
Abstract
Genome-wide association studies (GWAS) have identified numerous loci linked to late-onset Alzheimer’s disease (LOAD), but the pan-brain regional effects of these loci remain largely uncharacterized. To address this, we systematically analyzed all LOAD-associated regions reported by Bellenguez et al. using the FILER functional genomics catalog across 174 datasets, including enhancers, transcription factors, and quantitative trait loci. We identified 41 candidate causal variant-effector gene pairs and assessed their impact using enhancer–promoter interaction data, variant annotations, and brain cell-type-specific gene expression. Notably, the LOAD risk allele of rs74504435 at the SEC61G locus was computationally predicted to increase EGFR expression in LOAD-related cell types: microglia, astrocytes, and neurons. Functional validation using promoter-focused Capture C, ATAC-seq, and CRISPR interference in the HMC3 human microglia cell line confirmed this regulatory relationship. Our findings reveal a microglial enhancer regulating EGFR in LOAD, suggesting EGFR inhibitors as a potential therapeutic avenue for the disease.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the leading cause of dementia in the United States and currently lacks effective treatments or prevention strategies. The most common form, late-onset Alzheimer’s disease (LOAD), typically begins after age 60 and is highly heritable (60-80%), indicating a significant genetic component in its development1. While the APOE locus remains the strongest genetic risk factor2, LOAD is complex and highly polygenic3. Previous genome-wide association studies (GWAS) identified over 20 LOAD-associated loci2,4; recent studies using UK Biobank proxy-AD or proxy-control samples have expanded this list to 75 loci5,6. Although progress has been made in linking LOAD genetic risk to microglial-mediated innate immune processes7,8,9, the broader cellular contexts of these variants remain incompletely understood. Emerging evidence suggests that LOAD-associated variants also affect other brain cell types, including myeloid cells10, astrocytes, and neurons, but the mechanisms across these diverse cellular environments remain largely uncharacterized.
Over 90% of GWAS variants are located in non-coding regions of the genome, outside of protein-coding sequences10,11. These non-coding variants are widely hypothesized to affect gene regulatory elements, such as enhancers12,13,14, which can influence the expression of distant target genes15. The difficulty in identifying such distal genes arises from the challenges related to linkage disequilibrium (LD) with nearby non-causal variants and the variability in the biological contexts of the corresponding target ‘effector’ genes. Despite these, some studies have applied various statistical and computational methods, along with new data types, to analyze non-coding GWAS signals for AD10,16,17,18. This effort is important because drugs with genetic support are twice as likely to gain approval19,20,21.
However, these studies face limitations in their analytical strategies. First, they traditionally focus only on top GWAS signals (sentinel variants), restricting the understanding of AD’s full genetic landscape5,6,16. Since any variant in LD with a sentinel could be causal, comprehensive analyses should include both sentinel and nearby LD variants. Second, genome-wide functional genomics data for brain tissues or specific cell types remain limited and relatively small compared to data from other cell types or cell lines22,23,24. Third, prior computational analyses of non-coding variants often failed to integrate eQTLs from independent sources to confirm consistency and replication of effect alleles and signal direction5,6,10,11. Finally, no previous analyses have combined eQTLs and enhancer–promoter interactions (EPI) to prioritize variant-to-gene (V2G) pairs in LOAD.
To address these limitations, we developed an enhanced post-GWAS non-coding variant analysis framework25. This approach systematically identifies candidate causal variants and relevant regulatory genomic features to improve our understanding of genetic loci associated with LOAD.
Results
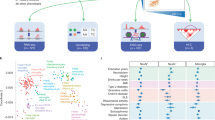
We summarize our strategy in Fig. 1, which comprises four key steps: “Preprocessing,” “Characterization,” “In silico validation,” and “Functional validation”.
-
1.
In the “Preprocessing” stage, we obtained pairwise-independent tag variants through LD-based pruning (1000 Genome panel) on all genome-wide significant LOAD GWAS variants (p < 5 × 10−8)5. To produce a larger pool of candidate causal variants, we included all proxy variants through LD-based expansion of the tag variant set (r2 > =0.7).
-
2.
In the “Characterization” step, these variants were annotated using FILER26 (a large-scale genomic data queryr tool), and their potential functional context(s) were predicted using SparkINFERNO25 (a scalable pipeline for inferring non-coding variants' molecular mechanisms). We identified candidate causal genes per variant using eQTL data. Additionally, we used HOMER27 to predict transcription factor binding site (TFBS) disruptions caused by the variants. Only variants within brain enhancers were retained in subsequent analysis steps. Together with their corresponding genes, this set formed the enhancer-based causal V2G pairs with TFBS.
-
3.
For the “In silico validation” step, we annotated and ranked the V2G pairs, putative causal variants, and effector genes using independent assays from new data sources. These include regulatory features (EPIs, QTLs, and open chromatin regions) and expression datasets (bulk RNA-seq, proteomics). We also performed consistency checks on QTLs across data sources.
-
4.
Finally, in the “Functional validation” step, we specifically contextualized one V2G pair using our existing datasets from multiple cell types, and in vitro validation using CRISPR interference (CRISPRi).

Our post-GWAS framework consists of the following steps: “Preprocessing,” “Characterization,” “In silico validation,” and “Functional validation.” Genome-wide significant variants from Bellenguez et al.5 were leveraged to identify regions of interest. Putative causal variants were analyzed for functional contexts and linked to potential causal genes using enhancers, eQTL data, and TFBS predictions. These variant-to-gene (V2G) pairs were characterized in brain tissues and cell types. Independent assays, including enhancer–promoter interactions (EPI), QTLs, and open chromatin regions from new data sources, were used for in silico validation. Functional validation included promoter-focused Capture C, ATAC-seq, RNA-seq, and CRISPRi in microglial cells.
Overall, we defined context-specific regulatory elements (variants and genes) across different cellular and tissue contexts, leveraging 174 datasets from 10 data sources. The number of regions, variants, and genes identified in each of the 4 steps in Fig. 1 is summarized in Supplementary Data 1. This enabled independent in silico validation of 41 variant-effector-gene (V2G) pairs. The rs74504435-EGFR V2G pair underwent further functional characterization, confirming it as a therapeutically tractable target.
Preprocessing: identification of genomic regions and candidate variants of interest
To define regions of interest and establish an initial discovery set of plausible candidate variants for further analyses, we leveraged the set of 5586 genome-wide significant variants (p < 5 × 10−8) from the full GWAS summary statistics5. We performed LD pruning using the 1000 genomes EUR panel28, obtaining 580 pairwise-independent (tag) variants (r² < 0.7). For each tag variant, we expanded its region of interest to include all proxy variants (r² ≥ 0.7) within 1 Mbps, with boundaries set by the most distant proxies. This LD-based expansion yielded a total of 9144 plausible candidate variants across all identified regions of interest (Preprocessing in Fig. 1, Methods: Preprocessing), increasing the candidate pool by 64% (from 5586 to 9144). Most candidate variants were non-coding, predominantly intergenic (38%) or intronic (24%), with 35% located in 5′ and 3′ UTR introns (Supplementary Fig. 1).
Characterization: identification of putative causal variants, genes, and variant-gene pairs
To explore the regulatory roles of candidate variants, we adapted the SparkINFERNO framework25 and overlapped variants with 174 brain-related functional genomics (FG) tracks in FILER26. These tracks cover 35 brain regions and 7 brain cell types across 10 data resources, representing five regulatory types: enhancers, histone modifications, QTLs, EPIs, and topologically associating domains (TADs) (Supplementary Fig. 2).
We quantified the probability of a candidate regulatory variant co-localizing with an eQTL signal. Among 9144 variants (including 1355 on chr19 in the APOE region), 229 had at least one colocalized signal (locus-level posterior colocalization probability (PP.H4.abf) >0.7), individual SNP-level posterior colocalization probability (SNP.PP.H4 > 0.5; see Supplementary Data 2 for the summary of colocalization results when using more stringent PP.H4.abf thresholds) in any of 13 GTEx29 brain tissue-specific eQTL tracks (see Supplementary Data 3 for the colocalized signals with FDR < 0.05, i.e., PP.H4.abf >0.95). These 229 variants defined 1601 tissue-V2G pairs, putatively regulating 232 genes. Notably, 14 variants were identified in ≥10 brain regions, while 103 variants (44.9%) were brain-region specific. Figure 2A highlights a subset of the top colocalization results (defined by locus-level posterior colocalization probability (PP.H4.abf) >0.99 and fewer than 100 SNPs per locus) across GTEx brain tissues.

A A subset of significant colocalization results across 59 loci and 13 GTEx brain regions. B All significant brain colocalization results of TSPAN14. C Significant brain colocalization results from multiple independent loci inside the ACE region. D All significant brain colocalization results of EGFR.
A single line of functional evidence is often considered insufficient to robustly implicate regulatory activity at a given locus. To address this limitation, we developed an unbiased confluent context identification strategy (Methods: “Steps for unbiased confluent context identification”) to integrate multiple lines of evidence to prioritize putative causal variants, genes, and V2G pairs for subsequent experimental validation. Of the 229 colocalized candidate variants, 68 (29.69%) overlap predicted HOMER27 TFBS (Method: Transcription factor binding site (TFBS) disruption). Among these, 15 (22%) and 23 (34%) fall within brain enhancers defined by ROADMAP30 and EpiMAP31, respectively (Method: Enhancers overlap). Overall, we identified 24 putatively causal variants (14 beyond the APOE locus) overlapping a brain eQTL, a brain enhancer (ROADMAP or EpiMAP), and a TFBS, forming 279 candidate tissue-V2G pairs involving 32 potential effector genes. Among non-APOE variants, 36% colocalized with >50% of GTEx brain tissues, with each variant interacting with an average of three (maximum 11) candidate effector genes. Notably, seven non-APOE variants did not affect the annotated (typically closest) genes in the original GWAS (Table 1)5. All selected colocalized candidate variants were common (non-reference allele frequency >0.05 in GWAS and 1000G). Table 1 summarizes the 14 candidate regulatory variants (beyond APOE) identified by this unbiased confluent context identification approach (see Supplementary Data 4 for robustness/sensitivity analysis of our V2G set under different/more stringent LD pruning thresholds). All eQTL effector genes and coloc results are detailed in Supplementary Data 5.
To further explore the genetic architecture of gene regulation in the brain, we examined loci with multiple candidate causal variants and their effects on gene expression across brain regions. Figure 2B highlights the significant colocalization results for the TSPAN14 locus, where three candidate causal variants were associated with the expression of four genes across nine GTEx brain regions. Figure 2C shows three independent signals, L1, L2, and L3 (pairwise r2 = 0.501, 0.364, and 0.709, respectively) at the ACE locus, each correlating with ACE gene expression across five GTEx brain regions. Figure 2D shows the two variants (r2 = 1.00) inside the EGFR locus. Together, these figures illustrate the complexity and diversity of genetic regulation, demonstrating how multiple independent variants can influence gene expression across diverse brain regions.
Potential biological roles of the identified transcription factor binding motifs
We found that the candidate regulatory variants are predicted to disrupt binding sites for 12 transcription factors (TFs) based on PWM analyses (Table 1). Several of these TFs are involved in mechanisms related to AD. Notably, GLIS3 (GLIS family zinc finger 3) is the only motif strongly linked to tau and amyloid pathology through transcriptional regulation32,33,34. SMAD2 (Mothers against decapentaplegic homolog 2), a key intracellular protein in the SMAD family, transduces signals from TGF-β ligands and mediates cellular responses. The TGF-β/SMAD2 pathway plays a complex role in AD, potentially affecting cell growth, differentiation, and immune responses35. Dysfunction in TGF-β signaling may lead to blood-brain barrier breakdown, and blocking TGF-β-SMAD2/3 signaling in peripheral macrophages has been proposed as a therapeutic strategy for AD36.
In silico validation: identification of putative causal signals with further support
Outside the APOE region, we identified 41 candidate V2G pairs comprising 14 variants and 26 protein-coding genes that are likely functional in brain tissues or cell types. For additional in silico support, we leveraged independent FG datasets. Recent FG data from large consortia37, harmonized studies38,39,40,41, and individual publications42,43 provided complementary or orthogonal evidence for in silico validation. We processed these datasets using hipFG44 and integrated them into FILER26, including two QTL (MetaBrain39, eQTL Catalogue45) and three EPI datasets (3DGenome41, 4DGenome40, Nott et al.43), standardizing all with metadata for efficient querying.
We first assessed the consistency of V2G pairs identified in brain regions across different data sources. Consistency in directionality means that a genetic variant’s effect on an eQTL (i.e., increasing or decreasing target gene expression) is the same across brain regions and data sources. Such consistency suggests shared genetic regulatory mechanisms across brain regions. However, eQTL directionality, represented by Z-scores, can vary between datasets due to (a) sampling differences, (b) QTL generation methods, or (c) statistical approaches. Since QTL datasets lack standardized presentation, harmonizing them with tools like hipFG44 can reduce these biases and improve interpretation.
In Fig. 3, the left panel shows hipFG-normalized Z-scores for each brain region (“System” in legend: Frontal Cortex, Limbic System, Basal Ganglia, Brain Stem) from two data sources (MetaBrain or eQTL Catalogue), along with average Z-scores across 13 GTEx brain regions (middle panel). The original, non-normalized Z-scores (Supplementary Fig. 3) show that 35 out of 41 V2G pairs had inconsistent directionality over >50% of the grouped brain regions. Strikingly, after Z-score normalization, inconsistencies dropped to only 6, an 83% improvement (McNemar’s Test, p < 0.0001), as shown in Fig. 3 (left panel).

All functional genomics data were processed using hipFG44, with effect directionality normalized. For both the left and middle panels, triangles pointing upwards (and in red) mean a positive Z-score, indicating that the alternative allele of a variant increases the gene expression, while those pointing downwards (and in green) mean the opposite effect. The size of the triangles represents the absolute value of the Z-score. The left panel shows the directionality (Z-scores) based on 23 QTL datasets from two data sources. The middle panel presents the average GTEx Z-scores, while the right panel displays the orthogonal support of EPI data for the V2G pairs (with a red square indicating the presence of EPI for that particular data source). The six loci with inconsistent directionalities were shown in yellow.
A ranking system integrating in silico evidence on genes, variants, and V2G pairs for selected putative causal signals
When an eQTL and an EPI co-occur for a given variant-to-gene (V2G) pair, it provides stronger evidence that the variant causally regulates gene expression46,47. To prioritize V2G pairs, we developed a ranking system based on four components: eQTL evidence (V2G_eQTL tier, assesses consistency of V2G pairs across gene expression datasets), EPI evidence (V2G_EPI tier, checks for V2G pairs in brain EPIs), variant properties (V_tier, evaluates variant characteristics), and gene expression (G_tier, considers gene relevance and expression). Each tier was generated by integrating data from multiple sources (Methods: Ranking of variant-gene pairs (Tier system). This structured approach helps prioritize V2G pairs based on robust, multi-source evidence.
The V2G_eQTL and V2G_EPI information are presented in Fig. 3, while overall tier rankings are displayed in Fig. 4. The top-ranked V2G pair was chr1:161186243:C:A_NDUFS2 (tier 11), followed by four pairs at tier 9 (chr7:54881563:A:G_EGFR, chr1:161186243:C:A_FCER1G, chr7:100373690_PVRIG, and chr17:63492371:G:A_ACE). These five pairs are hypothesized to have the greatest likelihood of functional study success.

To prioritize variant-to-gene (V2G) pairs for functional validation, we developed a ranking system based on four components: eQTL evidence (V2G_eQTL tier), EPI evidence (V2G_EPI tier), variant properties (V_tier), and gene expression (G_tier). Each tier was generated by integrating data from multiple sources.
Functional validation: an enhancer region harboring rs74504435 influences EGFR expression
We selected the second-highest-ranking V2G pair - rs74504435-EGFR - for further validation due to two main reasons. The LOAD risk allele at rs74504435 is associated with increased EGFR expression in multiple eQTL datasets (Fig. 5a), suggesting EGFR as a promising candidate for therapeutic targeting with existing EGFR inhibitors. This pair ranked highest in both the V2G_eQTL and V tiers, indicating robust support from diverse eQTL datasets and consistent functional annotations predicting a regulatory (enhancer) function for rs74504435 (Fig. 5a). Figure 5a visualizes this V2G pair using FILER tracks and Bellenguez GWAS summary statistics. rs74504435 regulates EGFR in 4 eQTLs: ROSMAP DLPFC, CommonMind DLPFC, GTEx Frontal Cortex, and GTEx cortex. It is also located within four brain-related enhancers from chromHMM and EpiMAP. We note that this top rs74504435 variant is also detected when using the most stringent pruning (LD = 0.1) and colocalization (PP.H4.abf >0.99) thresholds (Supplementary Data 4).

A Genome browser plot showing the functional annotation of the selected V2G pair in GRCh38 using FILER tracks at the discovery phase, including enhancers from chromHMM and EpiMAP, eQTLs from GTEx and eQTL Catalogue. Bellenguez GWAS identified the sentinel SNP rs76928645 (chr7:54873635:C:T) (p = 1.6 × 10−10), in high LD (r2 = 0.94) with our variant of interest rs74504435, which is annotated to reside in a brain enhancer in four different data sources. Plot is generated using NIAGADS genomicsDB. B ATAC-seq, promoter-focused Capture C, and Hi-C data in brain-relevant cell types showing chromatin state and looping between chr7:54881563:A:G (rs74504435) and EGFR. rs74504435 (highlighted by a yellow line) resides in open chromatin in our own iPSC-derived neural progenitors, neurons, and microglia, and in primary astrocytes. It is involved in a chromatin loop with EGFR in our own iPSC-derived neurons, primary astrocytes, and the microglial cell line HMC3 (promoter-focused Capture C data); as well as in our own iPSC-derived neurons and oligodendrocytes (Hi-C data). C CRISPRi results in a human microglia cell line. We performed CRISPRi in a human microglial cell line (HMC3) expressing dCas9-ZIM3-KRAB using lentiviral delivery of three sgRNA guides targeting the rs74504435 region (G1, G2, and G4) and two non-targeting guides (NTC: mean of control guides). Bar plots show the mean EGFR relative expression compared to a no-guide control as assessed by qPCR; error bars are SEM; N = 3. Statistical analysis via one-way ANOVA followed by Tukey test, **p < 0.001; *p < 0.05.
To investigate a potential regulatory role for rs74504435 on EGFR expression, we next validated this V2G pair by leveraging our collection of promoter-focused Capture C, ATAC-seq, and RNA-seq datasets from human brain-relevant cell types48,49,50, as well as Hi-C data from iPSC-derived astrocytes, microglia51, neurons, and oligodendrocytes52 (Supplementary Data 6). Via the ATAC-seq dataset, we observed that rs74504435 lies within open chromatin in several brain-relevant cell types, including iPSC-derived cortical neural progenitors and neurons49, primary astrocytes50, and iPSC-derived microglia48. We also observed a chromatin conformation capture contact (Capture C data) between this variant and EGFR in iPSC-derived cortical progenitors and neurons, primary astrocytes, and the microglial cell line HMC3. Using our RNA-seq datasets, we observed that EGFR was indeed expressed in these cell types48,49,50. From the Hi-C data, we observed that interactions between rs74504435 and EGFR exist in iPSC-derived neurons and oligodendrocytes. These findings are illustrated in Fig. 5b. Results for the other four top-ranked V2G pairs are summarized in Supplementary Data 7. While one of the three additional candidate SNPs (rs11585858) did show an open chromatin signature in iMg, the other 2 (rs4351 and rs2405442) resided in closed chromatin. Furthermore, none of the four V2G pairs was validated by Capture C in any of the cell types investigated; specifically, rs2405442 did not show any loop in any cell type, while rs11585858 had a loop to CFAP126 in HMC3 cells and rs4351 had loops to CYB561 in HMC3 and NPC, to KCNH6 in NPC, and to MARCHF10 in iMg. Therefore, we prioritized the V2G pair involving rs74504435 and EGFR for further experimental validation.
To validate the regulatory role for the region harboring rs74504435 and its influence on EGFR expression, we leveraged CRISPR interference (CRISPRi). We engineered the human microglial cell line HMC3 to stably express dCas9-KRAB (tagged with GFP). We transduced this line with lentivirus containing one of three sgRNAs targeting this region (G1, G2, and G4) or two control non-targeting sgRNAs (tagged with mCherry). After double selection for the presence of the guides and the dCas9-KRAB by FACS, we performed qPCR to assess EGFR levels. We found that two out of three targeting guides led to a consistent and significant decrease in EGFR expression levels compared to controls (one-way ANOVA p = 0.0002). G1 significantly decreased EGFR levels by 47% compared to the mean of the non-targeting controls (Tukey test p = 0.0004), and G2 by 27% (p = 0.04). G4 did not affect EGFR levels. Non-targeting guides did not show any effect when compared to a no-guide control. These results are shown in Fig. 5c.
Overall, our results support the hypothesis that the AD-associated variant rs74504435 is located within an enhancer region that regulates EGFR expression levels.
Discussion
Functionally characterizing non-coding AD GWAS loci is crucial for successful drug target discovery, and FG datasets can aid in this challenging task. However, FG data are often sparse and unharmonized, which hinders progress in this field. Here, we leveraged hipFG44, a tool that integrates FG data into FILER, and selected brain-profiled FG data to systematically validate non-coding AD signals for their regulatory potential. Our new framework combines confluent FG evidence and directionality checks to prioritize V2G pairs for functional validation.
Starting with AD GWAS variants from Bellenguez et al.5, we defined independent loci using the 1000G28 LD structure. For each locus, we evaluated functional contexts, conducted Bayesian colocalization of GWAS and eQTL signals, and identified putative causal variants, genes, and V2G pairs. We performed in silico validation using independent assays and data sources, followed by experimental validation with promoter-focused Capture C, ATAC-seq, and CRISPRi. This approach identified five plausible V2G pairs with highest tiers: chr1:161186243:C:A_NDUFS2 (tier 11), chr7:54881563:A:G_EGFR (tier 9), chr1:161186243:C:A_FCER1G (tier 9), chr7:100373690_PVRIG (tier 9), and chr17:63492371:G:A_ACE (tier 9). We successfully validated one of these pairs and its regulatory effect in a human microglia cell line.
Unlike prior post-GWAS methods, which rely on pre-selected top GWAS variants11,25, our framework utilized full GWAS summary statistics. We implemented QC steps to normalize genetic data against FG datasets in FILER, enabling a more systematic and comparable analysis of potential causal variants, genes, and V2G pairs. Our tiering system, which integrates support from eQTL, enhancer–promoter interactions, variant effects, and gene/protein expression, enabled improved prioritization of V2G pairs.
By leveraging full GWAS summary statistics and LD expansion in post-GWAS analyses, we uncovered additional candidate causal signals that were missed in previous studies5. When we compared the number of colocalization signals (the first step in our pipeline, GTEx brain data alone) to analyses that did not perform LD pruning or expansion11, we found that using the full summary statistics, rather than only top variants, yielded 4.5 times more candidate variants (9144 vs. 2024), 5 times more unique colocalized variants (2040 vs. 408), and twice as many candidate target genes (1529 vs. 762) across all GTEx data. Details for the candidate variants, colocalized variants, and corresponding targets from both the top variant analyses as well as the genome-wide approach we used in our current analyses can be found in Supplementary Data 8-11.
In our multi-tiered framework, colocalization served as an initial, relatively permissive filter rather than the final criterion. We then applied additional validation steps and external datasets to refine these signals. While others have used more stringent cutoffs as final criteria, we selected a >0.7 threshold at the first stage to capture a broader set of candidate variant-gene pairs (6971 at >0.7 vs 3404 at >0.9; Supplementary Data 3, 9). Importantly, over half of the signals confirmed in our in silico validation (23/41) would have been missed had we used stricter thresholds (>0.95 or >0.99) at the outset.
Using eQTL data for colocalization explained only a small fraction of the GWAS signals53. In our approach, we did not restrict analysis to signals with EPI support. Instead, we first prioritized eQTL colocalization and then sought additional in silico validation. 41 eQTLs were validated using independent eQTL datasets, and 37 out of 41 also had EPI support. EPI data can help prioritize V2G pairs when eQTL signals are weaker (e.g., loci chr7:100374211:A:G_CNPY4 and chr7:100373690:T:C_AP4M1). However, V2G pairs supported by both types of evidence are not always superior, as discrepancies may arise from biological differences or the uneven availability of FG assays, which limits cross-cell-type comparisons. More cell-type-specific assays, currently unavailable, could further improve V2G pair identification.
A potential concern when integrating multiple QTL datasets is the risk of circularity, where validation may inadvertently rely on overlapping information. In our study, we minimized this risk by strictly separating discovery from validation. Candidate loci were defined exclusively using GTEx v829 eQTL data, and validation was performed using independent datasets (MetaBrain39, MiGA42, and the eQTL Catalogue45), which are based on distinct cohorts, tissue sources, and analytical pipelines. Because these datasets do not share individuals or genotype data with GTEx, concordant colocalization signals represent independent replication rather than re-analysis of the same association. Importantly, we also incorporated orthogonal functional data types, including chromatin interaction datasets (3DGenome41, 4DGenome40, Nott et al.43), enhancer annotations (Nott et al.43), and TFBS information27, which are methodologically distinct from QTL-based approaches. This multi-tiered framework reduces the likelihood of circularity and increases the robustness of our gene prioritization strategy.
Using CRISPRi in a microglial cellular setting, we successfully validated EGFR as a target gene whose regulation is influenced by the AD variant rs74504435. The EGFR (Epidermal Growth Factor Receptor) gene product is a receptor tyrosine kinase that controls cell proliferation, survival, differentiation, and inflammation. In AD, it has been connected to disease progression54, with elevated levels associated with increased Aβ plaque formation. Additionally, EGFR inhibition modulates neuroinflammation and cognitive function in AD animal models55.
Interestingly, we found evidence supporting a role for the rs74504435-EGFR V2G pair in multiple brain-relevant cell types: (1) rs74504435 resides in open chromatin regions of neurons, microglia, and astrocytes in ATAC-seq data; (2) rs74504435 contacts the 3′ UTR region of EGFR in promoter-focused Capture C data generated from microglia, neurons and astrocytes; (3) rs74504435 also contacts the EGFR promoter in Hi-C data derived from iPSC-derived oligodendrocytes and neurons; (4) rs74504435 is associated with EGFR expression in single-nucleus RNA-seq data from astrocytes (p = 3.2 × 10−24, Z-score = −12.53) and oligodendrocyte progenitor cells (p = 0.003, Z-score = −2.95) derived from the dorsolateral prefrontal cortex in ROSMAP samples (Supplementary Fig. 4)56. In Bellenguez et al., another variant, rs76928645, was reported to interact with EGFR. Using our promoter-focused Capture C data and ATAC-seq data, we observed a loop via the Capture C experiment in NPC cells, but the loop does not reside in the open chromatin region. As a consequence, we conclude that EGFR’s functional role in AD likely involves several cell types, and further research is required to investigate whether the underlying mechanisms are distinct or shared across different brain cell types and whether pathogenesis is driven by a specific cell type.
Because APOE has an outsized effect on AD genetics and a complex haplotype structure, we removed this locus and the entire chr19 in the main analyses. To assess its contribution, we compared colocalization signals with and without chr19 (Supplementary Data 10). chr19 contributed 15% of candidate variants (1355/9144), covering 40% of candidate regions (417/1043), and accounted for 26% of colocalized signals. These results confirm the APOE region as a major driver of colocalization but also highlight substantial signals outside APOE and other chr19 loci, indicating additional genes and pathways likely contribute to AD pathogenesis.
There are two limitations to our study that can be addressed in future research. First, we restricted our analyses to SNPs. Indels were excluded because they are not consistently represented in most QTL datasets currently available, which limited our ability to integrate them across resources. Additionally, out of the three QTL data sources used for validation (eQTL Catalogue45, MetaBrain39, and MiGA42), only eQTL Catalogue had indels. Furthermore, indel detection and quantification vary substantially across genotyping arrays, in contrast to SNPs, which are more uniformly assayed. As QTL studies increasingly leverage whole-genome sequencing and standardized approaches for indel calling, incorporating indels into future pipelines will be important, especially given evidence for their role in LOAD (e.g., the TOMM40 region57). Second, our analyses were performed on GWAS data from European-ancestry populations. We did not include non-European GWAS in these analyses because of their small sample sizes. Our modular framework could, in principle, be adapted to diverse cohorts by incorporating ancestry-matched LD reference panels (e.g., 1000 Genomes Project28, Alzheimer’s Disease Sequencing Project58) and population-specific QTL datasets. The current scarcity of functional genomics resources from non-European populations is a major barrier. As more multi-ancestry GWAS and QTL datasets become available, extending this framework to diverse cohorts will be essential for improving the generalizability of genetic discoveries and for reducing health disparities in AD.
In conclusion, by combining an unbiased, confluent context identification framework, in silico V2G pair validation using QTL directionality, and a comprehensive scoring system that considers variant, gene, and V2G pair effects, we identified 41 AD-associated V2G pairs. The FILER-curated brain FG datasets and hipFG-harmonized FG data were instrumental to this success. Among the top findings, five V2G pairs achieved tier 9 or higher, and we demonstrated that AD-associated variant rs74504435 (chr7:54881563:A:G) resides in a regulatory region influencing EGFR expression, as validated by promoter-focused Capture C and CRISPRi functional experiments. Given that EGFR inhibitors are already approved for cancer therapy, EGFR could represent a promising candidate for repurposing as a therapeutic target for LOAD. Our framework and results provide valuable insights for future AD research.
Methods
Description of the AD GWAS
We analyzed Stage 1 genome-wide summary stats from Bellenguez et al.5, (downloaded from GWAS catalog http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90027001-GCST90028000/GCST90027158/). This dataset aggregates samples from International Genomics of Alzheimer’s Project (IGAP), with the inclusion of cohorts from European Alzheimer’s & Dementia BioBank (EADB), Alzheimer’s Disease Genetics Consortium (ADGC), and others, including clinically defined AD cases/controls and UK Biobank dementia samples, totaling 788,989 individuals with an effective sample size of 382,472. It contains 19,767,628 SNPs and 1,333,486 indels; our analysis focused on SNPs. For details, refer to the original publication5.
Identifying regions of interest for downstream analyses
We performed LD pruning using the 1000 Genomes Phase 3 EUR reference panel on all genome-wide significant variants (p < 5 × 10−8), identifying pairwise-independent tag variants (r2 < 0.7) using a 500 kb window. For each tag variant, we defined an analysis region by including variants that are in LD with the tag variant (r2 > =0.7), are within 1 M base pairs (bps), and are within 1000 variants of the tag. These variants, along with the tag variants, are considered candidate regulatory variants. Each analysis region is bounded by the outermost variants in LD with the tag. We note that not only the candidate variants, but all variants within these regions, even if some have no association with AD, will be included in colocalization analyses.
The LD-based thresholds used in our analyses are adjustable. Here, we use an r2 < 0.7 threshold for pruning the set of genome-wide significant (p < 5 × 10−8) variants. This threshold effectively removes variants in high LD (r > 0.83) and allows for ensuring the relative pairwise independence between the variants in the pruned set. At the LD expansion step, using r2 > 0.7 adds all the other variants that are in high LD (r = 0.83 or more) with the tag variants from the pruned set. Similar LD thresholds were used in previous studies (e.g., non-coding variant analysis59; or selecting a maximally informative set of SNPs60).
Genome partition analysis of all candidate regulatory variants
Variants were categorized into different genomic categories using the UCSC knownGene61,62, UCSC RepeatMasker62,63, and GENCODE v43 lncRNA annotations64 for the GRCh38/hg38 genome build. The 5′ UTR exons and introns, exons, introns, and 3′ UTR exons and introns were extracted from the knownGene annotation for each protein-coding gene. Promoter annotations were defined as 1000 bps genomic regions upstream of the transcription start site. To create a hierarchical genomic partition into disjoint 5′ UTR exon, 5′ UTR intron, 3′ UTR exon, 3′ UTR intron, promoter, exonic, and intronic regions (in this order), each region set was obtained by subtraction of the merged regions higher in the genomic hierarchy. For example, starting from merged 5′ UTR exonic regions, distinct 5′ UTR intronic regions were obtained by subtraction of 5′ UTR exonic regions from the merged 5′ UTR intronic regions, and 3′ UTR exon regions were obtained by subtracting both 5′ UTR exonic and intronic regions.
During analysis, GWAS variants were then assigned to mutually exclusive genomic element annotations using the created hierarchy: 5′ UTR exon > 5′ UTR intron > 3′ UTR exon > 3′ UTR intron > promoter > mRNA exon > mRNA intron. Overlaps, if any, with repeat element annotations (e.g., SINE, LINE) were also reported for all variants. Variants overlapping any of GENCODE lncRNA annotations were additionally classified into lncRNA exonic and/or lncRNA intronic variants. A variant not overlapping with any class of elements above (mRNA, lncRNA, repeat) was classified as intergenic.
Functional genomic annotations of all candidate regulatory variants
Genomic annotations from the FILER (functional genomic database which contains harmonized genomic annotation data across >30 primary data sources26) were used. 140 of these are used in the “Characterization”, i.e., discovery phase (including genome partition analyses, colocalization analyses, and unbiased confluent context identification), while 34 are used in “In silico validation”. See Supplementary Data 11 for details. In the discovery phase, the datasets included fundamental genome annotations and reference variant information (dbSNP65, GENCODE gene annotations64), genome-wide HOMER27 transcription factor binding tracks, and 140 brain-related FILER tracks (tissues and cells only) for variant characterization, including enhancers (10 from ROADMAP30, 55 from EpiMAP31), QTLs (26 from GTEx v829), and epigenetics (49 from ENCODE66).
Variant-Gene (V2G) pair identification (colocalization)
We hypothesized that putative causal variants affect gene expression in a cis-regulatory manner. To do so, we have performed a colocalization analysis within each genomic region to identify a shared (AD+eQTL) causal variant, if any. The colocalization analysis considers all possible causal configurations when computing the posteriors for colocalization (H4), including a null (H0) probability of no association for both AD and eQTL in the region, and the probabilities for AD-only association (H1), eQTL-only association (H2), and different/non-colocalized causal variants (H3). P(H4) provides an accurate measure of the colocalization probability, with the FDR corresponding to the cumulative probability of the alternative scenarios, P(H0) + P(H1) + P(H2) + P(H3) = 1 − P(H4), i.e., the probability that the AD and eQTL do not share the same causal variant in the region. A PP.H4.abf value of >0.95 is equivalent to FDR < 5%.
By aligning GWAS and eQTL signals via colocalization, we can identify genes most likely affected by disease-associated variants. Using the COLOC R package v5.2.367, we performed Bayesian colocalization on 9144 candidate variants against nominally significant eQTLs from 13 brain tissues in the GTEx v8 dataset. We defined a V2G pair as colocalized if the candidate variant was the most likely causal variant in the locus (SNP.H4.abf >0.5), the posterior probability for colocalization was greater than 0.7 (PP.H4.abf), and the locus contained more than one variant.
Transcription factor binding site (TFBS) disruption analyses of all candidate regulatory variants
HOMER (Hypergeometric Optimization of Motif EnRichment)27 is a custom motif database derived from high-quality ChIP-Seq data. A positional weight matrix (PWM) represents transcription factor (TF) DNA binding specificities. The delta PWM score (difference between reference and alternate alleles) estimates binding activity changes due to nucleotide variation. A candidate causal variant is selected for the next steps if it disrupts a TF binding site with a delta PWM score >|2| for any TF.
Enhancers overlap analyses of all candidate regulatory variants
Enhancers are DNA regulatory elements that activate gene transcription by forming chromatin loops to interact with target genes in a cell-type-specific manner. Databases like ROADMAP30 and EpiMAP31 catalog enhancers across various cell types and tissues. A candidate causal variant is considered potentially regulatory if it overlaps a brain enhancer found in either ROADMAP30 or EpiMAP31.
Unbiased confluent context identification of a putative causal V2G
We define a putative causal V2G pair as one with strong colocalization (“Variant-Gene (V2G) pair identification (colocalization)” section). The associated genes are considered putative causal genes. The putative causal variant is predicted to disrupt a TFBS (“Transcription factor binding site (TFBS) disruption analyses of all candidate regulatory variants” section) and overlap a tissue- or cell-type-specific enhancer (“Enhancers overlap analyses of all candidate regulatory variants” section) in any brain FG data. To identify which AD genetic signals may function in the brain-specific confluent context, we included all relevant tracks in FILER. We then required the colocalization tissue context to match the enhancers’, forming the final set of putative causal V2G pairs. A pair is excluded if it fails to meet any of these criteria.
Harmonizing in silico datasets by hipFG
In the validation phase, selected brain tracks include chromatin interactions (5 from 3DGenome41, 1 from 4DGenome40, 2 from Nott et al.43), QTLs (18 from eQTL Catalogue45, 4 from MetaBrain39, 4 from MiGA42). These tracks were generated from primary tissues/cell types but not cell lines. Each dataset was processed using hipFG (Harmonization and Integration Pipeline for Functional Genomics)44, an automated pipeline that standardizes, indexes, and integrates diverse functional genomics data (e.g., EPI, genomic intervals, QTLs) for scalable, searchable analysis.
In silico validation on candidate variant-gene pairs and genes
We validate selected V2G pairs and genes in silico using a set of independent FG resources (Fig. 1, “In silico validation”). Validation requires evidence from at least one, but not all, of the following categories. For V2G pairs, topologically associating domain (TAD) validation was based on TADs shared by the 3DGenome41, where a V2G pair was considered in silico validated if it overlapped with both anchors of the interaction. Enhancer–promoter interaction (EPI) validation included data profiled by PLAC-seq, 3C, 4C-Seq, 5C, Hi-C, ChIA-PET, and promoter-focused Capture C from Nott et al.43, 3DGenome41, and 4DGenome40 using only brain-related datasets; a V2G pair was in silico validated if it overlapped with both anchors of the interaction. Bulk tissue eQTL validation used brain tissue or region-related eQTLs from the eQTL Catalogue45 and the MetaBrain39, covering four brain regions. A V2G pair was in silico validated if it overlapped with, contained the same effect allele, and carried the same effect direction on the same gene as any eQTL profiled in these resources.
For genes, validation was based on two sources of evidence. Biological context was derived from the Human Protein Atlas (HPA)68,69; a gene was in silico validated if it was identified in the HPA as a protein-coding gene expressed in any brain region or cell type. Disease specificity was assessed using information from the Agora AMP-AD platform37, where a gene was in silico validated if it was included in the nominated list of genes.
Independent regulatory evidence and potential functions of selected variants
In addition to functional evidence from in silico validation, annotations from other sources can further support a variant’s regulatory or functional potential. As with previous analyses, evidence from any (not all) of the following categories is sufficient for confirming a variant as in silico functional.
Functional annotation based on active histone marks (H3K27ac) was derived from ENCODE66, where a variant was considered functional if it significantly overlapped any active histone mark peak (q-value < 5%). Open chromatin regions were analyzed using ATAC-seq data from ENCODE66, and a variant was considered functional if it was located within an ATAC-seq peak. Variant effect prediction was assessed using the Combined Annotation Dependent Depletion (CADD) score70 and RegulomeDB2 score71. CADD scores estimate variant deleteriousness, with higher scores indicating greater impact, while RegulomeDB2 integrates functional genomic assays to assign heuristic rankings for regulatory potential. A variant was considered functional if it had a CADD score >10 or a RegulomeDB2 score of 1a-1e. Genetic association evidence was evaluated using the GWAS Catalog72 and NIAGADS GenomicsDB73 to determine whether a putative causal variant was linked to Alzheimer’s disease (AD)-related traits. The GWAS Catalog contains variant-trait associations from over 130,000 GWASs across more than 18,000 traits (as of February 2025), and NIAGADS GenomicsDB is an interactive AD genetics database with 476.9K annotated variants from over 80 AD GWASs. A variant was considered functional if, in addition to being reported in Bellenguez et al.5 AD GWAS, it was also associated with a phenotype in any other AD GWAS.
Ranking of variant-gene (V2G) pairs (tier system)
Given the large number of variant-gene pairs identified after the confluence analyses, it remains challenging for wet-lab scientists to prioritize pairs for further functional work. We introduced a tiered system that integrates evidence from eQTLs (V2G_eQTL tier), EPIs (V2G_EPI tier), variants (V_tier), and genes (G_tier), each combining data from multiple sources (Methods: Ranking of variant-gene pairs (Tier system)).
The V2G_eQTL tier assessed four features: directionality across non-GTEx eQTLs (Metabrain39, eQTL Catalogue45), the presence of non-GTEx brain eQTLs, consistency between non-GTEx and GTEx brain-region eQTLs, and a negative, consistent Z-score across brain regions (we note that our directionality/Z-score consistency checks are not using formal hypothesis testing). Each criterion was assigned a score of 1, and the total V2G_eQTL tier score ranged from 1 to 4.
The V2G_EPI tier evaluated whether a V2G pair appeared in brain enhancer–promoter interaction datasets from 3DGenome41, 4DGenome40, or Nott et al.43. A score of 1 was assigned for each presence, and the total V2G_EPI tier ranged from 0 to 2.
The V_tier evaluates variant-level properties, including overlap with active histone marks, open chromatin regions, CADD scores70, RegulomeDB2 rankings71, and statistical significance in GWAS (GWAS Catalog72, NIAGADS GenomicsDB73). Variants meeting the ‘functional’ definition in each category were assigned a score of 1, resulting in a total V_tier range of 0–4.
The G_tier considered gene-level evidence, including nomination by the AMP-AD Agora37 and expression in brain regions or cell types according to the Human Protein Atlas (HPA)68,69. A score of 1 was assigned for each presence, and the total G_tier ranged from 0 to 2.
The overall tier for the V2G pair is the sum of all four components. The overall tier ranges from 1 to 11.
Hi-C data generation from iPSC cells for functional validation
Details of the iPSC cells used in this analysis can be found in the previous two studies51,74. To understand the potential function of the intergenic SNP rs74504435, we examined chromatin interactions using Hi-C analysis in iPSC-derived astrocytes, microglia cells, oligodendrocytes, and neurons52. An in situ Hi-C library was prepared using the protocol adapted from Rao et al.75. For each library, 450~550 million paired-end reads at 150 bps length were obtained. Sequencing data were processed using BWA76 to map each read end separately to the GRCh38 reference genomes. Duplicate and non-uniquely mapped reads were removed. For each library, over 270 million non-redundant, uniquely mapped, paired reads were used for further analysis. For robust enhancer–promoter interaction mapping, Chromatin loops were called using HiCorr77 to correct bias and LoopEnhance78 to remove noise.
Chromatin interaction analysis
We queried our existing genomic datasets (see Supplementary Data 12), including high-resolution promoter-focused Capture C, ATAC-seq, and RNA-seq from brain-relevant cell types (iPSC-derived neural progenitors and neurons48, iPSC-derived microglia and the human microglia cell line HMC349, and primary astrocytes50) to assess whether candidate variants were residing in open chromatin regions and contacting the promoter of an expressed gene.
Chromatin interactions calls obtained from promoter-focused Capture C datasets were available from the references provided for each cell type: briefly, paired-end reads were pre-processed using the HiCUP pipeline79 and aligned with bowtie280 to the reference genome, and significant interactions at 1-DpnII and 4-DpnII fragment resolutions were called using CHiCAGO80 with default parameters except for binsize, which was set to 2500. Open chromatin region calls from ATAC-seq datasets were also available from the same referenced studies: peaks were called using the ENCODE ATAC-seq pipeline (https://www.encodeproject.org/atac-seq/), selecting the resulting IDR optimal peaks. When more than two technical replicates were available for one cell type (i.e., for primary astrocytes, iPSC-derived microglia, and HMC3 cells), peaks were also called using a custom pipeline (“reproducible peaks”), where a peak is called if it’s present in the majority of the technical replicates available, and the union of IDR optimal peaks and reproducible peaks was used in the analysis. The queries to intersect these annotations with the candidate variants of interest were performed in GRCh37/hg19, and the results were lifted over to GRCh38 with the UCSC tool liftOver80 for comparison with other annotations.
EGFR sgRNA design
The genomic coordinate location for rs74504435 was obtained using the UCSC genome browser (build GRCh37/hg19). This genomic coordinate for rs74504435 plus and minus 200 bps was then entered into the software CHOPCHOP81 to generate a table of possible single guide RNAs (sgRNAs) for CRISPRi (repression) using Cas9. The Cas-OffFinder software82 was used to access the off-target mismatches of the possible sgRNAs generated from CHOPCHOP. After assessing the efficiency and off-target mismatches, three sgRNAs targeting rs7450443 (G1, G2, G4) were selected. Two non-targeting control guides (N2 and NTC3, Millipore Sigma) were used as negative controls for the CRISPRi experiments. As a positive control, we utilized a guide targeting an enhancer of the gene TSPAN14, since we previously validated this construct for CRISPRi experiments in HMC3.
Cloning the sgRNAs in a lentiviral plasmid
We leveraged a lentiviral vector created in the Chesi lab (SL33 Lenti-sgRNA(Tp2)-mCherry) to generate the backbone and the insert required for NEB HiFi DNA Assembly cloning. This lentiviral vector contains a U6 promoter driving the expression of one sgRNA; it also contains an sgRNA scaffold region and mCherry as a selection marker. To clone the sgRNA of interest into this vector, forward and reverse primers were designed and ordered through Azenta. The forward primer included: the sequence of the sgRNA of interest, complementary bases to the sgRNA scaffold, and overhanging bases (complementary to the vector - which is needed for Hifi cloning). The reverse primer was designed in such a way that when used with the forward primer in PCR, the amplicon would be the HiFi insert, i.e., the sequence of the sgRNA of interest, the sgRNA scaffold, and overhanging bases complementary to parts of the HiFi backbone. The SL33 Lenti-sgRNA(Tp2)-mCherry vector was digested with restriction enzymes XhoI and BsrG1 and run on a gel to isolate and extract the HiFi backbone. The forward and reverse primers were used in a PCR cycle with the SL33 Lenti-sgRNA(Tp2)-mCherry as template in order to obtain the HiFi insert. The backbone and the insert were combined in HiFi cloning to generate the respective sgRNA lentiviral plasmids. The plasmids were then transformed using NEB 5 alpha competent bacteria. After colony picking and miniprep, plasmids were submitted to Plasmidsaurus for Nanopore sequencing for validation. We cloned three sgRNAs designed to target rs74504435 (G1, G2, G4) and two non-targeting sgRNAs (N2 and NTC3).
Virus generation
On day 0, 400,000 HEK 293T cells per well were plated on PDL-coated 6-well plates. On day 1, cells were transfected with the respective sgRNA lentiviral plasmid in addition to envelope (Addgene plasmid #12259) and packaging (Addgene plasmid #12260) plasmids using the Lipofectamine 3000 reagent. On day 2, complete media changes were performed. On day 4, media from the HEK Cells (Virus Day Two) were collected, filtered through a 0.45-micron filter, aliquoted, and frozen/stored in the −80 °C.
Transducing HMC3 helper line cells
We generated a CRISPRi helper line by transducing the human microglial line HMC3 (ATCC #CRL-3304) with a lentiviral vector encoding Zim3 Krab dCas9 and GFP as a selection marker (Addgene plasmid #188778). This line (HMC3-zim3) was plated in a 6-well format - 200,000 cells per well on day 0. On day 1, cells were transduced with Day Two virus, and polybrene was used to aid in transduction efficiency. Due to the toxic nature of polybrene, if cells are exposed to it for too long, a complete media change was performed 18 h later (day 2). On the same day, cells were later moved from the 6-well format to a 100 mm plate in order to expand them for fluorescence-activated cell sorting (FACS). After cells reached about 80–100% confluency on the 100 mm plate, FACS was performed on these transduced cells as well as naïve HMC3 cells (used as a baseline for fluorescence) at the Flow Cytometry Core Laboratory at CHOP. Double positive cells were selected, i.e., the top 50% of cells that had both GFP fluorescence (indicating that these cells have dCas9) and mCherry fluorescence (indicating that these cells had the sgRNA lentiviral vector) were chosen. After recovering from flow sorting, cells were cultured and expanded until there were enough to perform qPCR experiments.
qPCR experiment
qPCR primers for EGFR (spanning exons 7–9), TSPAN14 (spanning exons 8–9), and GAPDH (spanning exons 2–3) were ordered through IDT. After cells were expanded post-FACS, cells were plated in a 6-well format (220,000 cells per well). 24 h after plating, cells were pelleted, and RNA extraction was immediately performed using the Qiagen RNeasy Plus micro kit and QIAShredder kit. Using Applied Biosystems’ High-Capacity cDNA reverse transcription kit, cDNA synthesis was also performed on the same day as RNA extraction. Power SYBRgreen PCR master mix from Applied Biosystems was used to perform standard comparative qPCR with primers for the two target and one housekeeping gene (EGFR, TSPAN14, and GAPDH). Three biological replicates in total were performed for each condition.
Extra information on the sgRNAs
Sequences of sgRNAs targeting rs7450443 w/o PAM sequence:
G1: 5′ TAGGCCTGAATGTCAATCAC 3′
G2: 5′ AGTGTGTTGAGTGTGAACAC 3′
G4: 5′ GTGTCAGCTCTCACTGAAAG 3′
Sequences of non-targeting guides
N2: 5′ CGCTTCCGCGGCCCGTTCAA 3′ was ordered from Millipore Sigma and was delivered in the form of a virus
NTC3: 5′ CCCGAGCAGTGGCTCGCTA 3′ is a non-targeting guide ordered from Millipore Sigma
Sequence of positive control - sgRNA targeted to the enhancer of TSPAN14 (Tspan14_enh): 5′ CTTAGGCGCTGCATACCGTA 3′
Data availability
The analyzed AD GWAS summary statistics data is available in the GWAS catalog ([https://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90027001-GCST90028000/GCST90027158/]). The selected FG datasets used in this study are available from the FILER database and in Supplementary Data 12.
Code availability
Reported analyses were performed using the adapted INFERNO pipeline (https://bitbucket.org/wanglab-upenn/bash-INFERNO).
References
Gatz, M. et al. Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. J. Gerontol. A Biol. Sci. Med. Sci. 52, M117–M125 (1997).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993).
Escott-Price, V. et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 138, 3673–3684 (2015).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015).
Heppner, F. L., Ransohoff, R. M. & Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 (2015).
Sims, R. et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384 (2017).
Novikova, G. et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 12, 1610 (2021).
Amlie-Wolf, A. et al. Inferring the molecular mechanisms of noncoding Alzheimer’s disease-associated genetic variants. J. Alzheimers Dis. 72, 301–318 (2019).
Bulger, M. & Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 144, 327–339 (2011).
Wittkopp, P. J. & Kalay, G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 13, 59–69 (2011).
Ong, C. T. & Corces, V. G. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 12, 283–293 (2011).
Corradin, O. & Scacheri, P. C. Enhancer variants: evaluating functions in common disease. Genome Med. 6, 85 (2014).
Schwartzentruber, J. et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 53, 392–402 (2021).
Bryois, J. et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat. Neurosci. 25, 1104–1112 (2022).
Kosoy, R. et al. Genetics of the human microglia regulome refines Alzheimer’s disease risk loci. Nat. Genet. 54, 1145–1154 (2022).
King, E. A., Davis, J. W. & Degner, J. F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15, e1008489 (2019).
Razuvayevskaya, O., Lopez, I., Dunham, I. & Ochoa, D. Genetic factors associated with reasons for clinical trial stoppage. Nat. Genet. 56, 1862–1867 (2024).
Minikel, E. V., Painter, J. L., Dong, C. C. & Nelson, M. R. Refining the impact of genetic evidence on clinical success. Nature 629, 624–629 (2024).
Schilder, B. M. & Raj, T. Fine-mapping of Parkinson’s disease susceptibility loci identifies putative causal variants. Hum. Mol. Genet. 31, 888–900 (2022).
Li, M. et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018).
Chen, Z. et al. Functional genomics provide key insights to improve the diagnostic yield of hereditary ataxia. Brain 146, 2869–2884 (2023).
Kuksa, P. P. et al. SparkINFERNO: a scalable high-throughput pipeline for inferring molecular mechanisms of non-coding genetic variants. Bioinformatics 36, 3879–3881 (2020).
Kuksa, P. P. et al. FILER: a framework for harmonizing and querying large-scale functional genomics knowledge. NAR Genom. Bioinform. 4, lqab123 (2022).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Hoon, D. S. B., Rahimzadeh, N. & Bustos, M. A. EpiMap: Fine-tuning integrative epigenomics maps to understand complex human regulatory genomic circuitry. Signal Transduct. Target. Ther. 6, 179 (2021).
Scoville, D. W., Kang, H. S. & Jetten, A. M. GLIS1-3: emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig. 4, 80 (2017).
Bu, S., Lv, Y., Liu, Y., Qiao, S. & Wang, H. Zinc finger proteins in neuro-related diseases progression. Front. Neurosci. 15, 760567 (2021).
Calderari, S. et al. Molecular genetics of the transcription factor GLIS3 identifies its dual function in beta cells and neurons. Genomics 110, 98–111 (2018).
von Bernhardi, R., Cornejo, F., Parada, G. E. & Eugenin, J. Role of TGFbeta signaling in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 9, 426 (2015).
Town, T. et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 14, 681–687 (2008).
AMP-AD AGORA.
Kerimov, N. et al. eQTL Catalogue 2023: new datasets, X chromosome QTLs, and improved detection and visualisation of transcript-level QTLs. PLoS Genet. 19, e1010932 (2023).
de Klein, N. et al. Brain expression quantitative trait locus and network analyses reveal downstream effects and putative drivers for brain-related diseases. Nat. Genet. 55, 377–388 (2023).
Teng, L., He, B., Wang, J. & Tan, K. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics 31, 2560–2564 (2015).
Wang, Y. et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151 (2018).
Lopes, K. P. et al. Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nat. Genet. 54, 4–17 (2022).
Nott, A. et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019).
Cifello, J. et al. hipFG: high-throughput harmonization and integration pipeline for functional genomics data. Bioinformatics 39, btad673 (2023).
Kerimov, N. et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 53, 1290–1299 (2021).
Nasser, J. et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243 (2021).
Selvarajan, I. et al. Integrative analysis of liver-specific non-coding regulatory SNPs associated with the risk of coronary artery disease. Am. J. Hum. Genet. 108, 411–430 (2021).
Burton, E.A. et. al. Variant‐to‐function mapping of late‐onset Alzheimer’s disease GWAS loci in human microglial models implicates RTFDC1 as an effector gene at the CASS4 locus. Alzheimers Dement. 20(Suppl 1), 089683 (2025).
Su, C. et al. 3D promoter architecture re-organization during iPSC-derived neuronal cell differentiation implicates target genes for neurodevelopmental disorders. Prog. Neurobiol. 201, 102000 (2021).
Littleton, S. H. et al. Variant-to-function analysis of the childhood obesity chr12q13 locus implicates rs7132908 as a causal variant within the 3’ UTR of FAIM2. Cell Genom. 4, 100556 (2024).
Moura, S. et al. Comparing Alzheimer’s genes in African, European, and Amerindian induced pluripotent stem cell-derived microglia. Alzheimers Dement. 21, e70031 (2025).
Akgun, B. et al. A genome-wide association study in Peruvians suggests new risk loci for Alzheimer disease. Preprint at medRxiv https://doi.org/10.1101/2023.11.29.23299201 (2023).
Mostafavi, H., Spence, J. P., Naqvi, S. & Pritchard, J. K. Systematic differences in discovery of genetic effects on gene expression and complex traits. Nat. Genet. 55, 1866–1875 (2023).
Choi, H. J., Jeong, Y. J., Kim, J. & Hoe, H. S. EGFR is a potential dual molecular target for cancer and Alzheimer’s disease. Front. Pharmacol. 14, 1238639 (2023).
Chiang, H. C., Wang, L., Xie, Z., Yau, A. & Zhong, Y. PI3 kinase signaling is involved in Abeta-induced memory loss in Drosophila. Proc. Natl. Acad. Sci. USA 107, 7060–7065 (2010).
Fujita, M. et al. Cell subtype-specific effects of genetic variation in the Alzheimer’s disease brain. Nat. Genet. 56, 605–614 (2024).
Chiba-Falek, O., Gottschalk, W. K. & Lutz, M. W. The effects of the TOMM40 poly-T alleles on Alzheimer’s disease phenotypes. Alzheimers Dement. 14, 692–698 (2018).
Leung, Y. Y. et al. Alzheimer’s Disease Sequencing Project release 4 whole genome sequencing dataset. Alzheimers Dement. 21, e70237 (2025).
Amlie-Wolf, A. et al. INFERNO: inferring the molecular mechanisms of noncoding genetic variants. Nucleic Acids Res. 46, 8740–8753 (2018).
Carlson, C. S. et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 74, 106–120 (2004).
Hsu, F. et al. The UCSC known genes. Bioinformatics 22, 1036–1046 (2006).
Perez, G. et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. 53, D1243–D1249 (2025).
Jurka, J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16, 418–420 (2000).
Mudge, J. M. et al. GENCODE 2025: reference gene annotation for human and mouse. Nucleic Acids Res. 53, D966–D975 (2025).
Sherry, S. T., Ward, M. & Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9, 677–679 (1999).
Luo, Y. et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Sjostedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, eaay5947 (2020).
Karlsson, M. et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 7, eabh2169 (2021).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Dong, S. et al. Annotating and prioritizing human non-coding variants with RegulomeDB v.2. Nat. Genet. 55, 724–726 (2023).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Greenfest-Allen, E. et al. NIAGADS Alzheimer’s GenomicsDB: A resource for exploring Alzheimer’s disease genetic and genomic knowledge. Alzheimers Dement. 20, 1123–1136 (2024).
Ramirez, A. M. et al. Ancestral genomic functional differences in oligodendroglia: implications for Alzheimer’s disease. Alzheimers Dement. 21, e70593 (2025).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Lu, L. et al. Robust Hi-C maps of enhancer-promoter interactions reveal the function of non-coding genome in neural development and diseases. Mol. Cell 79, 521–534.e515 (2020).
Zhang, S. et al. DeepLoop robustly maps chromatin interactions from sparse allele-resolved or single-cell Hi-C data at kilobase resolution. Nat. Genet. 54, 1013–1025 (2022).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Acknowledgements
We would like to thank the members of the Alzheimer’s Disease Genetics Consortium for their feedback. The results published here are in whole or in part based on data obtained from Agora, a platform initially developed by the NIA-funded AMP-AD consortium that shares evidence in support of AD target discovery. Agora is available at: https://doi.org/10.57718/agora-adknowledgeportal. Y.Y.L., P.P.K., L.C., J.C., E.G.-A., O.V., G.D.S., L.-S.W. were supported by the U24-AG041689, U54-AG052427, U01-AG032984. Y.Y.L. was funded by R01-AG085518, R01-AG086467, Biomarkers Across Neurodegenerative Diseases (BAND 3) (award number 18062), co-funded by Michael J Fox Foundation, Alzheimer’s Association, Alzheimer’s Research UK, and the Weston Brain institute. L.B., S.L., N.T., and A.C. was funded by R35-HG011959. S.F.A.G. was funded by R01-HL143790, R01-AG057516, R01-HD056465 and the Daniel B. Burke Endowed Chair for Diabetes Research. S.M., A.R., K.C., L.W. and J.M.V. were supported by the U01-AG072579. F.J. was supported by R01-HG009658. G.W .was supported by R01-AG076901 and the Urbut Family Foundation. R.F. and P.D.J. were supported by U01-AF072572.
Author information
Authors and Affiliations
Contributions
Y.Y.L., P.P.K., A.C., G.D.S., and L.-S.W. conceived and designed the experiments. Y.Y.L., P.P.K., J.C., and L.C. performed data analyses. L.C., J.C., L.B., S.L., N.T., S.M., A.R., K.C., and R.F. carried out the data production and data generation under the supervision of Y.Y.L., S.F.A.G., A.C., F.J., G.W., P.D.J., J.M.V., and L.W. P.P.K., E.G.-A., and O.V. provided IT support. Y.Y.L., P.P.K., S.F.A.G., A.C., and L.-S.W. wrote and edited the manuscript. Y.Y.L., P.D.J., J.M.V., S.F.A.G., A.C., G.D.S., and L.-S.W. secured funding. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leung, Y.Y., Kuksa, P.P., Carter, L. et al. Integrated genomic analysis and CRISPRi implicates EGFR in Alzheimer’s disease risk. npj Dement. 1, 42 (2025). https://doi.org/10.1038/s44400-025-00049-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44400-025-00049-5
