^{N}\), such that we can generate new samples \(\widehat{x} \sim {p}_{\theta }(x)\) that resemble real data. These models underpin advances in biology tasks ranging from molecular generation to protein design and cellular imaging67,68,71,72,73, with AlphaFold11,12,74 standing out as one of the most prominent and transformative examples, recognized with the Nobel Prize in 2024. AlphaFold leverages deep generative principles to predict protein 3D structures directly from amino acid sequences, a task that had challenged the field for decades13,60,75. By effectively modeling the conditional distribution over protein conformations, AlphaFold not only revolutionized protein structure prediction but also highlighted the broader potential of generative models to capture complex, structured biological phenomena at scale. In biology domains, data is often high-dimensional, multimodal, and governed by physical or biochemical constraints76,77,78,79, requiring generative models to strike a careful balance between validity, diversity, and interpretability. In this section, we provide a brief overview of the major paradigms in generative modeling, with the goal of establishing a conceptual and mathematical foundation for understanding more recent developments such as FM. For clarity and consistency, all symbols used throughout this paper are summarized in Table 2. We also briefly compare different generative modeling paradigms and FM in Table 3. To further enhance accessibility for readers from diverse scientific backgrounds, we provide a glossary of key technical terms in the Supplementary Information Section “Technical Terms”.
Variational autoencoder (VAE)
Variational autoencoders (VAEs)80,81,82,83,84 are a class of latent-variable generative models that aim to model the data distribution pdata(x) through a learned probabilistic decoder pθ(x∣z), where z is a latent variable drawn from a prior p(z), typically a standard Gaussian. Since the true posterior p(z∣x) is often intractable, VAEs introduce an approximate posterior qϕ(z∣x), known as the encoder, and optimize the model using variational inference. The training objective is to maximize a variational lower bound, known as the evidence lower bound (ELBO), on the marginal log-likelihood of the data:
The first term encourages accurate reconstruction of the input data from the latent variable z, while the second term regularizes the approximate posterior to stay close to the prior distribution. During training, the reparameterization trick is used to allow gradients to backpropagate through the sampling process, typically by expressing z ~ qϕ(z∣x) as z = μ(x) + σ(x) ⊙ ϵ, where \(\epsilon \sim {\mathcal{N}}(0,I)\). However, VAEs often suffer from over-regularization and produce blurred outputs, especially in high-dimensional domains such as images and molecular graphs85,86,87.
Generative adversarial network (GAN)
Generative adversarial networks (GANs)18 are a class of implicit generative models that learn to generate realistic data by playing a two-player minimax game between two neural networks: a generator Gθ and a discriminator Dϕ. The generator maps noise samples z ~ p(z), typically drawn from a simple prior such as a Gaussian, into synthetic data samples Gθ(z). The discriminator attempts to distinguish between real samples x ~ pdata and generated samples Gθ(z). The original GAN objective is formulated as:
GANs are known to suffer from several practical challenges, including training instability, sensitivity to hyperparameters, and mode collapse Numerous variants have been proposed to improve training dynamics and sample diversity, such as Wasserstein GANs88, Least-Squares GANs89, and conditional GANs90. In biological applications, GANs have been used for generating realistic cell images91, synthesizing gene expression profiles20,92, and augmenting scarce datasets93. Despite their limitations, their ability to capture complex data distributions without explicit density estimation makes them a compelling choice for modeling high-dimensional biological data94.
Flow-based model
Flow-based models (also known as normalizing flows)95,96 are a family of generative models that construct complex data distributions by applying a sequence of invertible transformations to a simple base distribution, typically a standard Gaussian distribution. Given a base variable z ~ pz(z), a flow model learns an invertible mapping x = fθ(z) such that the model distribution pθ(x) can be computed exactly via the change-of-variables formula:
The goal is to train the parameters θ to maximize the log-likelihood of the observed data under this model. The invertibility of fθ allows for exact and tractable likelihood computation, efficient sampling, and deterministic inference. To ensure both tractability and expressivity, flow models are often constructed as a composition of multiple simple bijective transformations:
Each component fk is designed to allow efficient computation of the Jacobian determinant and its inverse. Representative architectures include NICE97, RealNVP98, Glow99, and Masked Autoregressive Flows (MAF)100, which utilize affine coupling layers or autoregressive transforms to maintain invertibility.
However, the invertible constraint on fθ along with the need to compute the determinant of the Jacobian \(\frac{\partial {f}_{\theta }(x)}{\partial x}\) imposes significant constraints on model expressiveness and design flexibility. Continuous normalizing flow (CNF)101 address these limitations by replacing the discrete sequence of transformations (Eq. (4)) with a continuous-time dynamic system \(\frac{dx}{dt}=f(x(t),t)\). This formulation leads to a more efficient computation of the log-density change:
Notably, the vector field f is not required to be invertible.
CNFs serve as a foundational building block for FM. While CNFs allow for more expressive modeling, their training via maximum likelihood still demands computationally expensive ODE solvers. A core motivation behind flow matching is to simplify the training of ODE-based generative models, without sacrificing the benefits of continuous-time formulations.
Diffusion models (DM)
Diffusion models25,102,103,104,105 are a family of likelihood-based generative models that generate data by reversing a gradual noising process. They define a forward process that incrementally transforms data into noise, and parameterize a neural network to fit the groundtruth reverse process, recovering data from noise step by step.
Forward process
The forward process defines a sequence of latent variables \({\{{x}_{t}\}}_{t=0}^{T}\), which are the gradually corrupted version of the clean data x0 ~ pdata. A typical forward process is formulated as a set of Gaussian distributions conditioned on the previous step:
where {βt} is called noise schedule. Usually, the distribution of the corrupted data at any time t has a closed form:
Training
Similar to many likelihood-based models, negative log-likelihood is a canonical choice of the loss function25,102,106. Beyond that, cross-entropy or square error are also widely used25,107. Based on that, neural networks (NNs) are used to parameterize various components of the diffusion process, such as to predict the data108, predict the noise25, and predict the score105. The following unweighted regression loss for predicting the noise is a popular example:
Generation
Equipped with the NN-parameterized component, the reverse process of the diffusion process is used for generation. For example, the reverse process with the NN-predicted noise ϵθ can denoise the Gaussian noise \({x}_{T} \sim {\mathcal{N}}(0,I)\) gradually:
A well-known limitation of diffusion models is their slow sampling process, which often requires hundreds of iterative steps. To address this inefficiency, several acceleration techniques have been proposed, including the adoption of tailored numerical solvers109, model distillation108, and continuous-time formulations105,106. Notably, Probability flow ODE104 and DDIM105 demonstrate that there exists a deterministic ODE whose solution shares the same marginal distributions as the reverse-time stochastic differential equation (SDE) used in diffusion models. This observation is conceptually aligned with the idea behind flow matching (FM), although both probability flow ODE and DDIM remain trained using the standard loss functions of diffusion models, such as the evidence lower bound (ELBO).
Consistency models
Consistency models (CMs)110 are a recent family of generative models built upon the diffusion models. They aim to bypass the slow iterative denoising procedure of diffusion sampling by learning a direct mapping from noise to data.
Forward process
A consistency model is a neural function fθ(xt, t) that approximates the solution of the Probability flow ODE (PF-ODE) in closed form. Given a noisy sample xt at time t, fθ predicts its corresponding clean data x0. A defining property of CMs is self-consistency: all points on the same diffusion trajectory should map to the same output.
Training
CMs are trained from two main paradigms: Consistency distillation and Consistency training.
Consistency distillation (CD)110 distills a pretrained diffusion teacher into fθ. Given adjacent states (xt, xt+Δ) along the teacher’s PF-ODE trajectory, the student minimizes
Consistency training (CT)110,111 trains fθ from scratch without a teacher by sampling two noisy versions (xs, xt) of the same data x0 via a shared noise realization z: xt = x0 + σ(t)z, xs = x0 + σ(s)z:
Beyond the original formulation110, several variants have extended this idea. Multi-step CMs112 refine generation by repeatedly evaluating fθ over decreasing times (tn → 0). In addition, diffusion models are integrated with consistency models113,114. Some recent approaches further emphasize later noise stages during training115.
Flow-matching basics
In this section, we provide background knowledge on flow-matching (FM) models, including general FM and discrete FM.
General flow-matching
Flow-matching is a continuous-time generative framework that generalizes diffusion models by regressing a vector field that transports one distribution into another116. In general, FM aims to construct a velocity field uθ(x, t) to transport a source p0 to a target p1 via the continuity equation:
An FM can be trained by minimizing the squared loss between the neural velocity field uθ(x, t) and a reference velocity field \({u}_{t}^{* }(x,t)\) as follows:
Promising as it might be, directly optimizing the objective in Eq. (15) is impractical: the optimal velocity field u*(x, t) encodes a highly complex joint transformation between two high-dimensional distributions117. To overcome this challenge, conditional FM variants have been introduced to enable more tractable training (Paragraph -0a). Concurrently, rectified FM methods propose improved noise couplings along the straight-line probability path (Paragraph -0b). Finally, non-Euclidean FM extensions generalize the framework from flat Euclidean space to curved manifolds, accommodating data with intrinsic geometric structure (Paragraph -0c).
Conditional FM116,118,119,120
To resolve the intractable u*(x, t), conditional FM introduces a conditioning variable z, e.g., class label, and define a conditional path p(x∣t, z) such that the induced global path p(x∣t) = ∫zp(x∣t, z)p(z)dz transforms p0 to pdata and the corresponding conditional velocity field has analytical form. A conditional FM can be trained by minimizing the quadratic loss between the neural velocity field uθ(x, t) and the conditional velocity field \({u}_{t}^{* }(x,t,z)\) as follows:
The training procedure involves sampling a conditioning variable z, e.g., via linear interpolation119,121 or Gaussian path116, and random time t, constructing xt along the prescribed path, and minimizing the corresponding loss. Once the model is trained, the sampling/generation process is done by solving the learned ODE dx/dt = uθ(x, t) using an ODE solver from t = 0 (noise) to t = 1 (data). The key theoretical foundation of conditional FM is that the gradient of the FM objective in Eq. (15) is equivalent to gradient of the CFM objective in Eq. (16). Building upon the conditioning variable z, one can define velocity field in analytical forms with tractable training.
Rectified FM49,120,121,122,123
Infinite probability path exist between source and target distributions that can be leveraged by conditional FM, rectified FM prefers the linear transport trajectory that best connect two distributions121. proposes to train a velocity field carrying each sample x0 to its paired target x1 along nearly-straight lines via:
where pi is a coupling of p0 and p1. It is shown that the optimal transport (OT) coupling provides a straight coupling for p0 and p1, simplifying the flow and reducing curliness120,122.
Non-Euclidean FM50,124,125,126,127
Non-Euclidean flows extend continuous flows to curved data spaces. For example127, introduce Riemannian Continuous Normalizing Flows, defining the generative flow by an ODE on the manifold to model flexible densities on spheres, tori, hyperbolic spaces, etc.126. propose Neural Manifold ODEs, integrating dynamics chart-by-chart (e.g. via local coordinate charts) so that the learned velocity field stays tangent to the manifold. More recently124, propose Riemannian FM by using geodesic distances as a “premetric” they derive a closed-form target vector field pushing a base distribution to the data without any stochastic diffusion or divergence term. On simple manifolds (e.g. spheres or hyperbolic space where geodesics are known) Riemannian FM is completely simulation-free, and even on general geometries it only requires solving a single ODE without calculating expensive score or density estimates125. introduce Fisher FM, treating categorical distributions as points on the probability simplex with the Fisher-Rao metric and transporting them along spherical geodesics. In general, Riemannian flows replace straight-line interpolations with intrinsic geodesics and explicitly account for the manifold’s metric (e.g. via the Riemannian divergence in the change-of-density). These works tackle the challenges of defining tangent vector fields and volume corrections on curved spaces via chart-based integration, metric-adjusted log-density formulas, or flow-matching losses that avoid divergence estimates. Overall, they enable scalable generative modeling on curved domains (spheres, Lie groups, statistical manifolds, etc.), respecting curvature in ways standard Euclidean FM cannot.
Discrete flow-matching
Discrete FM has emerged as a powerful paradigm for generative modeling over discrete data domains, such as sequences, graphs, and categorical structures, covering a wide range of biological objects4,107. By extending the principles of continuous FM to discrete spaces, DFM enables the design of efficient, non-autoregressive generative models. This section delves into two principal frameworks: Continuous-Time Markov Chain (CTMC)-based methods (Paragraph -0a) and simplex-based methods (Paragraph -0b).
Continuous-time Markov chain (CTMC)
CTMC-based approaches model the generative process as a continuous-time stochastic evolution over discrete states, leveraging the mathematical framework of continuous-time Markov chains to define and learn probability flows128. utilizes CTMCs to model flows over discrete state spaces. This approach allows for the integration of discrete and continuous data, facilitating applications like protein co-design by enabling multimodal generative modeling. Fisher Flow125 adopts a geometric perspective by considering categorical distributions as points on a statistical manifold endowed with the Fisher-Rao metric. This approach leads to optimal gradient flows that minimize the forward Kullback-Leibler divergence, improving the quality of generated discrete data129. expanded the design space of discrete generative models by allowing arbitrary discrete probability paths within the CTMC framework. This holistic approach enables the use of diverse corruption processes, providing greater flexibility in modeling complex discrete data distributions. DeFog130 is a discrete FM framework tailored for graph generation. By employing a CTMC-based approach, DeFoG achieves efficient training and sampling, outperforming existing diffusion models in generating realistic graph.
Simplex-based discrete FM
Simplex-based methods operate within the probability simplex, modeling flows over continuous relaxations of discrete distributions. These approaches often employ differentiable approximations to handle the challenges posed by discrete data. SimplexFlow131 combines continuous and categorical flow matching for 3D de novo molecule generation, where intermediate states are guaranteed to reside on the simplex. Dirichlet FM51 utilizes mixtures of Dirichlet distributions to define probability paths over the simplex, addressing discontinuities in training targets and enables efficient. α-flow132 unifies various continuous-state discrete FM models under the lens of information geometry. By operating on different α-representations of probabilities, this framework optimizes the generalized kinetic energy, enhancing performance in tasks such as image and protein sequence generation. STGFlow133 employs a Gumbel-Softmax interpolant with a time-dependent temperature for controllable biological sequence generation, which includes a classifier-based guidance mechanism that enhances the quality and controllability of generated sequences.
Sequence modeling
FM has emerged as a powerful framework for biological sequence generation, offering deterministic and controllable modeling of discrete structures such as DNA, RNA, and whole-genome data. In this section, we survey different FM models designed for biological sequence generation, including DNA sequence, RNA sequence, whole-genome modeling, and antibody design. By leveraging continuous transformations, flow matching enables efficient generation of sequences conditioned on various biological constraints and properties.
DNA sequence generation
Early deep generative models, e.g. GANs or autoregressive models, struggled to satisfy the complex constraints of functional genomics sequences. FM models provide natural solutions to bridge this gap by mapping discrete nucleotide sequences into continuous probabilistic spaces for training51. Instead of simulating a stochastic diffusion51, FM models directly train a continuous vector field that transports a simple base distribution, e.g., uniform distribution over nucleotides, into the empirical DNA data distribution.
Fisher-Flow125 introduces a geometry-based flow matching approach, which treats discrete DNA sequences as points on a statistical manifold endowed with the Fisher-Rao metric. By allowing for continuous reparameterization of discrete data, probability mass is transported along optimal geometric paths on the positive orthant of a hypersphere, achieving state-of-the-art performance on DNA promoter and enhancer sequence generation benchmarks compared to earlier diffusion-based and flow-based models.
Besides categorical distribution, Dirichlet distribution is adopted to handle discrete sequences. Dirichlet Flow51 utilizes mixtures of Dirichlet distributions to define probability paths on the simplex, addressing discontinuities and pathologies in naive linear flow matching. Dirichlet Flow enables one-step DNA sequence generation and achieves superior distributional metrics and target-specific design performance compared to prior models on complex DNA design tasks.
In addition, STGFlow133 proposes straight-through guidance, combining Gumbel-Softmax flows with classifier-based guidance to steer the generation process toward desired sequence properties, facilitating controllable de novo DNA sequence generation. MOG-DFM134 generalizes discrete flow matching guidance into a multi-objective paradigm. It leverages multiple scalar objectives and computes a hybrid rank-directional score at each sampling step.
RNA sequence generation
Flow matching has recently been applied to RNA sequence and structure design. Rather than focusing solely on sequence generation, existing FM methods prioritize structural fidelity, enabling advanced applications in inverse folding, protein-conditioned design, and ensemble backbone sampling. RNACG135 introduces a versatile flow-matching framework for conditional RNA generation that supports tasks ranging from 3D inverse folding to translation efficiency prediction. RNAFlow136 couples an RNA inverse-folding module with a pretrained structure predictor to co-generate RNA sequences and their folded structures in the context of bound proteins. RiboGen137 develops the first deep network to jointly synthesize RNA sequences and all-atom 3D conformations via equivariant multi-flow architectures. RNAbpFlow138 presents a SE(3)-equivariant flow-matching model that conditions on both sequence and base-pair information to sample diverse RNA backbone ensembles. More recently, RiboFlow139 proposes to synergize the design of RNA structure and sequence by integrating RNA backbone frames, torsion angles and sequence features for an explicit modeling on RNA’s dynamic conformation.
Whole-genome modeling
At the whole-genome level, flow matching has been applied to model single-cell genomics data. GENOT140 employs entropic Gromov-Wasserstein flow matching to learn mappings between cellular states in single-cell transcriptomics, facilitating studies of cell development and drug response. cellFlow141 proposes a generative flow-based model for single-cell count data that operates directly in raw transcription count space, preserving the discrete nature of the data. CFGen142 introduces a flow-based conditional generative model capable of generating multi-modal and multi-attribute single-cell data, addressing tasks such as rare cell type augmentation and batch correction.
Antibody sequence generation
FM has also been utilized for antibody sequence generation. IgFlow143 proposes a SE(3)-equivariant FM model for de novo antibody variable region generation (heavy/light chains and CDR loops). IgFlow supports unconditional antibody sequence-structure generation and conditional CDR loop inpainting, producing structures comparable to those from a diffusion-based model while achieving higher self-consistency in conditional designs; it also offers efficiency benefits like faster inference and better sample efficiency than the diffusion counterpart. dyAb144 proposes a flexible antibody design FM, which integrates coarse-grained antigen-antibody interface alignment with fine-grained flow matching on both sequences and structures. By explicitly modeling antigen conformational changes (via AlphaFold2 predictions) before binding, dyAb significantly improves the design of high-affinity antibodies in cases where target antigens undergo dynamic structural shifts.
These advancements demonstrate the versatility of flow matching in modeling complex biological sequences and structures, providing a unified framework for deterministic and controllable generation across various biological domains.
Molecule generation
Molecule generation is a fundamental task in biological modeling, playing a crucial role in drug discovery, material design, and understanding molecular interactions145,146,147. The ability to generate novel molecules with desired properties has significant implications for both theoretical and applied research in life sciences148,149. Traditional approaches, such as rule-based simulations and heuristic algorithms, often face challenges in scalability and diversity150,151. In contrast, generative models, including flow matching, offer a data-driven approach to efficiently explore the vast chemical space26,152,153.
In this section, we review recent advancements in molecule generation using flow matching techniques. We focus on methods that leverage continuous probability flow trajectories to generate novel molecular structures and properties, highlighting how flow matching has enhanced molecule generation.
2D molecule generation
Although real-world molecules are inherently three-dimensional objects, as illustrated in Fig. 4, researchers often simplify the problem by using 2D graph-based molecular modeling when the 3D structure is not the primary focus154,155,156. This approach offers several advantages, including increased computational efficiency and reduced information requirements during inference.

Each molecule is visualized as a 2D graph, where atoms are nodes and chemical bonds are edges, capturing both structural and topological properties.
Flow matching on graph data remains relatively unexplored, as the concept of flow matching itself is still under development. Nevertheless, existing studies often use 2D molecule generation as a preliminary test case to evaluate newly proposed flow matching variants. For instance, Eijkelboom et al.157 combine flow matching with variational inference to introduce Variational Flow Matching for graph generation and CatFlow for handling categorical data. Additionally, GGFlow158 presents a discrete flow matching generative model that integrates optimal transport for molecular graphs. This model features an edge-augmented graph transformer, enabling direct communication among chemical bonds, thereby improving the representation of molecular structures. DeFoG159 introduces a discrete formulation of flow matching tailored to the graph domain, explicitly decoupling the training and sampling phases to overcome inefficiencies in traditional diffusion-based models. By leveraging permutation-invariant graph matching objectives and exploring a broader sampling design space, DeFoG achieves strong empirical results on molecular graph generation with significantly fewer refinement steps.
3D molecule generation
Generating accurate 3D molecular structures is a critical task in drug discovery and structural biology160. As illustrated in Fig. 5, unlike 2D graph-based approaches, which primarily capture atomic connectivity, 3D molecular representations inherently encode spatial information, including bond angles, torsions, and stereochemistry. This spatial fidelity is essential for modeling interactions such as molecular docking, binding affinity, and conformational stability. While 2D representations cannot distinguish between stereoisomers or capture geometric nuances, 3D methods accurately model spatial conformation, enabling a more precise understanding of molecular properties145,161,162.

Atoms are shown as nodes positioned in 3D Euclidean space, and bonds are represented as edges connecting them. These visualizations capture spatial geometry and stereochemistry important for molecular property prediction.
SE(3)-equivariant
To ensure physically meaningful and symmetry-consistent outputs, recent advancements have incorporated SE(3)-equivariant neural architectures into flow matching models. These models leverage the inherent symmetries of molecular systems, modeling graph generation as a continuous normalizing flow over node and edge features. For instance, Megalodon163 introduces scalable transformer models with basic equivariant layers, trained using a hybrid denoising objective to generate 3D molecules efficiently, achieving state-of-the-art results in both structure generation and energy benchmarks. EquiFM45 further improves the generation of 3D molecules by combining hybrid probability transport with optimal transport regularization, significantly speeding up sampling while maintaining stability. EquiFlow164 addresses the challenge of conformation prediction using conditional flow matching and an ODE solver for fast and accurate inference. By leveraging equivariant modeling, these methods improve the generation of valid and physically consistent molecular conformations, advancing the field of 3D molecule generation. Equivariant Variational Flow Matching165 frames flow matching as a variational inference problem and enables both end-to-end conditional generation and post-hoc controlled sampling without retraining. The model further provides a principled equivariant formulation of VFM, ensuring invariance to rotations, translations, and atom permutations, which are essential for molecular applications.
Efficiency
Generating high-quality 3D molecular structures efficiently is a major challenge in drug discovery and structural biology. While generative models have shown promise in modeling complex molecular structures, many existing approaches suffer from slow sampling speeds and computational inefficiency. Flow matching-based methods leverage optimal transport and equivariant architectures to achieve faster and more reliable generation. For instance, GOAT166 formulates a geometric optimal transport objective to map multi-modal molecular features efficiently, using an equivariant representation space to achieve a double speedup compared to previous methods. MolFlow167 introduces scale optimal transport, significantly reducing sampling steps while maintaining high chemical validity. SemlaFlow168 combines latent attention with equivariant flow matching, achieving an order-of-magnitude speedup with as few as 20 sampling steps. A recent work introduces SO(3)-Averaged Flow Matching with Reflow169, targeting both training and inference efficiency for 3D molecular conformer generation. The proposed SO(3)-averaged training objective leads to faster convergence and improved generalization compared to Kabsch-aligned or optimal transport baselines. ET-Flow170 leverages equivariant flow matching to generate low-energy molecular conformations efficiently, bypassing the need for complex geometric calculations.
Guided generation
Guided and conditional generation enables the creation of structures that align with specific biological properties or conditions. In the context of flow matching, guided generation incorporates domain-specific knowledge to steer the generative process, while conditional generation aims to produce diverse outputs based on given inputs or contexts. These approaches are especially valuable in applications where accurate constraints are available. Recent advancements in flow matching have introduced several methods to enhance guided and conditional generation. FlowDPO171 addresses the challenge of 3D structure prediction by combining flow matching with Direct Preference Optimization (DPO), minimizing hallucinations while producing high-fidelity atomic structures. In conditional generation, Extended Flow Matching (EFM)172 generalizes the continuity equation, enabling more flexible modeling by incorporating inductive biases. For mixed-type molecular data, FlowMol173 extends flow matching to handle both continuous and categorical variables, achieving robust performance in 3D de novo molecule generation. 3D energy-based flow matching174 further enhances conditional generation by explicitly incorporating energy signals into both training and inference, improving structural plausibility and convergence. Together, these advances highlight the growing adaptability of flow-based approaches in generating biologically meaningful 3D molecular structures under domain constraints. Additionally, OC-Flow175 leverages optimal control theory to guide flow matching without retraining, showing superior efficiency on complex geometric data, including protein design.
Conditional molecule design and applications
Recent advancements in flow matching for property-driven molecule design focus on not only generating the molecules themselves, but also predicting potential functionalities of the generated molecules. In scenarios requiring precise geometric control, GeoRCG176 enhances molecule generation by integrating geometric representation conditions, achieving significant quality improvements on challenging benchmarks. Additionally, conditional generation with improved structural plausibility has been addressed by integrating distorted molecules into training datasets, as demonstrated in Improving Structural Plausibility in 3D Molecule Generation177. This method leverages property-conditioned training to selectively generate high-quality conformations. Stiefel Flow Matching178 tackles the problem of structure elucidation under moment constraints by embedding molecular point clouds within the Stiefel manifold, allowing for efficient and accurate generation of 3D structures with precise physical properties. Finally, IDFlow179 adopts an energy-based perspective on flow matching for molecular docking, where the generative process learns a deep mapping function to transform random molecular conformations into physically plausible protein-ligand binding structures. PropMolFlow180 further advances property-guided molecule generation through a geometry-complete SE(3)-equivariant flow matching framework integrating five different property embedding methods with a Gaussian expansion of scalar properties. TemplateFM181 introduces a ligand-based generation framework that leverages flow matching for template-guided 3D molecular alignment.
Structure-Based Drug Design (SBDD) is a key task in AI-assisted drug discovery, aiming to design small-molecule drugs that can bind to a given protein pocket structure. The main challenges in this domain lie in modeling the target protein structure, capturing protein-ligand interactions, enabling multimodal generation, and ensuring the chemical validity of generated molecules. In recent years, generative models have shown great potential in addressing these challenges, with Flow Matching (FM) models demonstrating unique advantages in multimodal modeling and generation efficiency. MolFORM182 applies multimodal FM to the SBDD setting and employs DPO to optimize molecular binding affinity. FlexSBDD183 further introduces protein pocket flexibility into the model, making it more reflective of real-world binding scenarios. In addition, MolCRAFT184 adopts a Bayesian Flow Network (BFN) to model multimodal distributions in continuous parameter space, where BFN similarly defines a flow distribution. Moreover185, reveals the equivalence between BFN, diffusion models, and stochastic differential equations (SDEs). PocketXMol186 provides a unified generative model for handling a variety of protein-ligand tasks. PAFlow187 introduces prior-guided flow matching with a learnable atom-number predictor to steer generation toward high-affinity regions and aligning molecule size with pocket geometry.
Protein generation
"Protein generation” can encompass a variety of tasks. To avoid confusion, we provide a brief comparison in Table 4.
Unconditional generation
Backbone generation
Protein backbone generation aims to rapidly synthesize physically realizable 3D scaffolds that are diverse, designable, and functionally conditionable, while adhering to SE(3)-equivariance, local bond constraints, and global topological consistency. Recent efforts approach this challenge from two directions: enhancing the flow matching framework and improving protein feature representation learning. From the flow matching perspective, FrameFlow188 accelerates diffusion by reframing it as deterministic SE(3) flow matching, cutting sampling steps five-fold and doubling designability over FrameDiff. Rosetta Fold diffusion 2 (RFdiffusion2)189 uses the RosettaFold All-Atom neural network architecture and is trained with flow matching for improved training and generation efficiency. FoldFlow-SFM47 further extends this by introducing stochastic flows on SE(3) manifolds using Riemannian optimal transport, enabling the rapid generation of long backbones (up to 300 residues) with high novelty and diversity. Complementarily, recent work also advances architectural designs for protein representation learning. Yang et al.190 combine global Invariant Point Attention (IPA) with local neighborhood aggregation to extract meaningful features, and further use ESMFold and AlphaFold3 to filter the invalid generated backbones. Wagner et al.191 proposes Clifford frame attention (CFA), an extension of IPA by exploiting projective geometric algebra and higher-order message passing to capture residue-frame interactions, yielding highly designable proteins with richer fold topologies. FoldFlow-2192 augments SE(3) flows with PLM embeddings and a multi-modal fusion trunk, enabling sequence-conditioned generation with reinforced reward alignment and state-of-the-art diversity, novelty, and designability on million-scale synthetic-real datasets. Proteina193 scales unconditional FM to a 400 M-parameter non-equivariant transformer trained on 21 M synthetic backbones, using hierarchical CATH conditioning to transport isotropic noise to native-like Cα traces. ProtComposer194 augments a Multiflow128 backbone with SE(3)-invariant cross-attention to user-sketched 3-D ellipsoid tokens, steering the FM vector field toward compositional spatial layouts while preserving unconditional diversity.
Co-design generation
Recent work reframes sequence-structure co-design as learning a unified vector field that jointly models discrete amino acid identities and continuous 3D coordinates, bypassing the traditional two-stage pipeline that separately samples a backbone before fitting a compatible sequence. This co-generative setting is especially challenging due to the need to reconcile fundamentally different data manifolds, enforce SE(3) symmetry, and ensure bidirectional invertibility, all while scaling to the vast combinatorial space of long proteins. CoFlow195 proposes a joint discrete flow that models residue identities and inter-residue distances as CTMC states, augmented with a multimodal masked language module that allows structural flows and sequence tokens to condition each other. Discrete Flow Models (DFM)128 formalize flow matching on arbitrary discrete spaces by interpreting score-based guidance as CTMC generator reversal. Instantiated as MultiFlow, this framework enables sequence-only, structure-only, or joint generation within a single architecture-agnostic model, achieving state-of-the-art perplexity and TM-scores while being orders of magnitude faster than diffusion-based baselines. Finally, APM196 introduces a Seq&BB module that jointly learns continuous SE(3) flows for backbone frames and discrete token flows for sequences, leveraging protein language models, Invariant Point Attention, and Transformer encoders to capture residue-level and pairwise interactions. APM supports precise interchain modeling and de novo design of protein complexes with specified binding properties.
Conditional generation
Motif-scaffolding generation
Motif-scaffolding generation: conditional SE(3) flow-matching models embed fixed functional motifs into de-novo backbones by learning equivariant vector fields that respect both local motif geometry and global fold constraints, overcoming the diversity and fidelity limits of earlier diffusion approaches. FrameFlow-Motif197 augments FrameFlow188 with motif amortization and inference-time motif guidance, enabling scaffold generation around functional motifs with special-designed data augmentation and estimated conditional scores. EVA198 casts scaffolding as geometric inverse design, steering a pretrained flow along motif-aligned probability paths to accelerate convergence and boost structural fidelity. RFdiffusion2189 conducts catalytic site motif scaffolding at a much higher success rate, enabling de novo design of enzymes.
Pocket & binder design
Conditional pocket and binder design tackles the dual challenge of sculpting a protein interface that both accommodates a specific ligand conformation and retains global fold stability, all while respecting SE(3) symmetry and the rich geometric-chemical priors that govern non-covalent recognition. Flow-matching models address these hurdles by learning equivariant vector fields that map an easy base distribution to the manifold of ligand-compatible protein-ligand complexes in a single, differentiable pass, avoiding the slow guidance loops and hand-crafted potentials of earlier diffusion or docking pipelines. AtomFlow199 unifies protein and ligand atoms into “biotokens” and applies atomic-resolution SE(3) flow matching to co-generate ligand conformations and binding backbones directly from a 2-D molecular graph. Additionally, FLOWR200 frames structure-aware ligand design as SE(3)-equivariant flow matching on a mixed continuous-categorical space. It learns the manifold of pocket-compatible molecules by coupling continuous FM for 3D atomic coordinates with categorical FM for fragment/chemotype identities, using equivariant optimal transport and an efficient pocket-conditioning mechanism to enforce interaction-aware constraints in a single pass. Building on FLOWR200, FLOWR.root201 unifies de novo generation, pharmacophore/interaction-conditional sampling, and fragment elaboration with joint heads for multi-endpoint affinity prediction and confidence estimation, sharing the conditional vector field while supervising downstream properties for multi-purpose structure-aware design. FlowSite202 introduces a self-conditioned harmonic flow objective that first aligns apo proteins to a harmonic potential and then co-generates discrete residue types and 3-D ligand poses, supporting multi-ligand docking and outperforming prior generative and physics-based baselines on pocket-level benchmarks. PocketFlow203 incorporates protein-ligand interaction priors (e.g., hydrogen-bond geometry) directly into the flow, then applies multi-granularity guidance to produce high-affinity pockets that significantly improve Vina scores and generalize across small molecules, peptides, and RNA ligands. To efficiently recover all-atom structures from coarse-grained simulations, FlowBack204 utilizes flow matching to map coarse-grained representations to all-atom configurations, achieving high fidelity in protein and DNA structure reconstruction.
Structure prediction
Conformer prediction
Accurately sampling the conformational ensembles underlying protein function remains challenging due to the cost of exhaustive molecular dynamics. Recent work leverages sequence-conditioned, SE(3)-equivariant flow matching to efficiently generate diverse, physically consistent states aligned with experimental observables. AlphaFold Meets Flow Matching205 repurposes single-state predictors (AlphaFold, ESMFold) as generative engines by fine-tuning them under a harmonic flow-matching objective, yielding AlphaFlow/ESMFlow ensembles that surpass MSA-subsampled AlphaFold on the precision-diversity trade-off and reach equilibrium observables faster than replicate MD trajectories. P2DFlow206 augments SE(3) flow matching with a latent “ensemble” dimension and a physics-motivated prior, enabling it to reproduce crystallographic B-factor fluctuations and ATLAS MD distributions more faithfully than earlier baselines.
Side-chain packing
Predicting rotameric states for each residue requires joint compliance with steric constraints, energetic preferences, and SE(3)-equivariance. Recent work has explored constrained side-chain prediction through flow matching. FlowPacker207 formulates side-chain placement as torsional flow matching, coupling the learned vector field to EquiformerV2208, an SE(3)-equivariant graph attention backbone. PepFlow209 generalizes this approach to full-atom peptides using a multi-modal flow that captures joint distributions over backbone frames, side-chain torsions, and residue identities. Partial sampling from this flow achieves state-of-the-art results in fixed-backbone packing and receptor-bound refinement, while maintaining full differentiability for downstream design applications.
Docking prediction
Recent work reframes protein-ligand docking as a flow-matching (FM) generative problem, replacing diffusion with a simulation-free objective that learns a bijective map from unbound receptors (apo) to bound complexes (holo). FlowSite202 introduces a self-conditioned FM objective that harmonically couples translational, rotational and torsional degrees of freedom. By leveraging GAT and TFN layers for ligand-protein interaction modeling, it further extends to jointly generate contact residues and ligand coordinates, substantially improving sample quality, simplicity, and generality in pocket-level docking. Meanwhile, FlowDock210 learns a geometric flow mapping unbound to bound structures, while predicting per-complex confidence and binding affinity estimates. ForceFM211 reframes protein-ligand docking as force-guided manifold flow matching, injecting physics-based energy gradients into translational, rotational, and torsional flows to steer generation toward low-energy, physically realistic conformations.
Peptide and antibody generation
Recent work206,209,212,213,214 formulates peptide design as conditional flow matching over multiple geometric and categorical manifolds, explicitly modeling residue type, spatial position, orientation, and angles in a unified generative framework. PepFlow209 introduces the first multi-modal flow matching framework for protein structure design, jointly modeling residue positions via Euclidean CFM, orientations via Spherical CFM, angles via Toric CFM, and types via Simplex CFM. This unified approach achieves excellent performance on sequence recovery and side-chain packing in receptor-conditioned design tasks. D-Flow206 extends this paradigm to D-peptides by augmenting limited training data through a chirality-aware mirror transformation and incorporating a lightweight structural adapter into a pretrained protein language model. PPFlow212 formulates peptide torsion generation as flow matching on a (3n − 3)-torus with n being the number of amino acids, while modeling global transitions and residue types via Euclidean flows and employing SO(3)-CFM for rotations. This formulation enables effective conditional sampling for diverse tasks such as peptide optimization and docking. Finally, NLFlow213 pioneers non-linear conditional vector fields by employing polynomial interpolation over the position manifold, enabling faster convergence toward binding pockets and effectively addressing temporal inconsistencies across modalities. This approach leads to improvements in structural stability and binding affinity compared to prior linear flow models. Collectively, these studies underscore the importance of manifold-specific flows, conditioning strategies, and geometric priors for scalable, high-fidelity peptide generation. In contrast to these geometry-intensive approaches, ProtFlow214 treats peptides as amino acid sequences and bypasses non-Euclidean representations by embedding each residue using a pretrained protein language model (PLM). In the embedding space of PLMs, ProtFlow trains a reflow-enabled sequence flow model that supports both single-step generation and multi-chain co-design. Collectively, these studies highlight the critical role of manifold-specific flows, conditioning strategies, and geometric priors in enabling scalable and high-fidelity peptide generation.
The study of antibody structure design with flow matching is emerging as well. For instance, FlowAB215 utilizes energy-guided SE(3) flow matching to improve antibody structure refinement, integrating physical priors to enhance CDR accuracy with minimal computational overhead.
Other bio applications
Dynamic cell trajectory prediction
Dynamic cell trajectory: generative trajectory models seek to reconstruct the continuously branching, stochastic evolution of cells from high-dimensional, sparsely sampled single-cell readouts, which is an endeavor hampered by severe noise, irregular time points, and the risk that straight Euclidean interpolants stray outside the biological manifold. CellFlow216 tackles this by framing morphology evolution under perturbations as an image-level flow-matching problem on cellular masks, enabling realistic, perturbation-conditioned movies of shape change that outperform diffusion and GAN baselines in both faithfulness and diversity. GENOT-L140 introduces an entropic Gromov-Wasserstein flow that couples gene-expression geometry across time points, producing probabilistic lineage trajectories that capture heterogeneity and branching better than optimal-transport predecessors while remaining simulation-free. Metric Flow Matching217 instead learns geodesic vector fields under a data-induced Riemannian metric, yielding smoother interpolations that respect the manifold’s curvature and achieving state-of-the-art accuracy on single-cell trajectory benchmarks with fewer artifacts than Euclidean flows. Diversified Flow Matching218 extends this line of work by ensuring translation identifiability across diverse conditional distributions, a key challenge in modeling heterogeneous cellular states. Unlike prior GAN-based solutions, this work formulates the problem within an ODE-based flow matching framework, offering stable training and explicit transport trajectories. Collectively, these works highlight the importance of geometry-aware objectives and probabilistic conditioning for faithful dynamic cell-state generation.
Bio-image generation and enhancement
Leveraging continuous probability flow to efficiently model biological structures, flow matching has shown great potential for bio-image generation and enhancement, enabling faster and more accurate modeling of complex biological data. One notable application is FlowSDF219, which introduces image-guided conditional flow matching for medical image segmentation. By modeling signed distance functions (SDF) instead of binary masks, FlowSDF achieves smoother and more accurate segmentation. This method also generates uncertainty maps, enhancing robustness in prediction tasks. For medical image synthesis, an optimal transport flow matching approach220 addresses the challenge of balancing generation speed and image quality. By creating a more direct mapping between distributions, this method reduces inference time while maintaining high-quality outputs, and supports diverse imaging modalities, including 2D and 3D. In MR image reconstruction, Multi-Modal Straight Flow Matching (MMSFlow)221 significantly reduces the number of inference steps by forming a linear path between undersampled and reconstructed images. Leveraging multi-modal information with low- and high-frequency fusion layers, MMSFlow achieves state-of-the-art performance in fastMRI and Brats-2020 benchmarks.
Cellular microenvironments from spatial transcriptomics
Flow matching has also emerged as a powerful framework for modeling spatial transcriptomics (ST) data, which captures gene expression levels across spatial locations within a tissue. The core task in ST involves reconstructing or generating spatially-resolved gene expression maps that reflect underlying cellular microenvironments and tissue organization. One such method is STFlow222 which introduces a scalable flow matching framework for generating spatial transcriptomics data from whole-slide histology images. It models the joint distribution of gene expression across all spatial spots in a slide, thereby explicitly capturing cell-cell interactions and tissue organization. Complementarily, Wasserstein Flow Matching (WFM)223 generalizes flow-based generative modeling to families of distributions. It introduces a principled way to model both 2D and 3D spatial structures of cellular microenvironments, and leverages the geometry of Wasserstein space to better match distributional characteristics across biological contexts. Together, these methods highlight the utility of flow matching in capturing the spatially-aware, high-dimensional distributions characteristic of modern transcriptomics datasets.
Neural activities
Flow matching has recently shown promise in modeling and aligning neural activity, particularly for time series and brain-computer interface (BCI) applications, where neural signals are often stochastic and nonstationary. Stream-level Flow Matching with Gaussian Processes224 extends conditional flow matching by introducing streams, which are latent stochastic paths modeled with Gaussian processes. This reduces variance in vector field estimation, enabling more accurate modeling of correlated time series such as neural recordings. Flow-Based Distribution Alignment225 tackles inter-day neural signal shifts in BCIs through source-free domain adaptation. By learning stable latent dynamics via flow matching and ensuring stability through Lyapunov analysis, it enables reliable few-trial neural adaptation across days. These approaches highlight the versatility of flow matching for neural data, supporting both high-fidelity generation and robust adaptation with limited supervision. DIFFEOCFM226 introduces Riemannian flow matching for brain connectivity matrices by leveraging pullback metrics to perform conditional FM on matrix manifolds, enabling efficient vector-field learning and fast sampling while preserving manifold constraints.
Evaluation tasks and datasets
In this section, we summarize evaluation tasks and datasets used for assessing flow matching methods in biology and life sciences. As listed in Tables 5 and 6, these tasks span a wide spectrum of domains, including genomics, transcriptomics, molecular chemistry, and structural biology. For each dataset, we also report its data scale or number of samples. Flow matching has been applied to a diverse set of generation and modeling problems, such as biological sequence generation, cell trajectory inference, molecule design, and protein structure modeling.
Sequence-level generation: flow matching models have been evaluated on tasks like DNA51,125,133, RNA227,228,229, and protein230,231,232 sequence generation. These datasets range from promoter and enhancer sequences to large-scale protein and metagenomic corpora, covering both canonical and noncoding regions of the genome.
Single-cell modeling and trajectory inference: flow matching has been used to model temporal or conditional transitions in high-dimensional single-cell gene expression data, including developmental trajectories233, perturbation responses234, and modality prediction235. Datasets such as PBMC236, dentate gyrus237, and Tabula Muris238 provide diverse experimental contexts for evaluating these tasks.
Molecular generation and conformation modeling: datasets such as QM9239, ZINC240, GEOM-Drugs241, and MOSES242 provide chemically diverse molecular structures, enabling evaluation of molecular validity, novelty, and 3D geometry. Flow matching models are tested on their ability to generate, edit, or align molecular graphs and conformers.
Protein and complex design: structural datasets like SCOPe243, ATLAS244, and curated PDB subsets support evaluation of flow-based models on protein backbone generation, folding, and structural refinement. Complementary datasets such as Binding MOAD245, CrossDocked246, BioLip2247, and PepBDB248 enable studies on molecular docking, peptide-protein interactions, and binder generation.
Notably, many datasets are reused across different tasks due to their structural richness and biological relevance. For instance, the Protein Data Bank (PDB)232 is used in tasks ranging from protein sequence design and backbone generation to modeling conformational dynamics and performing docking. Similarly, SAbDab249 supports antibody sequence generation, structural modeling, and binder discrimination.
Despite the growing adoption of flow matching in biology, the field still lacks unified benchmarks for many tasks. This is likely due to the inherent heterogeneity of biological problems, ranging from sequence to structure, from single-cell to population scale, which makes standardized evaluation more challenging. This stands in contrast to fields like computer vision or NLP, where well-defined benchmarks are more prevalent250,251,252,253. Continued efforts in dataset curation and task formulation are needed to support consistent and reproducible assessment of generative models in the life sciences.
Future direction
Flow matching for discrete sequence generation
Flow matching has recently emerged as a promising generative modeling paradigm, offering a compelling balance between generation quality and training stability. While its success in continuous domains like image and molecule generation has been widely documented, applying FM to discrete sequence generation, especially in domains such as natural language, genomics, and code, remains a vibrant and largely underexplored frontier.
One of the most intriguing directions lies in understanding the representational advantages of discrete Flow Matching compared to traditional paradigms such as Masked Language Modeling (MLM). Unlike MLM, which relies on partial observation and token masking, FM provides a direct mapping from a base distribution to the target sequence via a continuous probability flow. This raises the question: Can discrete FM yield more semantically coherent representations and facilitate better downstream performance in tasks such as classification? Recent advances, such as Fisher Flow125 and Dirichlet FM51, demonstrate that geometry-aware formulations over the probability simplex can encode meaningful geometric constraints and structure-aware trajectories, enabling more faithful modeling of discrete data distributions.
Another fundamental question concerns the generation capabilities of discrete FM relative to autoregressive (AR) models. While AR models remain the gold standard in natural language generation due to their strong likelihood modeling and contextual fluency, they suffer from slow sampling and exposure bias. In contrast, discrete FM supports parallel generation through ODE integration or sampling over learned Markov trajectories, offering substantial efficiency gains. However, its generation quality still lags behind state-of-the-art AR transformers in language generation125, prompting future research into architectural refinements and better training objectives.
Furthermore, the integration of FM with Transformer architectures remains an open challenge. Existing Transformer-based FM models either operate in latent embedding space or use discrete-continuous relaxations (e.g., Gumbel-Softmax) to approximate gradient flows. Yet, the Transformer’s causal attention structure may be suboptimal for non-autoregressive FM-based sequence generation, especially in domains where left-to-right order is arbitrary or non-existent (e.g., protein sequences, biological pathways). This invites research into order-agnostic architectures or the use of permutation-invariant encoders to better align with FM-based modeling.
Finally, flow matching may offer unique advantages in non-language sequence modeling tasks, such as biomolecular design and genome modeling, where biological constraints (e.g., base-pairing, structural motifs) must be enforced. Unlike language, these sequences often lack natural generation order and exhibit rich multi-modal dependencies. FM’s ability to incorporate conditioning, geometry-aware constraints, and structure-guided generation (e.g., via SE(3)-equivariant or manifold-aware flows) makes it a particularly attractive candidate. Future work may focus on developing discrete FM formulations that are not only domain-adaptive, but also biologically interpretable and sample-efficient.
Small molecule generation and modeling
Small molecule generation is a core task in cheminformatics and drug discovery, where FM has recently shown promising capabilities in both unconditional and conditional generation settings. By modeling continuous probability flows between simple priors and molecular distributions, FM offers an appealing alternative to diffusion models, with improved sample efficiency and the potential to integrate domain knowledge. However, due to the scarcity of molecular structure data and the complexity of structural constraints, several key challenges remain before FM can fully realize its potential for small molecule generation.
One fundamental limitation lies in the data scarcity and structural heterogeneity of small molecule datasets. Unlike macromolecules such as proteins, which benefit from large-scale structural repositories (e.g., PDB), small molecule datasets are often limited in size and diversity, especially for annotated 3D conformers. As a result, FM models trained on these datasets may struggle to generalize across different chemical scaffolds, limiting their utility in low-resource or out-of-distribution scenarios. Addressing this issue may require more effective data augmentation strategies (e.g., using force field simulations or generative conformer expansion), transfer learning pipelines, or semi-supervised flow matching objectives that make better use of unlabeled data.
To improve the physical plausibility and functional relevance of generated small molecules, a key direction lies in incorporating domain-specific inductive priors into both the training and sampling stages of flow matching. Small molecules are governed by well-defined chemical and physical constraints, such as bond lengths and angles, valence rules, charge distributions, and conformational energetics, which can be explicitly modeled to constrain the learned probability flow. Embedding such priors into the vector field design or generation trajectories (e.g., via energy-guided loss functions or structure-aware conditioning) can substantially improve the realism and synthesizability of generated compounds.
At the same time, enhancing the conditional generation capabilities of FM is essential for tasks that demand goal-directed molecular design, such as generating molecules with desired pharmacological properties, satisfying functional group templates, or fitting into predefined binding pockets. Conditional flow matching offers a natural framework for structure- and property-guided generation, enabling fine-grained control over outputs via learned trajectories that satisfy specific constraints. Future work may explore more expressive conditioning schemes, multi-property guidance, or interaction-aware control mechanisms, paving the way for FM-based models to support precision molecular design in high-stakes domains such as drug discovery and materials engineering.
A further challenge lies in modeling molecular interactions and dynamic processes. Molecular docking and binding affinity prediction remain critical tasks in early-stage drug design, requiring models to account for conformational flexibility in small molecules and the adaptive nature of protein binding pockets, particularly with respect to side-chain rearrangements. Even more challenging tasks, such as enzyme design, involve not just molecular recognition but also modeling of specific reaction mechanisms. Thus, leveraging the FM framework to capture inter-molecular interactions and reaction dynamics represents a crucial and promising direction for future research.
Protein
In the field of protein modeling, Flow Matching (FM) has emerged as an efficient approach for sequence and structure modeling, demonstrating complementary advantages to traditional methods. Proteins, as highly complex biological macromolecules, exhibit a unique combination of discrete primary sequences and continuous three-dimensional structures, which poses distinct challenges for the design and training of FM-based models.
One important future direction is to establish effective matching mechanisms across different protein modalities. For example, in mapping from amino acid sequences to 3D structures, FM could serve as a bridge between discrete and continuous spaces, enhancing the model’s expressiveness in structure prediction and generation tasks. Furthermore, in applications such as protein-protein docking and complex assembly modeling, FM offers a promising framework for capturing transformation paths in high-dimensional, complex spaces.
In addition, modeling protein dynamics, such as conformational changes or ligand-induced fit, remains a core challenge in structural biology. Future work may explore integrating FM with physical simulations (e.g., molecular dynamics) or diffusion-based processes, enabling the learning of natural transition paths between protein states and improving interpretability of their functional mechanisms.
Conclusion
Flow matching has become a compelling alternative to diffusion-based generative modeling, offering advantages in stability, efficiency, and control. In this survey, we provide a structured overview of its growing use in biology and life sciences, covering a diverse range of tasks from sequence generation and molecular design to protein modeling. We also compile a comprehensive list of datasets used for evaluation, including their scale and cross-task applicability. Despite promising progress, we also summarize the challenges that the field faces. We hope this survey could clarify current trends and motivate future research at the intersection of generative modeling and the life sciences.
Data availability
No datasets were generated or analyzed during the current study.
References
Lipman, Y., Chen, R. T. Q., Ben-Hamu, H., Nickel, M. & Le, M. Flow matching for generative modeling. In The Eleventh International Conference on Learning Representations, ICLR 2023, Kigali, Rwanda (Openreview.net, 2023).
Jin, Y. et al. Pyramidal flow matching for efficient video generative modeling. In The Thirteenth International Conference on Learning Representations, ICLR 2025, Singapore (OpenReview.net, 2025).
Hu, V. T. et al. Flow matching for conditional text generation in a few sampling steps. In Proc. 18th Conference of the European Chapter of the Association for Computational Linguistics, EACL 2024—Volume 2: Short Papers, St. Julian’s, Malta (eds Graham, Y. & Purver, M.) 380–392 (Association for Computational Linguistics, 2024).
Gat, I. et al. Discrete flow matching. Adv. Neural Inf. Process Syst. 37, 133345–133385 (2024).
Church, G. M. & Gilbert, W. Genomic sequencing. Proc. Natl. Acad. Sci. USA 81, 1991–1995 (1984).
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Pareek, C. S., Smoczynski, R. & Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 52, 413–435 (2011).
Luo, S., Guan, J., Ma, J. & Peng, J. A 3d generative model for structure-based drug design. Adv. Neural Inf. Process Syst. 34, 6229–6239 (2021).
Sadybekov, A. V. & Katritch, V. Computational approaches streamlining drug discovery. Nature 616, 673–685 (2023).
Mathur, S. & Hoskins, C. Drug development: lessons from nature. Biomed. Rep. 6, 612–614 (2017).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with alphafold 3. Nature 630, 493–500 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Baek, M. et al. Efficient and accurate prediction of protein structure using rosettafold2. bioRxiv https://doi.org/10.1101/2023.05.24.542179 (2023).
Robb, R. A. Biomedical Imaging, Visualization, and Analysis (John Wiley & Sons, Inc., 1999).
Tempany, C. M. & McNeil, B. J. Advances in biomedical imaging. JAMA 285, 562–567 (2001).
Webb, A. Introduction to Biomedical Imaging (John Wiley & Sons, 2022).
Rangayyan, R. M. BiomedicaL Image Analysis (CRC Press, 2004).
Goodfellow, I. et al. Generative adversarial networks. Commun. ACM 63, 139–144 (2020).
Lan, L. et al. Generative adversarial networks and its applications in biomedical informatics. Front. Public Health 8, 164 (2020).
Lee, M. Recent advances in generative adversarial networks for gene expression data: a comprehensive review. Mathematics 11, 3055 (2023).
He, K. et al. Masked autoencoders are scalable vision learners. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition. 16000–16009 (IEEE, 2022).
Kraus, O. et al. Masked autoencoders for microscopy are scalable learners of cellular biology. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition. 11757–11768 (IEEE, 2024).
Yuan, M. et al. Proteinmae: masked autoencoder for protein surface self-supervised learning. Bioinformatics 39, btad724 (2023).
Chien, H.-Y. S., Goh, H., Sandino, C. M. & Cheng, J. Y. Maeeg: Masked Auto-encoder for eeg representation learning. Preprint at arXiv https://doi.org/10.48550/arXiv.2211.02625 (2022).
Ho, J., Jain, A. & Abbeel, P. Denoising diffusion probabilistic models. Adv. Neural Inf. Process Syst. 33, 6840–6851 (2020).
Guo, Z. et al. Diffusion models in bioinformatics and computational biology. Nat. Rev. Bioeng. 2, 136–154 (2024).
Yang, L. et al. Diffusion models: a comprehensive survey of methods and applications. ACM Comput. Surv. 56, 1–39 (2023).
Faeder, J. R., Blinov, M. L., Goldstein, B. & Hlavacek, W. S. Rule-based modeling of biochemical networks. Complexity 10, 22–41 (2005).
Hwang, M., Garbey, M., Berceli, S. A. & Tran-Son-Tay, R. Rule-based simulation of multi-cellular biological systems—a review of modeling techniques. Cell Mol. Bioeng. 2, 285–294 (2009).
Faeder, J. R., Blinov, M. L. & Hlavacek, W. S. Rule-based modeling of biochemical systems with bionetgen. MIMB, Syst. Biol. 500, 113–167 (2009).
Chylek, L. A., Harris, L. A., Faeder, J. R. & Hlavacek, W. S. Modeling for (physical) biologists: an introduction to the rule-based approach. Phys. Biol. 12, 045007 (2015).
Willard, J., Jia, X., Xu, S., Steinbach, M. & Kumar, V. Integrating physics-based modeling with machine learning: a survey. Preprint at arXiv arXiv:2003.04919 1, 1–34 (2020).
Newman, J. Physics of the Life Sciences (Springer Science & Business Media, 2008).
Franklin, K., Muir, P., Scott, T. & Yates, P. Introduction to Biological Physics for the Health and Life Sciences (John Wiley & Sons, 2019).
Baverstock, K. Life as physics and chemistry: a system view of biology. Prog. Biophys. Mol. Biol. 111, 108–115 (2013).
Yelmen, B. & Jay, F. An overview of deep generative models in functional and evolutionary genomics. Annu. Rev. Biomed. Data Sci. 6, 173–189 (2023).
Anstine, D. M. & Isayev, O. Generative models as an emerging paradigm in the chemical sciences. J. Am. Chem. Soc. 145, 8736–8750 (2023).
Bilodeau, C., Jin, W., Jaakkola, T., Barzilay, R. & Jensen, K. F. Generative models for molecular discovery: recent advances and challenges. Wiley Interdiscip. Rev Comput. Mol. Sci. 12, e1608 (2022).
Xue, D. et al. Advances and challenges in deep generative models for de novo molecule generation. Wiley Interdiscip. Rev Comput. Mol. Sci. 9, e1395 (2019).
Fu, D. & He, J. DPPIN: a biological repository of dynamic protein-protein interaction network data. In IEEE International Conference on Big Data, Big Data 2022, Osaka, Japan (eds Tsumoto, S. et al.) 5269–5277 (IEEE, 2022).
Zheng, L. et al. Pyg-ssl: a graph self-supervised learning toolkit. CoRR abs/2412.21151 https://doi.org/10.48550/arXiv.2412.21151 (2024).
Fu, D. et al. Climatebench-m: a multi-modal climate data benchmark with a simple generative method. CoRR abs/2504.07394 https://doi.org/10.48550/arXiv.2504.07394 (2025).
Zheng, L., Jing, B., Li, Z., Tong, H. & He, J. Heterogeneous contrastive learning for foundation models and beyond. In Proc. 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, KDD 2024, Barcelona, Spain (eds Baeza-Yates, R. & Bonchi, F.) 6666–6676 (ACM, 2024).
Fu, D. et al. Parametric graph representations in the era of foundation models: a survey and position. CoRR abs/2410.12126 https://doi.org/10.48550/arXiv.2410.12126 (2024).
Song, Y. et al. Equivariant flow matching with hybrid probability transport for 3d molecule generation. In Advances in Neural Information Processing Systems 36: Annual Conference on Neural Information Processing Systems 2023, NeurIPS 2023, New Orleans, LA, USA (eds Oh, A. et al.) (NeurIPS, 2023).
Klein, L., Krämer, A. & Noé, F. Equivariant flow matching. Adv. Neural Inf. Process Syst. 36, 59886–59910 (2023).
Bose, A. J. et al. Se(3)-stochastic flow matching for protein backbone generation. In The Twelfth International Conference on Learning Representations, ICLR 2024, Vienna, Austria (OpenReview.net, 2024).
Cheng, C., Li, J., Peng, J. & Liu, G. Categorical flow matching on statistical manifolds. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems 2024, NeurIPS 2024, Vancouver, BC, Canada (eds Globersons, A. et al.) (NeurIPS, 2024).
Kornilov, N., Mokrov, P., Gasnikov, A. & Korotin, A. Optimal flow matching: learning straight trajectories in just one step. Adv. Neural Inf. Process Syst. 37, 104180–104204 (2024).
Chen, R. T. & Lipman, Y. Flow matching on general geometries. In The Twelfth International Conference on Learning Representations, ICLR 2024, Vienna, Austria (OpenReview.net, 2024).
Stark, H. et al. Dirichlet flow matching with applications to dna sequence design. In Proceedings of the 41st International Conference on Machine Learning (Vienna, Austria, 2024).
Ruth, M., Hannon, B., Ruth, M. & Hannon, B. Modeling Dynamic Biological Systems (Springer, 1997).
Edelman, G. M. & Gally, J. A. Degeneracy and complexity in biological systems. Proc. Natl. Acad. Sci. USA 98, 13763–13768 (2001).
Haefner, J. W. Modeling Biological Systems:: Principles and Applications (Springer Science & Business Media, 2005).
Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021).
et al, J. L.-M. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685 (2019).
Kim, D. et al. The architecture of sars-cov-2 transcriptome. Cell 181, 914–921 (2020).
Sahin, U., Karikó, K. & Türeci, Ö mrna-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Baker, D. & Sali, A. Protein structure prediction and structural genomics. Science 294, 93–96 (2001).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Li, P., Pei, Y. & Li, J. A comprehensive survey on design and application of autoencoder in deep learning. Appl. Soft Comput. 138, 110176 (2023).
Jabbar, A., Li, X. & Omar, B. A survey on generative adversarial networks: variants, applications, and training. ACM Comput. Surv. CSUR 54, 1–49 (2021).
Cao, H. et al. A survey on generative diffusion models. IEEE Trans. Knowl. Data Eng. 36, 2814–2830 (2024).
Croitoru, F.-A., Hondru, V., Ionescu, R. T. & Shah, M. Diffusion models in vision: a survey. IEEE Trans. Pattern Anal. Mach. Intell. 45, 10850–10869 (2023).
Liang, S., Pan, Z., Liu, W., Yin, J. & De Rijke, M. A survey on variational autoencoders in recommender systems. ACM Comput. Surv. 56, 1–40 (2024).
Xia, X. et al. Gan-based anomaly detection: a review. Neurocomputing 493, 497–535 (2022).
Du, Y. et al. Machine learning-aided generative molecular design. Nat. Mach. Intell. 6, 589–604 (2024).
Tang, X. et al. A survey of generative ai for de novo drug design: new frontiers in molecule and protein generation. Brief. Bioinforma. 25, bbae338 (2024).
Greener, J. G., Kandathil, S. M., Moffat, L. & Jones, D. T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 23, 40–55 (2022).
Morehead, A. et al. How to go with the flow: flow matching in bioinformatics and computational biology. Authorea Preprints (2025).
Mock, M., Langmead, C. J., Grandsard, P., Edavettal, S. & Russell, A. Recent advances in generative biology for biotherapeutic discovery. Trends Pharm. Sci. 45, 255–267 (2024).
Kell, D. B., Samanta, S. & Swainston, N. Deep learning and generative methods in cheminformatics and chemical biology: navigating small molecule space intelligently. Biochem. J. 477, 4559–4580 (2020).
Liu, M., Li, C., Chen, R., Cao, D. & Zeng, X. Geometric deep learning for drug discovery. Expert Syst. Appl. 240, 122498 (2024).
Yang, Z., Zeng, X., Zhao, Y. & Chen, R. Alphafold2 and its applications in the fields of biology and medicine. Signal Transduct. Target Ther. 8, 115 (2023).
Mariani, V., Biasini, M., Barbato, A. & Schwede, T. lddt: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 29, 2722–2728 (2013).
Berman, H., Henrick, K. & Nakamura, H. Announcing the worldwide protein data bank. Nat. Struct. Mol. Biol. 10, 980 (2003).
Shimoyama, M. et al. The rat genome database 2015: genomic, phenotypic and environmental variations and disease. Nucleic Acids Res. 43, D743–D750 (2015).
AlQuraishi, M. Proteinnet: a standardized data set for machine learning of protein structure. BMC Bioinforma. 20, 1–10 (2019).
Kim, S. et al. Pubchem substance and compound databases. Nucleic Acids Res. 44, D1202–D1213 (2016).
Kingma, D. P. et al. Auto-encoding variational Bayes (2013).
Kingma, D. P. et al. An introduction to variational autoencoders. Found. Trends Mach. Learn 12, 307–392 (2019).
Girin, L. et al. Dynamical variational autoencoders: a comprehensive review. Found. Trends Mach. Learn. 15, 1-175 (2022).
Pu, Y. et al. Variational autoencoder for deep learning of images, labels and captions. In Advances in Neural Information Processing Systems. Vol. 29 (NeurIPS, 2016).
Kusner, M. J., Paige, B. & Hernández-Lobato, J. M. Grammar variational autoencoder. In International Conference on Machine Learning, 1945–1954 (PMLR, 2017).
Bredell, G., Flouris, K., Chaitanya, K., Erdil, E. & Konukoglu, E. Explicitly minimizing the blur error of variational autoencoders. In The Eleventh International Conference on Learning Representations, ICLR 2023, Kigali, Rwanda (Openreview.net, 2023).
Takida, Y. et al. Preventing oversmoothing in vae via generalized variance parameterization. Neurocomputing 509, 137–156 (2022).
Dai, B., Wang, Z. & Wipf, D. The usual suspects? Reassessing blame for vae posterior collapse. In International Conference on Machine Learning, 2313–2322 (PMLR, 2020).
Arjovsky, M., Chintala, S. & Bottou, L. Wasserstein GAN. CoRR abs/1701.07875 http://arxiv.org/abs/1701.07875 (2017).
Mao, X. et al. Least squares generative adversarial networks. In Proc. IEEE International Conference on Computer Vision. 2794–2802 (IEEE, 2017).
Mirza, M. & Osindero, S. Conditional generative adversarial nets. Preprint at arXiv https://doi.org/10.48550/arXiv.1411.1784 (2014).
Bafti, S. M. et al. Biogan: an unpaired gan-based image to image translation model for microbiological images. Preprint at arXiv https://doi.org/10.48550/arXiv.2306.06217 (2023).
Chaudhari, P., Agrawal, H. & Kotecha, K. Data augmentation using mg-gan for improved cancer classification on gene expression data. Soft Comput. 24, 11381–11391 (2020).
Yang, H., Xiang, Z., Li, X. & Zhang, W. An improved gan-based data augmentation model for addressing data scarcity in srms. Meas. Sci. Technol. 36, 026129 (2025).
Osokin, A., Chessel, A., Carazo Salas, R. E. & Vaggi, F. Gans for biological image synthesis. In Proc. IEEE International Conference on Computer Vision. 2233–2242 (IEEE, 2017).
Rezende, D. & Mohamed, S. Variational inference with normalizing flows. In International Conference on Machine Learning, 1530–1538 (PMLR, 2015).
Kobyzev, I., Prince, S. J. & Brubaker, M. A. Normalizing flows: an introduction and review of current methods. IEEE Trans. Pattern Anal. Mach. Intell. 43, 3964–3979 (2020).
Dinh, L., Krueger, D. & Bengio, Y. NICE: non-linear independent components estimation. In 3rd International Conference on Learning Representations, ICLR 2015, San Diego, CA, USA, Workshop Track Proceedings (eds Bengio, Y. & LeCun, Y.) (Openreview.net, 2015).
Dinh, L., Sohl-Dickstein, J. & Bengio, S. Density estimation using real NVP. In 5th International Conference on Learning Representations, ICLR 2017, Toulon, France, Conference Track Proceedings (OpenReview.net, 2017).
Kingma, D. P. & Dhariwal, P. Glow: generative flow with invertible 1x1 convolutions. In Advances in Neural Information Processing Systems 31: Annual Conference on Neural Information Processing Systems 2018, NeurIPS 2018, Montréal, Canada (eds Bengio, S. et al.) 10236–10245 (NeurIPS, 2018).
Papamakarios, G., Murray, I. & Pavlakou, T. Masked autoregressive flow for density estimation. In Advances in Neural Information Processing Systems 30: Annual Conference on Neural Information Processing Systems, Long Beach, CA, USA (eds Guyon, I. et al.) 2338–2347 (NeurIPS, 2017).
Chen, R. T., Rubanova, Y., Bettencourt, J. & Duvenaud, D. K. Neural ordinary differential equations. In Advances in Neural Information Processing Systems 31 (NeurIPS, 2018).
Sohl-Dickstein, J., Weiss, E. A., Maheswaranathan, N. & Ganguli, S. Deep unsupervised learning using nonequilibrium thermodynamics. In Proc. 32nd International Conference on Machine Learning, ICML 2015, Lille, France, 37 of JMLR Workshop and Conference Proceedings (eds Bach, F. R. & Blei, D. M.) 2256–2265 (JMLR.org, 2015).
Song, Y. & Ermon, S. Generative modeling by estimating gradients of the data distribution. In Advances in Neural Information Processing Systems 32: Annual Conference on Neural Information Processing Systems 2019, NeurIPS, Vancouver, BC, Canada (eds Wallach, H. M. et al.) 11895–11907 (NeurIPS, 2019).
Song, Y. et al. Score-based generative modeling through stochastic differential equations. In 9th International Conference on Learning Representations, ICLR 2021, Virtual Event, Austria (OpenReview.net, 2021).
Song, J., Meng, C. & Ermon, S. Denoising diffusion implicit models. In 9th International Conference on Learning Representations, ICLR 2021, Virtual Event, Austria (OpenReview.net, 2021).
Campbell, A. et al. A continuous time framework for discrete denoising models. In Advances in Neural Information Processing Systems 35: Annual Conference on Neural Information Processing Systems 2022, NeurIPS 2022, New Orleans, LA, USA (eds Koyejo, S. et al.) (NeurIPS, 2022).
Austin, J., Johnson, D. D., Ho, J., Tarlow, D. & van den Berg, R. Structured denoising diffusion models in discrete state-spaces. In Advances in Neural Information Processing Systems 34: Annual Conference on Neural Information Processing Systems 2021, NeurIPS, Virtual (eds Ranzato, M., Beygelzimer, A., Dauphin, Y. N., Liang, P. & Vaughan, J. W.) 17981–17993. https://proceedings.neurips.cc/paper/2021/hash/958c530554f78bcd8e97125b70e6973d-Abstract.html (NeurIPS, 2021).
Salimans, T. & Ho, J. Progressive distillation for fast sampling of diffusion models. In The Tenth International Conference on Learning Representations, ICLR 2022, Virtual Event (OpenReview.net, 2022).
Lu, C. et al. Dpm-solver: a fast ODE solver for diffusion probabilistic model sampling in around 10 steps. In Advances in Neural Information Processing Systems 35: Annual Conference on Neural Information Processing Systems 2022 (eds Koyejo, S. et al.) (NeurIPS, New Orleans, 2022).
Song, Y., Dhariwal, P., Chen, M. & Sutskever, I. Consistency models (2023).
Song, Y. & Dhariwal, P. Improved techniques for training consistency models. In The Twelfth International Conference on Learning Representations, ICLR 2024, (OpenReview.net, Vienna, Austria, 2024).
Heek, J., Hoogeboom, E. & Salimans, T. Multistep consistency models. Preprint at arXiv https://doi.org/10.48550/arXiv.2403.06807 (2024).
Kim, D. et al. Consistency trajectory models: learning probability flow ode trajectory of diffusion. In The Twelfth International Conference on Learning Representations, ICLR 2024, (OpenReview.net, Vienna, Austria, 2024).
Geng, Z., Pokle, A., Luo, W., Lin, J. & Kolter, J. Z. Consistency models made easy. In The Thirteenth International Conference on Learning Representations, ICLR 2025, (Openrview.net, Singapore, 2025).
Lee, S. et al. Truncated consistency models. In The Thirteenth International Conference on Learning Representations, ICLR 2025 (Openreview.net, Singapore, 2025).
Lipman, Y., Chen, R. T., Ben-Hamu, H., Nickel, M. & Le, M. Flow matching for generative modeling. In The Eleventh International Conference on Learning Representations, ICLR 2023 (Openreview.net, Kigali, Rwanda, 2023).
Lipman, Y. et al. Flow matching guide and code. Preprint at arXiv https://doi.org/10.48550/arXiv.2412.06264 (2024).
Eijkelboom, F., Bartosh, G., Andersson Naesseth, C., Welling, M. & van de Meent, J.-W. Variational flow matching for graph generation. Adv. Neural Inf. Process Syst. 37, 11735–11764 (2024).
Albergo, M. S. & Vanden-Eijnden, E. Building normalizing flows with stochastic interpolants. In The Eleventh International Conference on Learning Representations, ICLR 2023 (Openeview.net, Kigali, Rwanda, 2023).
Tong, A. et al. Improving and generalizing flow-based generative models with minibatch optimal transport. Transactions on Machine Learning Research 1–34 (2024).
Liu, X., Gong, C. & Liu, Q. Flow straight and fast: learning to generate and transfer data with rectified flow. In The Eleventh International Conference on Learning Representations, ICLR 2023 (Openreview.net, Kigali, Rwanda, 2023).
Liu, X., Gong, C. & Liu, Q. Flow straight and fast: learning to generate and transfer data with rectified flow. In The Eleventh International Conference on Learning Representations https://openreview.net/forum?id=XVjTT1nw5z (2023).
Lee, S., Lin, Z. & Fanti, G. Improving the training of rectified flows. Adv. Neural Inf. Process Syst. 37, 63082–63109 (2024).
Chen, R. T. & Lipman, Y. Riemannian flow matching on general geometries. In The Twelfth International Conference on Learning Representations, ICLR 2024, Vienna, Austria (Openreview.net, 2024).
Davis, O. et al. Fisher flow matching for generative modeling over discrete data. Adv. Neural Inf. Process Syst. 37, 139054–139084 (2024).
Lou, A. et al. Neural manifold ordinary differential equations. Adv. Neural Inf. Process Syst. 33, 17548–17558 (2020).
Mathieu, E. & Nickel, M. Riemannian continuous normalizing flows. Adv. Neural Inf. Process Syst. 33, 2503–2515 (2020).
Campbell, A., Yim, J., Barzilay, R., Rainforth, T. & Jaakkola, T. Generative flows on discrete state-spaces: enabling multimodal flows with applications to protein co-design. In Proceedings of the 41st International Conference on Machine Learning. 5453–5512 (Vienna, Austria, PMLR, 2024).
Shaul, N. et al. Flow matching with general discrete paths: a kinetic-optimal perspective. In The Thirteenth International Conference on Learning Representations. (Openreview.net, Singapore, ICLR 2025, 2025).
Qin, Y., Madeira, M., Thanou, D. & Frossard, P. DeFOG: discrete flow matching for graph generation. In Proceedings of the 42nd International Conference on Machine Learning. (Vancouver, BC, Canada. PMLR, 2025).
Dunn, I. & Koes, D. R. Exploring discrete flow matching for 3d de novo molecule generation. Preprint at arXiv https://doi.org/10.48550/arXiv.2411.16644 (2024).
Cheng, C., Li, J., Fan, J. & Liu, G. α-flow: a unified framework for continuous-state discrete flow matching models. Preprint at arXiv https://doi.org/10.48550/arXiv.2504.10283 (2025).
Tang, S., Zhang, Y., Tong, A. & Chatterjee, P. Gumbel-softmax flow matching with straight-through guidance for controllable biological sequence generation. Preprint at arXiv https://doi.org/10.48550/arXiv.2503.17361 (2025).
Chen, T., Zhang, Y., Tang, S. & Chatterjee, P. Multi-objective-guided discrete flow matching for controllable biological sequence design. CoRR abs/2505.07086 https://doi.org/10.48550/arXiv.2505.07086 (2025).
Gao, L. & Lu, Z. J. RNACG: a universal RNA sequence conditional generation model based on flow-matching. Preprint at arXiv https://doi.org/10.48550/arXiv.2407.19838 (2024).
Nori, D. & Jin, W. RNAFlow: RNA structure & sequence design via inverse folding-based flow matching. In Proceedings of the 41st International Conference on Machine Learning. (Vienna, Austria, PMLR 235, 2024).
Rubin, D., Costa, A. d. S., Ponnapati, M. & Jacobson, J. RiboGen: RNA sequence and structure co-generation with equivariant multiflow. Preprint at arXiv https://doi.org/10.48550/arXiv.2503.02058 (2025).
Tarafder, S. & Bhattacharya, D. RNAbpFlow: base pair-augmented SE(3)-flow matching for conditional RNA 3d structure generation. bioRxiv https://doi.org/10.1101/2025.01.24.634669 (2025).
Ma, R. et al. RiboFlow: conditional de novo RNA co-design via synergistic flow matching. In The Thirty-ninth Annual Conference on Neural Information Processing Systems (NeurIPS, 2025).
Klein, D., Uscidda, T., Theis, F. & Cuturi, M. Genot: Entropic (gromov) wasserstein flow matching with applications to single-cell genomics. Adv. Neural Inf. Process Syst. 37, 103897–103944 (2024).
Palma, A., Richter, T., Zhang, H., Dittadi, A. & Theis, F. J. cellflow: a generative flow-based model for single-cell count data. In ICLR 2024 Workshop on Machine Learning for Genomics Explorations (ICLR 2024 Workshop, 2024).
Palma, A. et al. Multi-modal and multi-attribute generation of single cells with CFGen. In The Thirteenth International Conference on Learning Representations (Openreview.net, 2025).
Nagaraj, S., Shanehsazzadeh, A., Park, H., King, J. & Levine, S. Igflow: flow matching for de novo antibody design. In Advances in Neural Information Processing Systems (NeurIPS, 2024).
Tan, C. et al. dyab: Flow matching for flexible antibody design with alphafold-driven pre-binding antigen. In Proceedings of the AAAI Conference on Artificial Intelligence. Vol. 39, 782–790 (AAAI Press, 2025).
Hoogeboom, E., Satorras, V. G., Vignac, C. & Welling, M. Equivariant diffusion for molecule generation in 3d. In International Conference on Machine Learning, 8867–8887 (PMLR, 2022).
Liu, Q., Allamanis, M., Brockschmidt, M. & Gaunt, A. L. Constrained graph variational autoencoders for molecule design. In Advances in Neural Information Processing Systems 31: Annual Conference on Neural Information Processing Systems 2018, NeurIPS 2018 (eds Bengio, S. et al.) 7806–7815 https://proceedings.neurips.cc/paper/2018/hash/b8a03c5c15fcfa8dae0b03351eb1742f-Abstract.html (2018).
Vignac, C. et al. Digress: discrete denoising diffusion for graph generation. In The Eleventh International Conference on Learning Representations, ICLR 2023, Kigali, Rwanda (OpenReview.net, 2023).
Luo, S., Guan, J., Ma, J. & Peng, J. A 3d generative model for structure-based drug design. In Advances in Neural Information Processing Systems 34: Annual Conference on Neural Information Processing Systems 2021, NeurIPS 2021 (eds Ranzato, M., Beygelzimer, A., Dauphin, Y. N., Liang, P. & Vaughan, J. W.) 6229–6239 https://proceedings.neurips.cc/paper/2021/hash/314450613369e0ee72d0da7f6fee773c-Abstract.html (2021).
Peng, X. et al. Pocket2mol: efficient molecular sampling based on 3d protein pockets. In International Conference on Machine Learning, 17644–17655 (PMLR, 2022).
Noé, F., Tkatchenko, A., Müller, K.-R. & Clementi, C. Machine learning for molecular simulation. Annu. Rev. Phys. Chem. 71, 361–390 (2020).
Hollingsworth, S. A. & Dror, R. O. Molecular dynamics simulation for all. Neuron 99, 1129–1143 (2018).
Walters, W. P. & Barzilay, R. Applications of deep learning in molecule generation and molecular property prediction. Acc. Chem. Res. 54, 263–270 (2020).
Du, Y. et al. Machine learning-aided generative molecular design. Nat. Mac. Intell. 6, 589–604 (2024).
Guo, Z. et al. Graph-based molecular representation learning. In Proceedings of the Thirty-Second International Joint Conference on Artificial Intelligence, IJCAI, Macao, SAR, China, 6638–6646 (ijcai.org, 2023).
De Cao, N. & Kipf, T. Molgan: an implicit generative model for small molecular graphs. Preprint at arXiv https://doi.org/10.48550/arXiv.1805.11973 (2018).
Li, Y., Zhang, L. & Liu, Z. Multi-objective de novo drug design with conditional graph generative model. J. Cheminform. 10, 1–24 (2018).
Eijkelboom, F., Bartosh, G., Naesseth, C. A., Welling, M. & van de Meent, J. Variational flow matching for graph generation. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems 2024, NeurIPS 2024, Vancouver, BC, Canada (eds Globersons, A. et al.) http://papers.nips.cc/paper_files/paper/2024/hash/15b780350b302a1bf9a3bd273f5c15a4-Abstract-Conference.html (2024).
Hou, X. et al. Improving molecular graph generation with flow matching and optimal transport. CoRR abs/2411.05676 https://doi.org/10.48550/arXiv.2411.05676 (2024).
Qin, Y., Madeira, M., Thanou, D. & Frossard, P. Defog: Discrete flow matching for graph generation. CoRR abs/2410.04263 https://doi.org/10.48550/arXiv.2410.04263 (2024).
Baillif, B., Cole, J., McCabe, P. & Bender, A. Deep generative models for 3d molecular structure. Curr. Opin. Struct. Biol. 80, 102566 (2023).
Peng, X., Guan, J., Liu, Q. & Ma, J. Moldiff: addressing the atom-bond inconsistency problem in 3d molecule diffusion generation. In International Conference on Machine Learning, ICML, Honolulu, Hawaii, USA, Vol. 202 of Proceedings of Machine Learning Research (eds Krause, A. et al.) 27611–27629 (PMLR, 2023).
Huang, L., Zhang, H., Xu, T. & Wong, K. MDM: molecular diffusion model for 3d molecule generation. In Thirty-Seventh AAAI Conference on Artificial Intelligence, AAAI 2023, Thirty-Fifth Conference on Innovative Applications of Artificial Intelligence, IAAI 2023, Thirteenth Symposium on Educational Advances in Artificial Intelligence, EAAI (eds Williams, B., Chen, Y. & Neville, J.) 5105–5112 (AAAI Press, 2023).
Reidenbach, D., Nikitin, F., Isayev, O. & Paliwal, S. G. Applications of modular co-design for de novo 3d molecule generation. In NeurIPS 2024 Workshop on AI for New Drug Modalities (NeurIPS 2024 Workshop, 2024).
Tian, Q. et al. Equiflow: equivariant conditional flow matching with optimal transport for 3d molecular conformation prediction. CoRR abs/2412.11082 https://doi.org/10.48550/arXiv.2412.11082 (2024).
Eijkelboom, F. et al. Controlled generation with equivariant variational flow matching. In Forty-second International Conference on Machine Learning, ICML 2025 (OpenReview.net, 2025).
Hong, H., Lin, W. & Tan, K. C. Accelerating 3d molecule generation via jointly geometric optimal transport. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Irwin, R., Tibo, A., Janet, J. P. & Olsson, S. Efficient 3d molecular generation with flow matching and scale optimal transport. CoRR abs/2406.07266 https://doi.org/10.48550/arXiv.2406.07266 (2024).
Irwin, R., Tibo, A., Janet, J. P. & Olsson, S. Semlaflow–efficient 3d molecular generation with latent attention and equivariant flow matching. In The 28th International Conference on Artificial Intelligence and Statistics (PMLR, 2025).
Cao, Z. et al. Efficient molecular conformer generation with so (3) averaged flow-matching and reflow. In Forty-second International Conference on Machine Learning, ICML 2025 (PMLR, 2025).
Hassan, M. et al. Et-flow: Equivariant flow-matching for molecular conformer generation. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems 2024, NeurIPS 2024, Vancouver, BC, Canada (eds Globersons, A. et al.) http://papers.nips.cc/paper_files/paper/2024/hash/e8bd617e7dd0394ceadf37b4a7773179-Abstract-Conference.html (2024).
Jiao, R., Kong, X., Huang, W. & Liu, Y. 3d structure prediction of atomic systems with flow-based direct preference optimization. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems 2024, NeurIPS 2024, Vancouver, BC, Canada (eds Globersons, A. et al.) http://papers.nips.cc/paper_files/paper/2024/hash/c6fdc94aeb2cb3a426d510d970045dab-Abstract-Conference.html (2024).
Isobe, N., Koyama, M., Hayashi, K. & Fukumizu, K. Extended flow matching: a method of conditional generation with generalized continuity equation. CoRR abs/2402.18839 https://doi.org/10.48550/arXiv.2402.18839 (2024).
Dunn, I. & Koes, D. R. Mixed continuous and categorical flow matching for 3d de novo molecule generation. CoRR abs/2404.19739 https://doi.org/10.48550/arXiv.2404.19739 (2024).
Zhou, W. et al. Energy-based flow matching for generating 3d molecular structure. In Forty-second International Conference on Machine Learning, ICML 2025 (OpenReview.net, 2025).
Wang, L., Cheng, C., Liao, Y., Qu, Y. & Liu, G. Training free guided flow matching with optimal control. CoRR abs/2410.18070 https://doi.org/10.48550/arXiv.2410.18070 (2024).
Li, Z., Zhou, C., Wang, X., Peng, X. & Zhang, M. Geometric representation condition improves equivariant molecule generation. CoRR abs/2410.03655 https://doi.org/10.48550/arXiv.2410.03655 (2024).
Vost, L., Chenthamarakshan, V., Das, P. & Deane, C. M. Improving structural plausibility in 3d molecule generation via property-conditioned training with distorted molecules. bioRxiv https://doi.org/10.1101/2024.09.17.613136 (2024).
Cheng, A. H., Lo, A., Lee, K. L. K., Miret, S. & Aspuru-Guzik, A. Stiefel flow matching for moment-constrained structure elucidation. CoRR abs/2412.12540 https://doi.org/10.48550/arXiv.2412.12540 (2024).
Zhou, W., Sprague, C. I. & Azizpour, H. Energy-based flow matching for molecular docking (2025).
Zeng, C. et al. Propmolflow: property-guided molecule generation with geometry-complete flow matching. Preprint at arXiv https://doi.org/10.48550/arXiv.2505.21469 (2025).
Bergues, N. et al. Template-guided 3d molecular pose generation via flow matching and differentiable optimization. Preprint at arXiv https://doi.org/10.48550/arXiv.2506.06305 (2025).
Huang, J. & Zhang, D. Molform: multi-modal flow matching for structure-based drug design. Preprint at arXiv https://doi.org/10.48550/arXiv.2507.05503 (2025).
Zhang, Z., Wang, M. & Liu, Q. Flexsbdd: structure-based drug design with flexible protein modeling. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems 2024, NeurIPS 2024, Vancouver, BC, Canada (eds Globersons, A. et al.) http://papers.nips.cc/paper_files/paper/2024/hash/60fb8cf8000f0386063fb24ead366330-Abstract-Conference.html (2024).
Qu, Y. et al. Molcraft: structure-based drug design in continuous parameter space. In Forty-first International Conference on Machine Learning (PMLR, 2024).
Xue, K. et al. Unifying Bayesian flow networks and diffusion models through stochastic differential equations. In International Conference on Machine Learning, 55656–55681 (PMLR, 2024).
Peng, X. et al. Decipher fundamental atomic interactions to unify generative molecular docking and design. Preprint at bioRxiv https://doi.org/10.1101/2024.10.17.618827 (2024).
Zhou, J., Qian, H., Tu, S. & Xu, L. Prior-guided flow matching for target-aware molecule design with learnable atom number. Preprint at arXiv https://doi.org/10.48550/arXiv.2509.01486 (2025).
Yim, J. et al. Fast protein backbone generation with SE(3) flow matching. Preprint at arXiv https://doi.org/10.48550/arXiv.2310.05297 (2023).
Ahern, W. et al. Atom level enzyme active site scaffolding using rfdiffusion2. Nat. Methods 23, 96–105 (2026).
Yan, J. et al. Robust and reliable de novo protein design: a flow-matching-based protein generative model achieves remarkably high success rates. bioRxiv https://doi.org/10.1101/2025.04.29.651154 (2025).
Wagner, S. et al. Generating highly designable proteins with geometric algebra flow matching. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems (NeurIPS 2024, Vancouver, BC, Canada 2024).
Huguet, G. et al. Sequence-augmented SE(3)-flow matching for conditional protein backbone generation. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems (NeurIPS 2024, Vancouver, BC, Canada 2024).
Geffner, T. et al. Proteina: scaling flow-based protein structure generative models. In The Thirteenth International Conference on Learning Representations, ICLR 2025, (Openreview.net, Singapore, 2025).
Stark, H. et al. Protcomposer: compositional protein structure generation with 3d ellipsoids. In The Thirteenth International Conference on Learning Representations, ICLR 2025, (Openreview.net, Singapore, 2025).
Yang, S. et al. Co-design protein sequence and structure in discrete space via generative flow. Bioinformatics 41, btaf248 (2025).
Chen, R. et al. An all-atom generative model for designing protein complexes. In Forty-second International Conference on Machine Learning, ICML 2025 (OpenReview.net, 2025)
Yim, J. et al. Improved motif-scaffolding with SE(3) flow matching. arXiv https://doi.org/10.48550/arXiv.2401.04082 (2024).
Huang, Y. et al. Eva: geometric inverse design for fast protein motif-scaffolding with coupled flow. In The Thirteenth International Conference on Learning Representations (OpenReview.net, 2025).
Liu, J., Li, S., Shi, C., Yang, Z. & Tang, J. Design of ligand-binding proteins with atomic flow matching. Preprint at arXiv https://doi.org/10.48550/arXiv.2409.12080 (2024).
Cremer, J. et al. Flowr: flow matching for structure-aware de novo, interaction-and fragment-based ligand generation. Preprint at arXiv https://doi.org/10.48550/arXiv.2504.10564 (2025).
Cremer, J. et al. Flowr. root: a flow matching based foundation model for joint multi-purpose structure-aware 3d ligand generation and affinity prediction. Preprint at arXiv https://doi.org/10.48550/arXiv.2510.02578 (2025).
Stark, H., Jing, B., Barzilay, R. & Jaakkola, T. Harmonic self-conditioned flow matching for joint multi-ligand docking and binding site design. In Forty-first International Conference on Machine Learning (PMLR, 2024).
Zhang, Z., Zitnik, M. & Liu, Q. Generalized protein pocket generation with prior-informed flow matching. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems. (NeurIPS 2024, Vancouver, BC, Canada, 2024).
Jones, M. S., Khanna, S. & Ferguson, A. L. Flowback: a generalized flow-matching approach for biomolecular backmapping. J. Chem. Inf. Model 65, 672–692 (2025).
Jing, B., Berger, B. & Jaakkola, T. Alphafold meets flow matching for generating protein ensembles. In Proceedings of the 41st International Conference on Machine Learning, (PMLR 235, Vienna, Austria, 2024).
Jin, Y. et al. P2dflow: a protein ensemble generative model with se (3) flow matching. J. Chem. Theory Comput. 21, 3288–3296 (2025).
Lee, J. S. & Kim, P. M. Flowpacker: protein side-chain packing with torsional flow matching. Bioinformatics 41, btaf010 (2025).
Liao, Y.-L., Wood, B., Das, A. & Smidt, T. Equiformerv2: improved equivariant transformer for scaling to higher-degree representations. In The Twelfth International Conference on Learning Representations, (OpenReview.net, ICLR 2024, Vienna, Austria 2024).
Li, J. et al. Full-atom peptide design based on multi-modal flow matching. In Proceedings of the 41st International Conference on Machine Learning, (Vienna, Austria, PMLR 235, 2024).
Morehead, A. & Cheng, J. Flowdock: geometric flow matching for generative protein-ligand docking and affinity prediction. arXiv https://doi.org/10.48550/arXiv.2412.10966 (2025).
Guo, H., Liu, S. & Jing, B. Forcefm: enhancing protein-ligand predictions through force-guided flow matching. In The Thirty-Ninth Annual Conference on Neural Information Processing Systems (NeurIPS, 2025).
Lin, H. et al. Ppflow: target-aware peptide design with torsional flow matching. bioRxiv https://doi.org/10.1101/2024.03.07.583831 (2024).
Huang, D. & Tu, S. Non-linear flow matching for full-atom peptide design. Preprint at arXiv https://doi.org/10.48550/arXiv.2502.15855 (2025).
Kong, Z. et al. Protflow: fast protein sequence design via flow matching on compressed protein language model embeddings. Preprint at arXiv https://doi.org/10.48550/arXiv.2504.10983 (2025).
Zhang, J. et al. Efficient antibody structure refinement using energy-guided SE(3) flow matching. In IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2024, Lisbon, Portugal (eds Cannataro, M. et al.) 146–153 (IEEE, 2024).
Zhang, Y. et al. Cellflow: simulating cellular morphology changes via flow matching. In Forty-second International Conference on Machine Learning, ICML 2025 (OpenReview.net, 2025).
Kapusniak, K. et al. Metric flow matching for smooth interpolations on the data manifold. Adv. Neural Inf. Process Syst. 37, 135011–135042 (2024).
Shrestha, S. & Fu, X. Diversified flow matching with translation identifiability. In Forty-second International Conference on Machine Learning, ICML 2025 (OpenReview.net, 2025).
Bogensperger, L., Narnhofer, D., Falk, A., Schindler, K. & Pock, T. Flowsdf: flow matching for medical image segmentation using distance transforms. CoRR abs/2405.18087 https://doi.org/10.48550/arXiv.2405.18087 (2024).
Yazdani, M., Medghalchi, Y., Ashrafian, P., Hacihaliloglu, I. & Shahriari, D. Flow matching for medical image synthesis: bridging the gap between speed and quality. CoRR abs/2503.00266 https://doi.org/10.48550/arXiv.2503.00266 (2025).
Zhang, D., Han, Q., Xiong, Y. & Du, H. Mutli-modal straight flow matching for accelerated MR imaging. Comput. Biol. Med. 178, 108668 (2024).
Huang, T., Liu, T., Babadi, M., Jin, W. & Ying, R. Scalable generation of spatial transcriptomics from histology images via whole-slide flow matching. In Forty-second International Conference on Machine Learning, ICML 2025 (OpenReview.net, 2025).
Haviv, D., Pooladian, A.-A., Pe’er, D. & Amos, B. Wasserstein flow matching: generative modeling over families of distributions. In Forty-second International Conference on Machine Learning, ICML 2025 (OpenReview.net, 2025).
Wei, G. & Ma, L. Stream-level flow matching with Gaussian processes. In Forty-second International Conference on Machine Learning https://openreview.net/forum?id=qg9p1I5lmp (PMLR, 2025).
Wang, P., Qi, Y., Wang, Y. & Pan, G. Flow matching for few-trial neural adaptation with stable latent dynamics. In Forty-second International Conference on Machine Learning https://openreview.net/forum?id=nKJEAQ6JCY (2025).
Collas, A., Ju, C., Salvy, N. & Thirion, B. Riemannian flow matching for brain connectivity matrices via pullback geometry. Preprint at arXiv https://doi.org/10.48550/arXiv.2505.18193 (2025).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 (2003).
Adamczyk, B., Antczak, M. & Szachniuk, M. Rnasolo: a repository of cleaned pdb-derived rna 3d structures. Bioinformatics 38, 3668–3670 (2022).
Chu, Y. et al. A 5’ UTR language model for decoding untranslated regions of mRNA and function predictions. Nat. Mach. Intell. 6, 449–460 (2024).
Suzek, B. E. et al. Uniref clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Cornman, A. et al. The OMG dataset: an open metagenomic corpus for mixed-modality genomic language modeling. bioRxiv https://doi.org/10.1101/2024.08.14.607850 (2024).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Bastidas-Ponce, A. et al. Comprehensive single cell mRNA profiling reveals a detailed roadmap for pancreatic endocrinogenesis. Development 146, dev173849 (2019).
Srivatsan, S. R. et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 367, 45–51 (2020).
Luecken, M. D. et al. A sandbox for prediction and integration of DNA, RNA, and proteins in single cells. In Thirty-fifth Conference on Neural Information Processing Systems Datasets and Benchmarks Track (Round 2) (NeurIPS, 2021).
Derbois, C., Palomares, M.-A., Deleuze, J.-F., Cabannes, E. & Bonnet, E. Single cell transcriptome sequencing of stimulated and frozen human peripheral blood mononuclear cells. Sci. Data 10, 433 (2023).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Schaum, N. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris: the Tabula Muris Consortium. Nature 562, 367 (2018).
Ramakrishnan, R., Dral, P. O., Rupp, M. & Von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 1–7 (2014).
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S. & Coleman, R. G. Zinc: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768 (2012).
Axelrod, S. & Gomez-Bombarelli, R. Geom, energy-annotated molecular conformations for property prediction and molecular generation. Sci. Data 9, 185 (2022).
Polykovskiy, D. et al. Molecular sets (moses): a benchmarking platform for molecular generation models. Front. Pharm. 11, 565644 (2020).
Chandonia, J.-M. et al. Scope: improvements to the structural classification of proteins–extended database to facilitate variant interpretation and machine learning. Nucleic Acids Res. 50, D553–D559 (2022).
Vander Meersche, Y., Cretin, G., Gheeraert, A., Gelly, J.-C. & Galochkina, T. Atlas: protein flexibility description from atomistic molecular dynamics simulations. Nucleic Acids Res. 52, D384–D392 (2024).
Hu, L., Benson, M. L., Smith, R. D., Lerner, M. G. & Carlson, H. A. Binding moad (mother of all databases). Proteins Struct. Funct. Bioinforma. 60, 333–340 (2005).
Francoeur, P. G. et al. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model. 60, 4200–4215 (2020).
Zhang, C., Zhang, X., Freddolino, L. & Zhang, Y. Biolip2: an updated structure database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 52, D404–D412 (2024).
Wen, Z., He, J., Tao, H. & Huang, S.-Y. Pepbdb: a comprehensive structural database of biological peptide–protein interactions. Bioinformatics 35, 175–177 (2019).
Dunbar, J. et al. Sabdab: the structural antibody database. Nucleic Acids Res. 42, D1140–D1146 (2014).
Chang, Y. et al. A survey on evaluation of large language models. ACM Trans. Intell. Syst. Technol. 15, 1–45 (2024).
Wang, J. et al. Generalizing to unseen domains: a survey on domain generalization. IEEE Trans. Knowl. Data Eng. 35, 8052–8072 (2022).
Deng, J. et al. Imagenet: a large-scale hierarchical image database. In 2009 IEEE Conference on Computer Vision and Pattern Recognition, 248–255 (IEEE, 2009).
Dwivedi, V. P. et al. Benchmarking graph neural networks. J. Mach. Learn Res. 24, 1–48 (2023).
Saad, M. M., O’Reilly, R. & Rehmani, M. H. A survey on training challenges in generative adversarial networks for biomedical image analysis. Artif. Intell. Rev. 57, 19 (2024).
Zhang, Q. et al. Scientific large language models: a survey on biological & chemical domains. ACM Comput. Surv. 57, 1–38 (2025).
Sikkema, L. et al. An integrated cell atlas of the lung in health and disease. Nat. Med. 29, 1563–1577 (2023).
Vieira Braga, F. A. et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25, 1153–1163 (2019).
Moon, K. R. et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019).
Lance, C. et al. Multimodal single cell data integration challenge: results and lessons learned. bioRxiv https://doi.org/10.1101/2022.04.11.487796 (2022).
Dunn, I. & Koes, D. R. Mixed continuous and categorical flow matching for 3d de novo molecule generation. arXiv https://doi.org/10.48550/arXiv.2404.19739 (2024).
Brown, N., Fiscato, M., Segler, M. H. & Vaucher, A. C. Guacamol: benchmarking models for de novo molecular design. J. Chem. Inf. Model. 59, 1096–1108 (2019).
Buttenschoen, M., Morris, G. M. & Deane, C. M. Posebusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. 15, 3130–3139 (2024).
Wu, J. et al. Flowdesign: improved design of antibody cdrs through flow matching and better prior distributions. Cell Syst. 16, 101270 (2025).
Wang, R., Fang, X., Lu, Y. & Wang, S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 47, 2977–2980 (2004).
Agrawal, P. et al. Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinforma. 19, 105–124 (2019).
Yim, J. et al. Se (3) diffusion model with application to protein backbone generation. In Proceedings of the 40 th International Conference on Machine Learning, (Honolulu, Hawaii, USA. PMLR 202, 2023).
Jones, M. S., Khanna, S. & Ferguson, A. L. Flowback: a generalized flow-matching approach for biomolecular backmapping. J. Chem. Inf. Model. 65, 672–692 (2025).
Olsen, T. H., Boyles, F. & Deane, C. M. Observed antibody space: a diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. 31, 141–146 (2022).
Adolf-Bryfogle, J. et al. RosettaAntibodyDesign (RAbD): a general framework for computational antibody design. PLoS Comput. Biol. 14, e1006112 (2018).
UniProt Consortium, T. Uniprot: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699 (2018).
Bairoch, A. & Apweiler, R. The swiss-prot protein sequence database and its supplement trembl in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Wu, F. et al. D-flow: Multi-modality flow matching for d-peptide design. Preprint at arXiv https://doi.org/10.48550/arXiv.2411.10618 (2024).
Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K. & Moult, J. Critical assessment of methods of protein structure prediction (casp)—round xv. Proteins Struct. Funct. Bioinforma. 91, 1539–1549 (2023).
Morehead, A., Liu, J., Neupane, P., Giri, N. & Cheng, J. Protein-ligand structure and affinity prediction in casp16 using a geometric deep learning ensemble and flow matching. Proteins Struct. Funct. Bioinforma. 94, 295–301 (2025).
Abdin, O., Nim, S., Wen, H. & Kim, P. M. Pepnn: a deep attention model for the identification of peptide binding sites. Commun. Biol. 5, 503 (2022).
Ljosa, V., Sokolnicki, K. L. & Carpenter, A. E. Annotated high-throughput microscopy image sets for validation. Nat. Methods 9, 637 (2012).
Taylor, J., Earnshaw, B., Mabey, B., Victors, M. & Yosinski, J. RxRx1: an image set for cellular morphological variation across many experimental batches. In ICLR AI for Social Good Workshop (ICLR 2019 Workshop, 2019).
Chandrasekaran, S. N. et al. JUMP cell painting dataset: morphological impact of 136,000 chemical and genetic perturbations. Preprint at bioRxiv https://doi.org/10.1101/2023.03.23.534023 (2023).
Kumar, N. et al. A multi-organ nucleus segmentation challenge. IEEE Trans. Med. Imaging 39, 1380–1391 (2020).
Sirinukunwattana, K. et al. Gland segmentation in colon histology images: the glas challenge contest. Med. Image Anal. 35, 489–502 (2017).
Leclerc, S. et al. Deep learning for segmentation using an open large-scale dataset in 2d echocardiography. IEEE Trans. Med. Imaging 38, 2198–2210 (2019).
Antonelli, M. et al. The medical segmentation decathlon. Nat. Commun. 13, 4128 (2022).
Knoll, F. et al. fastMRI: a publicly available raw kspace and DICOM dataset of knee images for accelerated MR image reconstruction using machine learning. Radiology Artif. Intell. 2, e190007 (2020).
Menze, B. H. et al. The multimodal brain tumor image segmentation benchmark (brats). IEEE Trans. Med. Imaging. 34, 1993–2024 (2015).
Jaume, G. et al. HEST-1k: a dataset for spatial transcriptomics and histology image analysis. In Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems (NeurIPS 2024, Vancouver, BC, Canada 2024).
Chen, J. et al. STimage-1K4M: a histopathology image-gene expression dataset for spatial transcriptomics. Advances in Neural Information Processing Systems 38: Annual Conference on Neural Information Processing Systems (NeurIPS 2024, Vancouver, BC, Canada 2024).
Lohoff, T. et al. Integration of spatial and single-cell transcriptomic data elucidates mouse organogenesis. Nat. Biotechnol. 40, 74–85 (2022).
Stephenson, E. et al. Single-cell multi-omics analysis of the immune response in Covid-19. Nat. Med. 27, 904–916 (2021).
Steinmetz, N. A., Zatka-Haas, P., Carandini, M. & Harris, K. D. Distributed coding of choice, action and engagement across the mouse brain. Nature 576, 266–273 (2019).
Flint, R. D., Wright, Z. A., Scheid, M. R. & Slutzky, M. W. Long-term stability of neural prosthetic control signals from silicon cortical arrays in rhesus macaque motor cortex. J. Neural Eng. 9, 056009 (2012).
Churchland, M. M., Cunningham, J. P., Kaufman, M. T., Ryu, S. I. & Shenoy, K. V. Neural population dynamics during reaching. Nature 487, 51–56 (2012).
Cornblath, E. J., Heravi, E., Cunningham, J. P. & Sussillo, D. An empirical evaluation of neural population dynamics models for motor cortex. In Neural Latents Benchmark Workshop at NeurIPS (NeurIPS 2021 Workshop, 2021).
Acknowledgements
This work is supported by (1) The Agriculture and Food Research Initiative (AFRI) grant no. 2020-67021-32799/project accession no. 1024178 from the USDA National Institute of Food and Agriculture. The views and conclusions are those of the authors and should not be interpreted as representing the official policies of the funding agencies or the government. (2) The DOE Center for Advanced Bioenergy and Bioproducts Innovation (U.S. Department of Energy, Office of Science, Biological and Environmental Research Program under Award Number DE-SC0018420). Any opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the U.S. Department of Energy. The Agriculture and Food Research Initiative (AFRI) grant no. 2020-67021-32799/project accession no. 1024178 from the USDA National Institute of Food and Agriculture. U.S. Department of Energy, Office of Science, Biological and Environmental Research Program under Award Number DE-SC0018420.
Author information
Authors and Affiliations
Contributions
Zihao L., Z.Z., and X.L. drafted the main manuscript text. F.F. reviewed the datasets and benchmarks and assisted with publishing the GitHub resources repository. Y.Q. and Z.X. provided oversight of the biology-related and flow-matching-related sections, respectively, and contributed extensive feedback. Zhining L. prepared the figures and contributed to Section "Other bio applications". X.N. and T.W. contributed to Sections "Challenges of generative modeling for biology", "Connection to existing survey", and "Conclusion". G.L., H.T., and J.H. supervised the research. All authors reviewed the manuscript and provided valuable suggestions. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The corresponding author, J.H., serves as an Associate Editor for npj Artificial Intelligence. Aside from this editorial role, the authors declare no other competing financial or non-financial interests as defined by Nature Portfolio, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Zeng, Z., Lin, X. et al. Flow matching meets biology and life science: a survey. npj Artif. Intell. 2, 17 (2026). https://doi.org/10.1038/s44387-025-00066-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44387-025-00066-y
